The Histone Deacetylase Inhibitors MS-275 and SAHA Suppress the p38 Mitogen-Activated Protein Kinase Signaling Pathway and Chemotaxis in Rheumatoid Arthritic Synovial Fibroblastic E11 Cells



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. MS-275 and SAHA Suppressed p38 MAPK Signaling Pathway

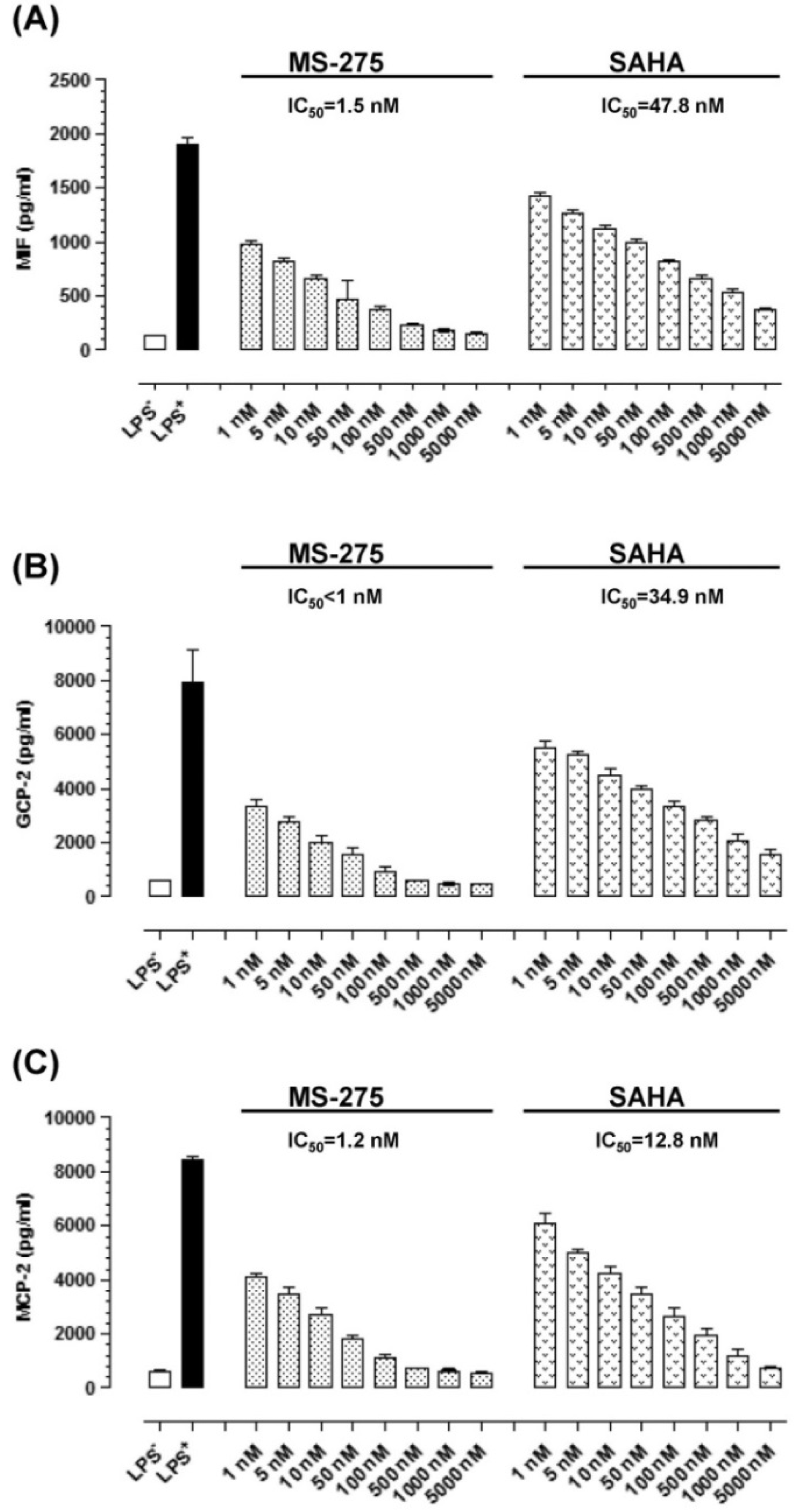
2.2. MS-275 and SAHA Suppressed Secretion of Granulocyte Chemotactic Protein-2 (GCP-2), Monocyte Chemotactic Protein-2 (MCP-2) and Macrophage Migration Inhibitory Factor (MIF) in E11 cells
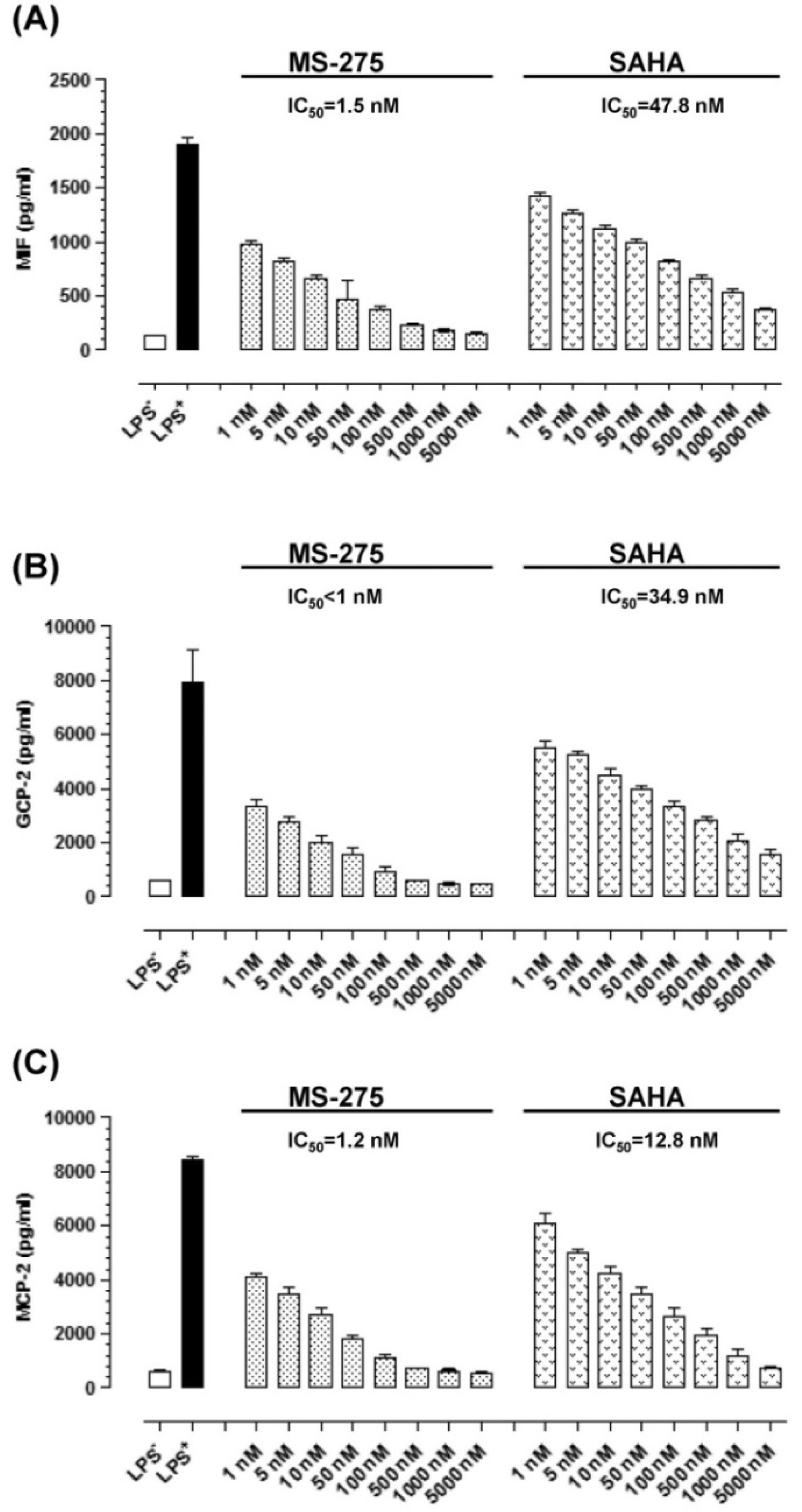
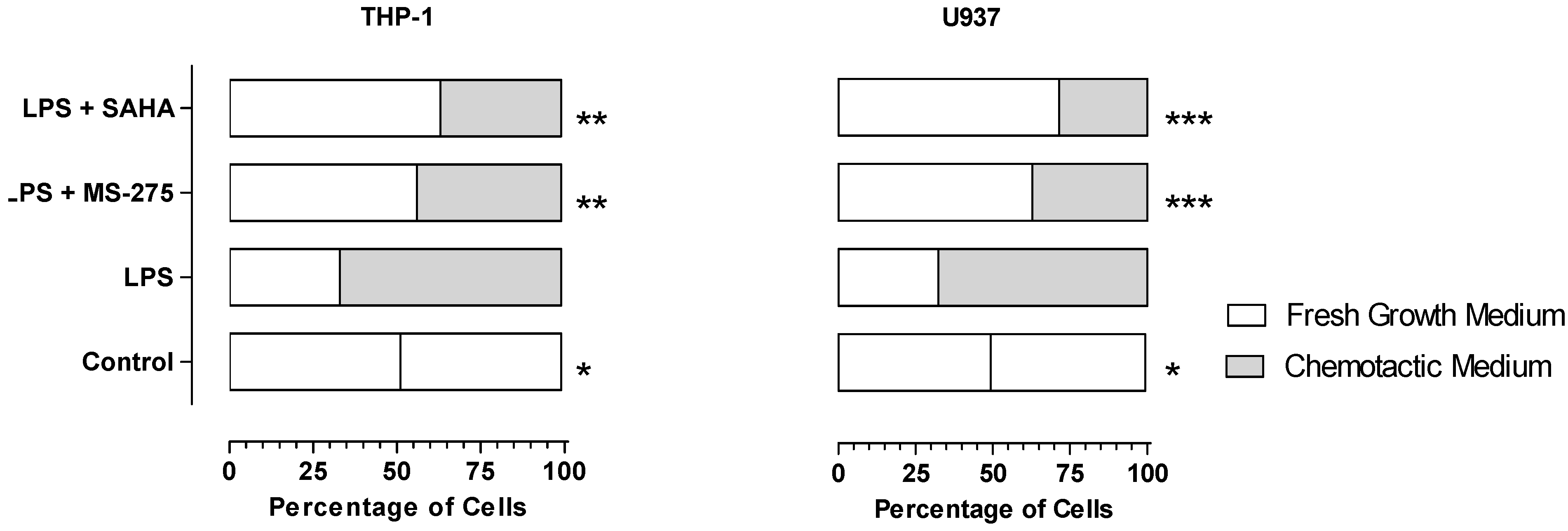
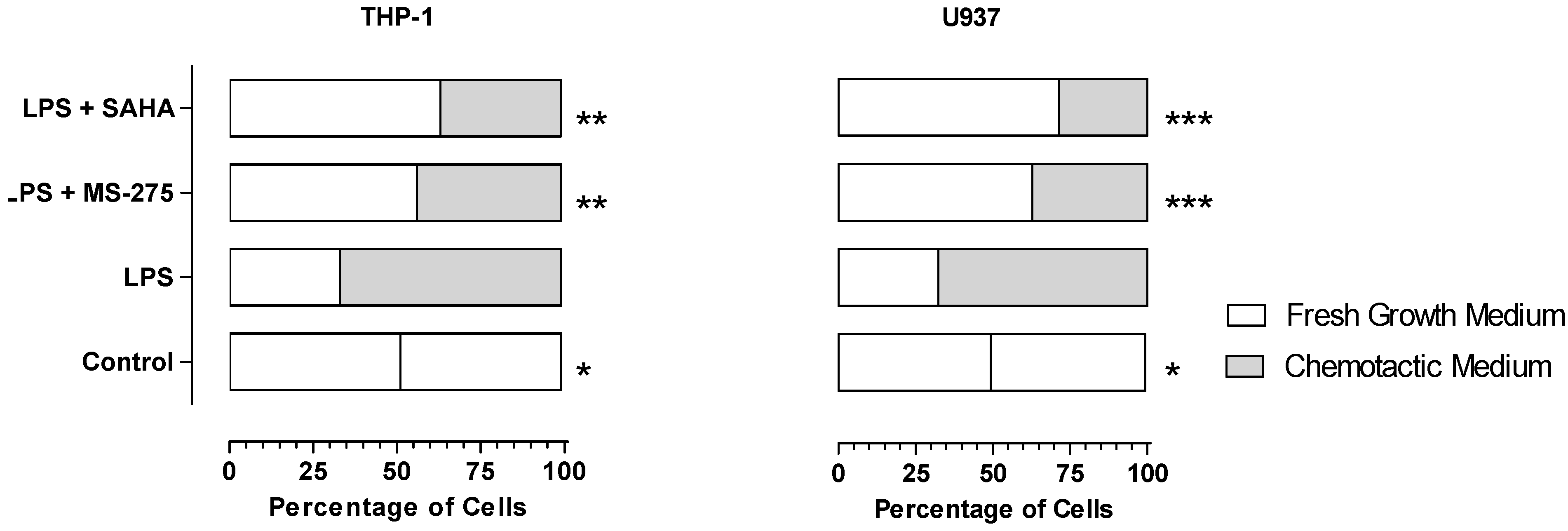
2.3. MS-275 and SAHA Inhibited E11-Driven Monocyte Migration
3. Experimental
3.1. HDAC Inhibitors
3.2. Cell Culture
3.3. Western Blotting Analyses of p38α, Phosphate-p38α (p-p38α) and MAPK Phosphatase (MKP)-1
3.4. Association of MKP-1 with p38α
3.5. Secretion of GCP-2, MCP-2 and MIF
3.6. Chemotaxis Assay
3.7. Data Analysis and Statistics
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef]
- Choo, Q.Y.; Ho, P.C.; Lin, H.S. Histone deacetylase inhibitors: New hope for rheumatoid arthritis? Curr. Pharm. Des. 2008, 14, 803–820. [Google Scholar] [CrossRef]
- Halili, M.A.; Andrews, M.R.; Sweet, M.J.; Fairlie, D.P. Histone deacetylase inhibitors in inflammatory disease. Curr. Top. Med. Chem. 2009, 9, 309–319. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef]
- Mai, A.; Rotili, D.; Valente, S.; Kazantsev, A.G. Histone deacetylase inhibitors and neurodegenerative disorders: Holding the promise. Curr. Pharm. Des. 2009, 15, 3940–3957. [Google Scholar] [CrossRef]
- Lin, H.S.; Hu, C.Y.; Chan, H.Y.; Liew, Y.Y.; Huang, H.P.; Lepescheux, L.; Bastianelli, E.; Baron, R.; Rawadi, G.; Clement-Lacroix, P. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharmacol. 2007, 150, 862–872. [Google Scholar] [CrossRef]
- O’Dell, J.R. Therapeutic strategies for rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2591–2602. [Google Scholar] [CrossRef]
- Smolen, J.S.; Steiner, G. Therapeutic strategies for rheumatoid arthritis. Nat. Rev. Drug Discov. 2003, 2, 473–488. [Google Scholar] [CrossRef]
- Makarov, S.S. NF-κB in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001, 3, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Firestein, G.S. NF-κB: Holy grail for rheumatoid arthritis? Arthritis Rheum. 2004, 50, 2381–2386. [Google Scholar] [CrossRef]
- Choo, Q.Y.; Ho, P.C.; Tanaka, Y.; Lin, H.S. Histone deacetylase inhibitors MS-275 and saha induced growth arrest and suppressed lipopolysaccharide-stimulated NF-κB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic e11 cells. Rheumatology 2010, 49, 1447–1460. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (map) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar]
- Schett, G.; Zwerina, J.; Firestein, G. The p38 mitogen-activated protein kinase (MAKP) pathway in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 909–916. [Google Scholar] [CrossRef]
- Thalhamer, T.; McGrath, M.A.; Harnett, M.M. MAKPs and their relevance to arthritis and inflammation. Rheumatology 2008, 47, 409–414. [Google Scholar] [CrossRef]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet 2001, 358, 903–911. [Google Scholar] [CrossRef]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef]
- Lefevre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnaker, E.M.; Tarner, I.H.; Robbins, P.D.; et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef]
- Hammaker, D.; Firestein, G.S. “Go upstream, young man”: Lessons learned from the p38 saga. Ann. Rheum. Dis. 2010, 69, i77–i82. [Google Scholar] [CrossRef]
- Sasakawa, Y.; Naoe, Y.; Sogo, N.; Inoue, T.; Sasakawa, T.; Matsuo, M.; Manda, T.; Mutoh, S. Marker genes to predict sensitivity to FK228, a histone deacetylase inhibitor. Biochem. Pharmacol. 2005, 69, 603–616. [Google Scholar]
- Cao, W.; Bao, C.; Padalko, E.; Lowenstein, C.J. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits toll-like receptor signaling. J. Exp. Med. 2008, 205, 1491–1503. [Google Scholar] [CrossRef]
- Heo, H.; Yoo, L.; Shin, K.S.; Kang, S.J. Suppression of caspase-11 expression by histone deacetylase inhibitors. Biochem. Biophys Res. Commun. 2009, 378, 79–83. [Google Scholar] [CrossRef]
- Choi, Y.; Park, S.K.; Kim, H.M.; Kang, J.S.; Yoon, Y.D.; Han, S.B.; Han, J.W.; Yang, J.S.; Han, G. Histone deacetylase inhibitor kbh-a42 inhibits cytokine production in raw 264.7 macrophage cells and in vivo endotoxemia model. Exp. Mol. Med. 2008, 40, 574–581. [Google Scholar] [CrossRef]
- Rahman, M.M.; Kukita, A.; Kukita, T.; Shobuike, T.; Nakamura, T.; Kohashi, O. Two histone deacetylase inhibitors, trichostatin a and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood 2003, 101, 3451–3459. [Google Scholar] [CrossRef]
- Portanova, P.; Russo, T.; Pellerito, O.; Calvaruso, G.; Giuliano, M.; Vento, R.; Tesoriere, G. The role of oxidative stress in apoptosis induced by the histone deacetylase inhibitor suberoylanilide hydroxamic acid in human colon adenocarcinoma ht-29 cells. Int. J. Oncol. 2008, 33, 325–331. [Google Scholar]
- Feng, R.; Oton, A.; Mapara, M.Y.; Anderson, G.; Belani, C.; Lentzsch, S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br. J. Haematol. 2007, 139, 385–397. [Google Scholar] [CrossRef]
- Schwab, M.; Reynders, V.; Ulrich, S.; Zahn, N.; Stein, J.; Schroder, O. Ppargamma is a key target of butyrate-induced caspase-3 activation in the colorectal cancer cell line caco-2. Apoptosis 2006, 11, 1801–1811. [Google Scholar] [CrossRef]
- Zhao, T.C.; Cheng, G.; Zhang, L.X.; Tseng, Y.T.; Padbury, J.F. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc. Res. 2007, 76, 473–481. [Google Scholar] [CrossRef]
- Morand, E.F.; Leech, M.; Bernhagen, J. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 2006, 5, 399–410. [Google Scholar] [CrossRef]
- Rubin, E.H.; Agrawal, N.G.; Friedman, E.J.; Scott, P.; Mazina, K.E.; Sun, L.; Du, L.; Ricker, J.L.; Frankel, S.R.; Gottesdiener, K.M.; et al. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 7039–7045. [Google Scholar] [CrossRef]
- Gore, L.; Rothenberg, M.L.; O’Bryant, C.L.; Schultz, M.K.; Sandler, A.B.; Coffin, D.; McCoy, C.; Schott, A.; Scholz, C.; Eckhardt, S.G. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, ms-275, in patients with refractory solid tumors and lymphomas. Clin. Cancer Res. 2008, 14, 4517–4525. [Google Scholar] [CrossRef]
- Blanchard, F.; Chipoy, C. Histone deacetylase inhibitors: New drugs for the treatment of inflammatory diseases? Drug Discov. Today 2005, 10, 197–204. [Google Scholar] [CrossRef]
- Abe, M.; Tanaka, Y.; Saito, K.; Shirakawa, F.; Koyama, Y.; Goto, S.; Eto, S. Regulation of interleukin (il)-1beta gene transcription induced by il-1beta in rheumatoid synovial fibroblast-like cells, e11, transformed with simian virus 40 large t antigen. J. Rheumatol. 1997, 24, 420–429. [Google Scholar]
- Nelson, R.D.; Quie, P.G.; Simmons, R.L. Chemotaxis under agarose: A new and simple method for measuring chemotaxis and spontaneous migration of human polymorphonuclear leukocytes and monocytes. J. Immunol. 1975, 115, 1650–1656. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Choo, Q.-Y.; Ho, P.C.; Tanaka, Y.; Lin, H.-S. The Histone Deacetylase Inhibitors MS-275 and SAHA Suppress the p38 Mitogen-Activated Protein Kinase Signaling Pathway and Chemotaxis in Rheumatoid Arthritic Synovial Fibroblastic E11 Cells. Molecules 2013, 18, 14085-14095. https://doi.org/10.3390/molecules181114085
Choo Q-Y, Ho PC, Tanaka Y, Lin H-S. The Histone Deacetylase Inhibitors MS-275 and SAHA Suppress the p38 Mitogen-Activated Protein Kinase Signaling Pathway and Chemotaxis in Rheumatoid Arthritic Synovial Fibroblastic E11 Cells. Molecules. 2013; 18(11):14085-14095. https://doi.org/10.3390/molecules181114085
Chicago/Turabian StyleChoo, Qiu-Yi, Paul C Ho, Yoshiya Tanaka, and Hai-Shu Lin. 2013. "The Histone Deacetylase Inhibitors MS-275 and SAHA Suppress the p38 Mitogen-Activated Protein Kinase Signaling Pathway and Chemotaxis in Rheumatoid Arthritic Synovial Fibroblastic E11 Cells" Molecules 18, no. 11: 14085-14095. https://doi.org/10.3390/molecules181114085