Cytotoxic and Radical Scavenging Nor-Dammarane Triterpenoids from Viburnum mongolicum

Abstract
:1. Introduction

2. Results and Discussion
2.1. Chemistry

{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1ax | 1.03 (ddd, 13.8, 13.3, 3.6) | 1.00 (ddd, 14.0, 13.5, 3.6) | 1.02 (ddd, 14.0, 13.2, 3.6) | 1.47 (ddd, 14.0, 13.3, 3.6) | 1.44 (ddd, 13.8, 13.6, 3.6) | 1.45 (ddd, 13.8, 13.6, 3.6) |
| 1eq | 1.69 (ddd, 13.3, 4.0, 3.6) | 1.66 (ddd, 13.5, 4.0, 3.6) | 1.68 (ddd, 13.2, 4.0, 3.6) | 1.95 (ddd, 13.3, 4.0, 3.6) | 1.94 (ddd, 13.3, 4.0, 3.6) | 1.93 (ddd, 13.5,3.8, 3.6) |
| 2ax | 1.59 (m) | 1.56 (m) | 1.59 (m) | 2.38 (ddd, 14.0, 13.3, 3.6) | 2.36 (ddd, 13.8, 13.6, 3.6) | 2.39 (ddd, 13.8, 13.6, 3.6) |
| 2eq | 1.67 (m) | 1.64 (m) | 1.66 (m) | 2.52 (ddd, 13.3, 4.0, 3.6) | 2.48 (ddd, 13.3, 4.0, 3.6) | 2.47 (ddd, 13.5, 3.8, 3.6) |
| 3 | 3.21 (dd, 14.0, 4.0) | 3.17 (dd, 14.0, 4.0) | 4.39 (dd, 14.0, 4.0) | - | - | - |
| 5 | 0.78 (dd, 14.0, 3.8) | 0.76 (dd, 13.8, 3.8) | 0.79 (dd, 13.8, 3.6) | 1.37 (dd, 13.8, 3.8) | 1.35 (dd, 13.5, 3.0) | 1.36 (dd, 13.0, 3.0) |
| 6ax | 1.47 (m) | 1.46 (m) | 1.48 (m) | 1.44 (m) | 1.42 (m) | 1.48 (m) |
| 6eq | 1.62 (m) | 1.58 (m) | 1.60 (m) | 1.58 (m) | 1.56 (m) | 1.59 (m) |
| 7ax | 1.58 (ddd, 14.0, 13.5, 3.8) | 1.55 (ddd, 14.0, 13.3, 3.8) | 1.57 (ddd, 14.0, 13.5, 3.8) | 1.54 (ddd, 13.8, 13.3, 3.8) | 1.53 (m) | 1.54 (m) |
| 7eq | 2.18 (ddd, 13.5, 4.0, 3.6) | 2.15 (ddd, 13.3, 4.0, 3.6) | 2.17 (ddd, 13.5, 4.0, 3.6) | 2.20 (m) | 2.18 (m) | 2.19 (m) |
| 9 | 1.55 (dd, 13.6, 3.0) | 1.53 (dd, 13.6, 3.2) | 1.55 (dd, 13.6, 3.2) | 1.51 (dd, 13.0, 3.0) | 1.49 (dd, 13.8, 3.0) | 1.52 (dd, 13.5, 3.0) |
| 11ax | 1.27 (m) | 1.26 (m) | 1.27 (m) | 1.27 (m) | 1.26 (m) | 1.24 (m) |
| 11eq | 1.84 (m) | 1.87 (m) | 1.89 (m) | 1.92 (m) | 1.91 (m) | 1.84 (m) |
| 12 | 3.62 (m) | 3.47 (m) | 3.49 (m) | 4.47 (m) | 4.44 (m) | 3.80 (m) |
| 13 | 1.44 (dd, 13.8, 3.2) | 2.16 (dd, 13.8, 3.2) | 2.08 (dd, 13.8, 3.2) | 2.12 (dd, 13.0, 3.0) | 2.11 (dd, 13.6, 3.0) | 2.78 (dd, 13.6, 8.0) |
| 15ax | 1.17 (ddd, 13.8, 13.0, 3.6) | 1.76 (ddd, 13.8, 13.2, 3.6) | 1.21 (ddd, 13.8, 13.3, 3.6) | 3.48 (dd, 13.0, 3.0) | 3.44 (dd, 13.6, 3.0) | 4.55 (d, 7.6) |
| 15eq | 1.56 (ddd, 13.0, 4.0, 3.6) | 1.77 (ddd, 13.2, 4.0, 3.6) | 1.68 (ddd, 13.2, 4.0, 3.6) | - | - | - |
| 16ax | 1.80 (m) | 1.70 (m) | 1.72 (m) | 2.02 (m) | 1.98 (m) | 5.63 (dd, 10.8, 7.6) |
| 16eq | 2.05 (m) | 2.02 (m) | 2.04 (m) | 2.35 (m) | 2.32 (m) | - |
| 17 | 2.29 (m) | 2.22 (m) | 2.88 (m) | 3.98 (m) | 4.23 (m) | 6.62 (dd, 10.8, 8.0) |
| 18 | 0.91 (s) | 0.96 (s) | 1.06 (s) | 0.87 (s) | 0.85 (s) | 0.89 (s) |
| 19 | 0.89 (s) | 0.93 (s) | 0.99 (s) | 0.99 (s) | 0.96 (s) | 0.98 (s) |
| 21 | 1.45 (s) | 1.35 (s) | 2.24 (s) | - | - | - |
| 22ax | 7.86 (d, 5.7) | 1.84 (m) | - | - | - | - |
| 22eq | - | 1.95(m) | - | - | - | - |
| 23ax | 6.06 (d, 5.7) | 1.98 (m) | - | - | - | - |
| 23eq | - | 2.05(m) | - | - | - | - |
| 24 | - | 4.93 (m) | - | - | - | - |
| 28 | 0.79 (s) | 0.77 (s) | 0.78 (s) | 0.79 (s) | 0.77 (s) | 0.78 (s) |
| 29 | 0.99 (s) | 0.97 (s) | 0.98 (s) | 1.01 (s) | 0.98 (s) | 1.03 (s) |
| 30 | 0.84 (s) | 0.89 (s) | 0.91 (s) | 1.24 (s) | 1.19 (s) | 1.26 (s) |
| Me | - | - | 2.01 (s) | 2.10 (s) | 2.08 (s) | - |
| O Me | - | 3.26 (s) | - | 3.21 (s) | - | - |
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 38.8 (t) | 38.8 (t) | 39.0 (t) | 40.1 (t) | 39.6 (t) | 39.6 (t) |
| 2 | 27.1 (t) | 27.1 (t) | 27.2 (t) | 34.2 (t) | 33.9 (t) | 33.8 (t) |
| 3 | 78.5 (d) | 78.4 (d) | 81.3 (d) | 218.8 (s) | 218.7 (s) | 219.2 (s) |
| 4 | 38.9 (s) | 38.8 (s) | 38.9 (s) | 47.6 (s) | 47.2 (s) | 47.7 (s) |
| 5 | 55.6 (d) | 56.0 (d) | 56.0 (d) | 55.5 (d) | 55.2 (d) | 55.8 (d) |
| 6 | 17.8 (t) | 17.9 (t) | 18.0 (t) | 19.7 (t) | 20.2 (t) | 20.1(t) |
| 7 | 32.8 (t) | 33.4 (t) | 33.3 (t) | 34.6 (t) | 34.5 (t) | 35.1 (t) |
| 8 | 39.8 (s) | 39.0 (s) | 39.0 (s) | 39.9 (s) | 39.8 (s) | 39.7 (s) |
| 9 | 50.1 (d) | 50.8 (d) | 51.0 (d) | 50.1 (d) | 49.9 (d) | 49.7 (d) |
| 10 | 37.4 (s) | 37.7 (s) | 37.5 (s) | 37.2 (s) | 36.7 (s) | 37.3 (s) |
| 11 | 32.3 (t) | 32.7 (t) | 32.4 (t) | 32.7 (t) | 32.1 (t) | 35.8 (t) |
| 12 | 70.7 (d) | 71.9 (d) | 71.2 (d) | 73.1 (d) | 72.9 (d) | 71.0 (d) |
| 13 | 49.6 (d) | 50.9 (d) | 50.9 (d) | 47.7 (d) | 47.4 (d) | 58.1 (d) |
| 14 | 51.7 (s) | 53.3 (s) | 51.1 (s) | 55.3 (s) | 54.8 (s) | 60.3 (s) |
| 15 | 31.1 (t) | 33.2 (t) | 31.8 (t) | 69.9 (d) | 69.4 (d) | 68.0 (d) |
| 16 | 26.4 (t) | 31.1 (t) | 26.8 (t) | 39.4 (t) | 39.0 (t) | 132.1 (d) |
| 17 | 47.0 (d) | 50.7 (d) | 52.9 (d) | 76.4 (d) | 71.5 (d) | 158.8 (d) |
| 18 | 14.8 (q) | 15.6 (q) | 15.2 (q) | 16.1 (q) | 16.2 (q) | 16.0 (q) |
| 19 | 15.6 (q) | 16.7 (q) | 16.0 (q) | 18.9 (q) | 18.8 (q) | 20.5 (q) |
| 20 | 91.7 (s) | 80.4 (s) | 215.4 (s) | - | - | - |
| 21 | 22.5 (q) | 30.5 (q) | 29.4 (q) | - | - | - |
| 22 | 161.3 (d) | 29.2 (t) | - | - | - | - |
| 23 | 120.5 (d) | 33.8 (t) | - | - | - | - |
| 24 | 172.1 (s) | 107.5 (d) | - | - | - | - |
| 28 | 15.4 (q) | 15.2 (q) | 15.3 (q) | 16.0 (q) | 15.9 (q) | 15.9 (q) |
| 29 | 28.0 (q) | 27.9 (q) | 28.0 (q) | 28.1 (q) | 28.0 (q) | 28.0 (q) |
| 30 | 17.2 (q) | 17.9 (q) | 16.7 (q) | 16.2 (q) | 14.9 (q) | 19.9 (q) |
| C=O | - | - | 172.9 (s) | 173.1 (s) | 172.6 (s) | - |
| Me | - | - | 21.7 (q) | 22.1 (q) | 21.3 (q) | - |
| OMe | - | 55.4 (q) | - | 58.6 (q) | - | - |
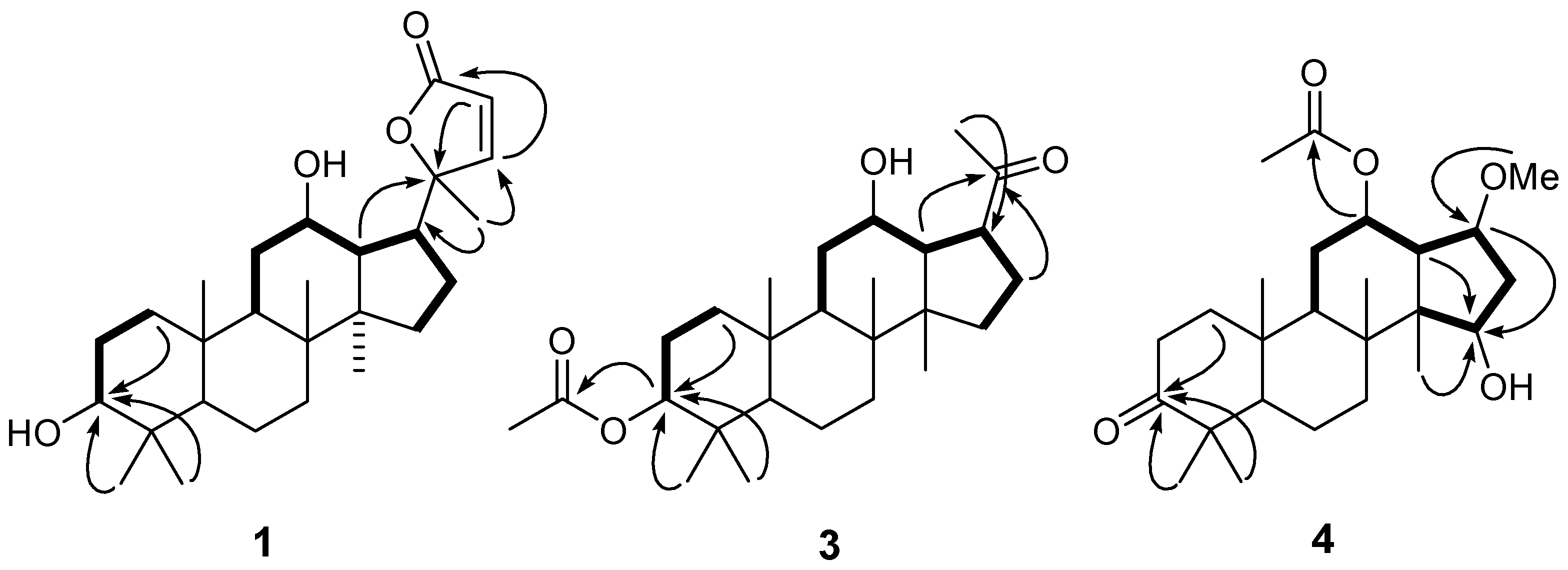
 ) and 1H-1H COSY (
) and 1H-1H COSY (  ) correlations of compounds 1, 3 and 4.
) correlations of compounds 1, 3 and 4.
2.2. Cytotoxic Activity
| Cell lines | |||||||
|---|---|---|---|---|---|---|---|
| A-549 | BGC-823 | HepG2 | HL-60 | MCF-7 | SMMC-7721 | W480 | |
| 1 | 79.4 | 80.3 | 80.2 | 78.8 | 81.8 | 80.5 | 81.6 |
| 2 | 29.7 | 29.6 | 29.4 | 29.4 | 27.1 | 30.1 | 24.9 |
| 3 | 63.2 | 66.8 | 51.1 | 68.2 | 53.5 | 50.1 | 59.7 |
| 4 | 12.3 | 15.9 | 14.3 | 17.0 | 15.1 | 14.7 | 17.1 |
| 5 | 12.7 | 12.2 | 12.8 | 13.8 | 11.3 | 11.7 | 18.3 |
| 6 | 14.3 | 9.4 | 10.1 | 11.1 | 10.4 | 9.7 | 19.1 |
| 7 | 31.9 | 31.2 | 30.7 | 32.2 | 28.1 | 29.9 | 27.6 |
| 8 | 76.3 | 68.7 | 66.9 | 72.3 | 76.2 | 70.8 | 69.4 |
| Doxorubicin | 18.3 | 14.7 | 22.0 | 31.7 | 24.9 | 35.4 | 15.9 |
2.3. Radical Scavenging Activity
| DPPH IC50 | ABTS·+ IC50 | |
|---|---|---|
| 1 | 276.8 ± 5 | 309.1 ± 6 |
| 2 | 253.1 ± 5 | 241.0 ± 5 |
| 3 | 244.7 ± 5 | 256.1 ± 5 |
| 4 | 101.3 ± 4 | 68.1 ± 4 |
| 5 | 99.7 ± 3 | 59.2 ± 3 |
| 6 | 94.1 ± 3 | 54.6 ± 3 |
| 7 | 261.4 ± 5 | 299.1 ± 5 |
| 8 | 241.0 ± 5 | 271.3 ± 6 |
| Trolox | 42.8 ± 1 | 80.1 ± 3 |
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
 = +0.4 (c = 0.40, MeOH). UV (CDCl3) λmax(log ε): 198 (0.10) nm. IR (KBr) νmax 3450, 2945, 2870, 1702, 1640, 1605, 1463, 1384, 1247, 1107 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 430 ([M]+). HR-ESI-MS (pos.) m/z: 453.2984 ([M+Na]+, C27H42O4Na. calcd. 453.2981). = +27.1 (c = 0.11, MeOH). UV (CDCl3) λmax(log ε): 205 (0.56) nm. IR (KBr) νmax 3425, 2945, 2876, 1630, 1383, 1276, 1033 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 448 ([M]+). HR-ESI-MS (pos.) m/z: 471.3454 ([M+Na]+, C28H48O4Na. calcd. 471.3450). = +46.3 (c = 0.15, MeOH). UV (CDCl3) λmax(log ε): 200 (0.72) nm. IR (KBr) νmax 3440, 2968, 2945, 2870, 1705, 1463, 1384, 1365, 1177, 1042, 1007 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 418 ([M]+). HR-ESI-MS (pos.) m/z: 441.2978 ([M+Na]+, C26H42O4Na. calcd. 441.2981). = +15.4 (c = 1.210, MeOH). UV (CDCl3) λmax(log ε): 206 (0.66) nm. IR (KBr) νmax 3450, 1735, 1635, 1345 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 420 ([M]+). HR-ESI-MS (pos.) m/z: 443.2774 ([M+Na]+, C25H40O5Na. calcd. 443.2773). = +11.4 (c = 1.130, MeOH). UV (CDCl3) λmax(log ε): 205 (0.69) nm. IR (KBr) νmax 3445, 1730, 1715, 1625, 1034 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 406 ([M]+). HR-ESI-MS (pos.) m/z: calcd. 429.2613 ([M+Na]+, C24H38O5Na. calcd. 429.2617). = +10.7 (c = 0.460, MeOH). UV (CDCl3) λmax(log ε): 202 (0.54) nm. IR (KBr) νmax 3444, 1736, 1630, 1034 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 346 ([M]+). HR-ESI-MS (pos.) m/z: 369.2405 ([M+Na]+, C22H34O3Na. calcd. 369.2406).
= +0.4 (c = 0.40, MeOH). UV (CDCl3) λmax(log ε): 198 (0.10) nm. IR (KBr) νmax 3450, 2945, 2870, 1702, 1640, 1605, 1463, 1384, 1247, 1107 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 430 ([M]+). HR-ESI-MS (pos.) m/z: 453.2984 ([M+Na]+, C27H42O4Na. calcd. 453.2981). = +27.1 (c = 0.11, MeOH). UV (CDCl3) λmax(log ε): 205 (0.56) nm. IR (KBr) νmax 3425, 2945, 2876, 1630, 1383, 1276, 1033 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 448 ([M]+). HR-ESI-MS (pos.) m/z: 471.3454 ([M+Na]+, C28H48O4Na. calcd. 471.3450). = +46.3 (c = 0.15, MeOH). UV (CDCl3) λmax(log ε): 200 (0.72) nm. IR (KBr) νmax 3440, 2968, 2945, 2870, 1705, 1463, 1384, 1365, 1177, 1042, 1007 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 418 ([M]+). HR-ESI-MS (pos.) m/z: 441.2978 ([M+Na]+, C26H42O4Na. calcd. 441.2981). = +15.4 (c = 1.210, MeOH). UV (CDCl3) λmax(log ε): 206 (0.66) nm. IR (KBr) νmax 3450, 1735, 1635, 1345 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 420 ([M]+). HR-ESI-MS (pos.) m/z: 443.2774 ([M+Na]+, C25H40O5Na. calcd. 443.2773). = +11.4 (c = 1.130, MeOH). UV (CDCl3) λmax(log ε): 205 (0.69) nm. IR (KBr) νmax 3445, 1730, 1715, 1625, 1034 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 406 ([M]+). HR-ESI-MS (pos.) m/z: calcd. 429.2613 ([M+Na]+, C24H38O5Na. calcd. 429.2617). = +10.7 (c = 0.460, MeOH). UV (CDCl3) λmax(log ε): 202 (0.54) nm. IR (KBr) νmax 3444, 1736, 1630, 1034 cm−1. 1H-NMR (CDCl3, 600 MHz) data see Table 1, 13C-NMR (CDCl3, 125 MHz) data see Table 2. EI-MS m/z: 346 ([M]+). HR-ESI-MS (pos.) m/z: 369.2405 ([M+Na]+, C22H34O3Na. calcd. 369.2406).3.4. Cytotoxicity Assay in Vitro
3.5. Microplate Assay for Radical Scavenging Activity DPPH
3.6. 2,2'-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) Radical Cation Decolorization Assay

4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 1–8 are available from the authors.
References
- Lobstein, A.; Haan-Archipoff, G.; Englert, J.; Kuhry, J.G.; Anton, R. Chemotaxonomical investigation in the genus Viburnum. Phytochemistry 1999, 50, 1175–1180. [Google Scholar] [CrossRef]
- Jordheim, M.; Giske, N.H.; Andersen, Ø.M. Anthocyanins in Caprifoliaceae. 2007, 35, 153–159.
- Kubo, M.; Chen, I.S.; Fukuyama, Y. Vibsane-type diterpenes from Taiwanese Viburnum odoratissimum. Chem. Pharm. Bull. 2001, 49, 242–245. [Google Scholar] [CrossRef]
- Lobstein, A.; Weniger, B.; Malécot, V.; Um, B.H.; Alzate, F.; Anton, R. Polyphenolic content of two Colombian Viburnum species (Caprifoliaceae). Biochem. Syst. Ecol. 2003, 31, 95–97. [Google Scholar] [CrossRef]
- Wang, L.Q.; Chen, Y.G.; Xu, J.J.; Liu, Y.; Li, X.M.; Zhao, Y. Compounds from Viburnum species and their biological activities. Chem. Biodivers. 2008, 5, 1879–1899. [Google Scholar] [CrossRef]
- Parveen, M.; Khan, M.S.; Shafiullah; Ilyas, M. Luteolin 3'-xylosyl(1→2) glucoside from viburnum grandifolium. Phytochemistry 1998, 49, 2535–2538. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Marzouk, M.S.A.; Moharram, F.A.; El-Sayed, M.M.; Baiuomy, A.R. Phytochemical constituents and hepatoprotective activity of Viburnum tinus. Phytochemistry 2005, 66, 2780–2786. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Minoshima, Y.; Kishimoto, Y.; Chen, I.S.; Takahashi, H.; Esumi, T. Iridoid glucosides and p-coumaroyl iridoids from Viburnum luzonicum and their cytotoxicity. J. Nat. Prod. 2004, 67, 1833–1838. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Minoshima, Y.; Kishimoto, Y.; Chen, I.S.; Takahashi, H.; Esumi, T. Cytotoxic iridoid aldehydes from Taiwanese Viburnum luzonicum. Chem. Pharm. Bull. 2005, 53, 125–127. [Google Scholar] [CrossRef]
- Tomassini, L.; Brkic, D. Iridoid glucosides from Viburnum lantana var. discolor. Planta Med. 1997, 63, 485–486. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Minami, H.; Ichikawa, R.; Takuchi, K. Hydroperoxylated guaiane-type sesquiterpenes from Viburnum awabuki. Phytochemistry 1996, 42, 741–746. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Fujii, H.; Minami, H.; Takahashi, H.; Kubo, M. Neovibsanin F and its congeners, rearranged vibsane-type diterpenes from Viburnum suspensum. J. Nat. Prod. 2006, 69, 1098–1100. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Kubo, M.; Minami, H.; Yuasa, H.; Matsuo, A.; Fujii, T.; Morisaki, M.; Harada, K. Rearranged vibsane-type diterpenes from Viburnum awabuki and photochemical reaction of vibsanin B. Chem. Pharm. Bull. 2005, 53, 72–80. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Minami, H.; Kagawa, M.; Kodama, M.; Kawazu, K. Chemical conversion of vibsanin C to vibsanin E and structure of 3-hydroxyvibsanin E from viburnum awabuki. J. Nat. Prod. 1999, 62, 337–339. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Minami, H.; Matsuo, A.; Kitamura, K.; Akizuki, M.; Kubo, M.; Kodama, M. Seven-membered vibsane-type diterpenes with a 5,10-cis relationship from Viburnum awabuki. Chem. Pharm. Bull. 2002, 50, 368–371. [Google Scholar] [CrossRef]
- Shen, Y.C.; Lin, C.L.; Chien, S.C.; Khalil, A.T.; Ko, C.L.; Wang, C.H. Vibsane diterpenoids from the leaves and flowers of Viburnum odoratissimum. J. Nat. Prod. 2004, 67, 74–77. [Google Scholar] [CrossRef]
- El-Gamal, A.A.; Wang, S.K.; Duh, C.Y. New diterpenoids from Viburnum awabuki. J. Nat. Prod. 2004, 67, 333–336. [Google Scholar] [CrossRef]
- Shen, Y.C.; Prakash, C.V.; Wang, L.T.; Chien, C.T.; Hung, M.C. New vibsane diterpenes and lupane triterpenes from Viburnum odoratissimum. J. Nat. Prod. 2002, 65, 1052–1055. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Minami, H.; Fujii, H.; Tajima, M. Triterpenoids from Viburnum suspensum. Phytochemistry 2002, 60, 765–768. [Google Scholar] [CrossRef]
- Kagawa, M.; Minami, H.; Nakahara, M.; Takahashi, H.; Takaoka, S.; Fukuyama, Y. Oleanane-type triterpenes from Viburnum awabuki. Phytochemistry 1998, 47, 1337–1341. [Google Scholar] [CrossRef]
- Rios, M.Y.; González-Morales, A.; Villarreal, M.L. Sterols, triterpenes and biflavonoids of Viburnum jucundum and cytotoxic activity of ursolic acid. Planta Med. 2001, 67, 683–684. [Google Scholar] [CrossRef]
- Kim, M.Y.; Iwai, K.; Onodera, A.; Matsue, H. Identification and antiradical properties of anthocyanins in fruits of Viburnum dilatatum thunb. J. Agric. Food Chem. 2003, 51, 6173–6137. [Google Scholar] [CrossRef]
- Zhu, X.D.; Dong, X.J.; Luo, S.D. Phenolic Compounds from Viburnum cylindricum. Helv. Chim. Acta 2005, 88, 339–342. [Google Scholar] [CrossRef]
- Machida, K.; Nakano, Y.; Kikuchi, M. Phenolic glycosides from Viburnum dilatatum. Phytochemistry 1991, 30, 2013–2014. [Google Scholar] [CrossRef]
- Tu, L.; Zhao, Y.; Yu, Z.Y.; Cong, Y.W.; Xu, G.; Peng, L.Y.; Zhang, P.T.; Cheng, X.; Zhao, Q.S. Six New Dammarane Triterpenoids from Viburnum cylindricum. Helv. Chim. Acta 2008, 91, 1578–1587. [Google Scholar] [CrossRef]
- Wang, F.; Guan, Y.J. Cytotoxic nor-dammarane triterpenoids from Dysoxylum hainanense. Fitoterapia 2012, 83, 13–17. [Google Scholar] [CrossRef]
- Tundis, R.; Bonesi, M.; Deguin, B.; Loizzo, M.R.; Menichini, F.; Conforti, F.; Menichini, F. Cytotoxic activity and inhibitory effect on nitric oxide production of triterpene saponins from the roots of Physospermum verticillatum (Waldst and Kit) (Apiaceae). Bioorg. Med. Chem. 2009, 17, 4542–4547. [Google Scholar]
- Jirapast, S.; Serm, S.; Pongpun, S.; Suttira, K.; Jonkolnee, J.A.; Santi, T.P. Two new cytotoxic isomeric indole alkaloids from the roots of Nauclea orientalis. Fitoterapia 2010, 81, 830–833. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Wei, D.; Wang, X.; Luo, J.; Kong, L. Cytotoxic tirucallane C26 triterpenoids from the stem barks of Aphanamixis grandifolia. Phytochemistry 2010, 71, 2199–2204. [Google Scholar] [CrossRef]
- Brem, B.; Seger, C.; Pacher, T.; Hartl, M.; Hadacek, F.; Hofer, O.; Vajrodaya, S.; Greger, H. Antioxidant dehydrotocopherols as a new chemical character of Stemona species. Phytochemistry 2004, 65, 2719–2729. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, X.; Wang, W. Cytotoxic and Radical Scavenging Nor-Dammarane Triterpenoids from Viburnum mongolicum. Molecules 2013, 18, 1405-1417. https://doi.org/10.3390/molecules18021405
Wang X, Wang W. Cytotoxic and Radical Scavenging Nor-Dammarane Triterpenoids from Viburnum mongolicum. Molecules. 2013; 18(2):1405-1417. https://doi.org/10.3390/molecules18021405
Chicago/Turabian StyleWang, Xiaohua, and Wei Wang. 2013. "Cytotoxic and Radical Scavenging Nor-Dammarane Triterpenoids from Viburnum mongolicum" Molecules 18, no. 2: 1405-1417. https://doi.org/10.3390/molecules18021405