Design and Synthesis of a Biotinylated Chemical Probe for Detecting the Molecular Targets of an Inhibitor of the Production of the Pseudomonas aeruginosa Virulence Factor Pyocyanin


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
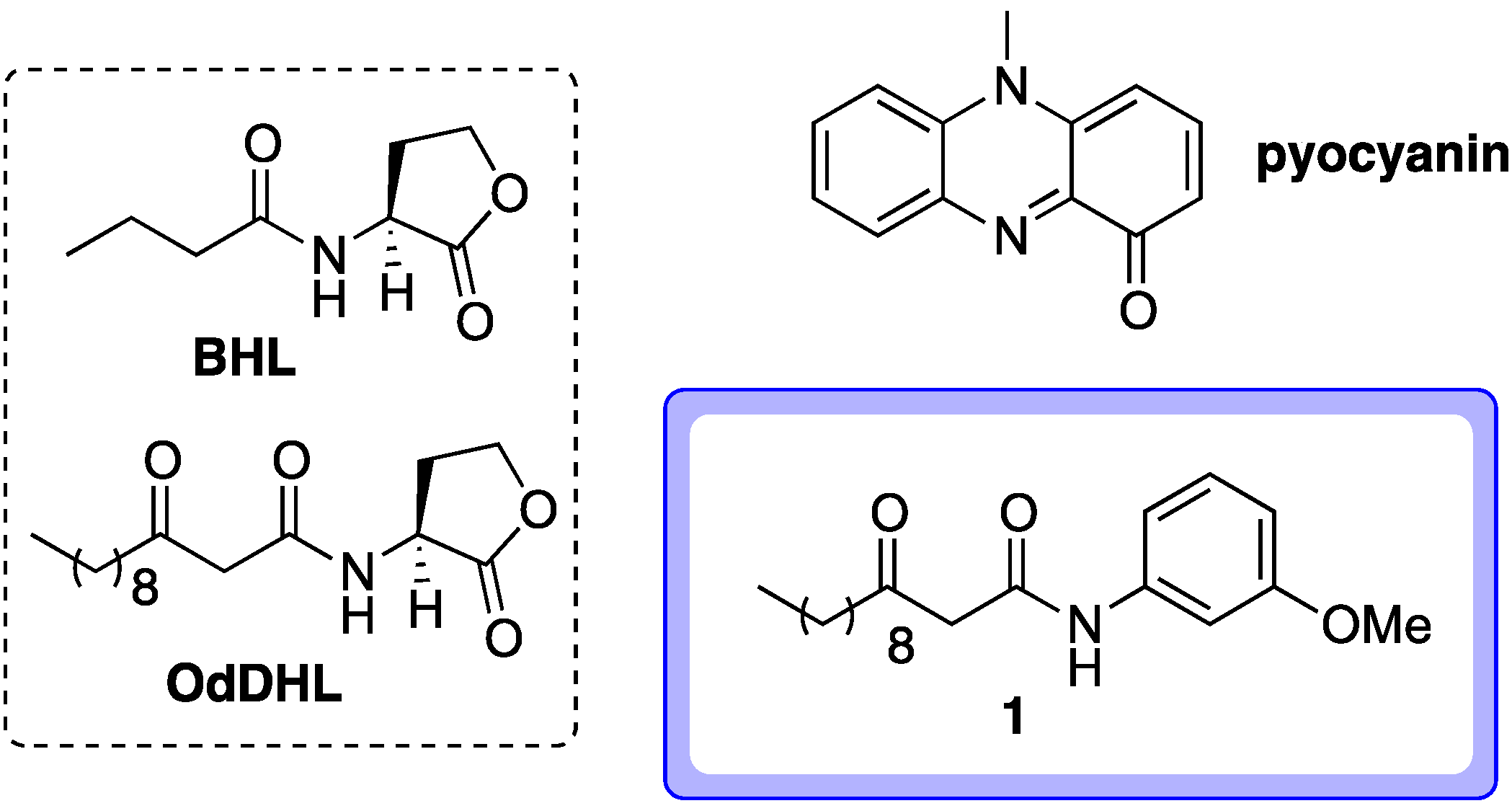
2. Results and Discussion
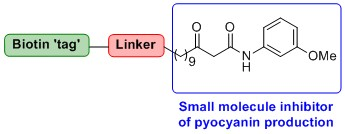
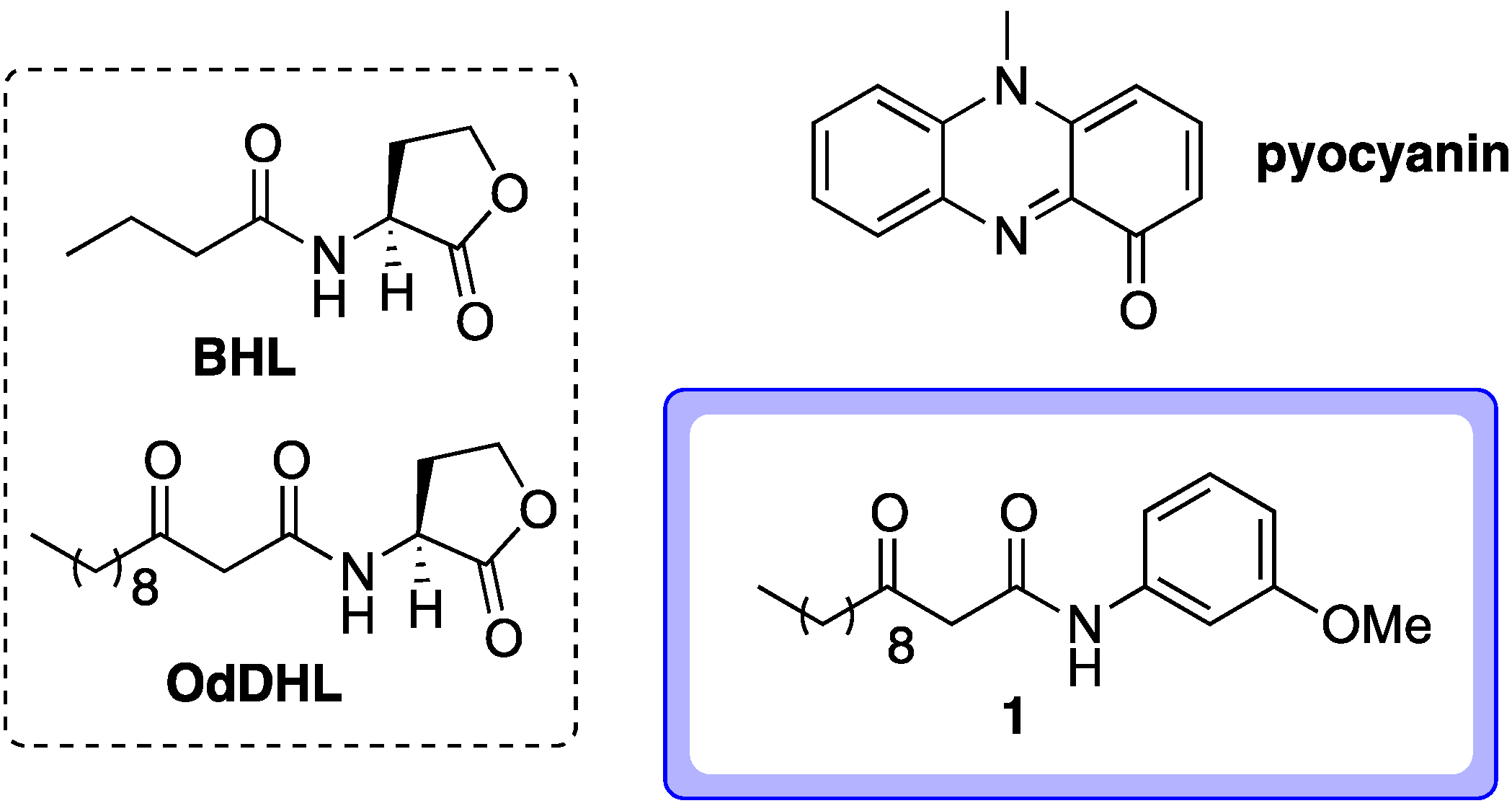
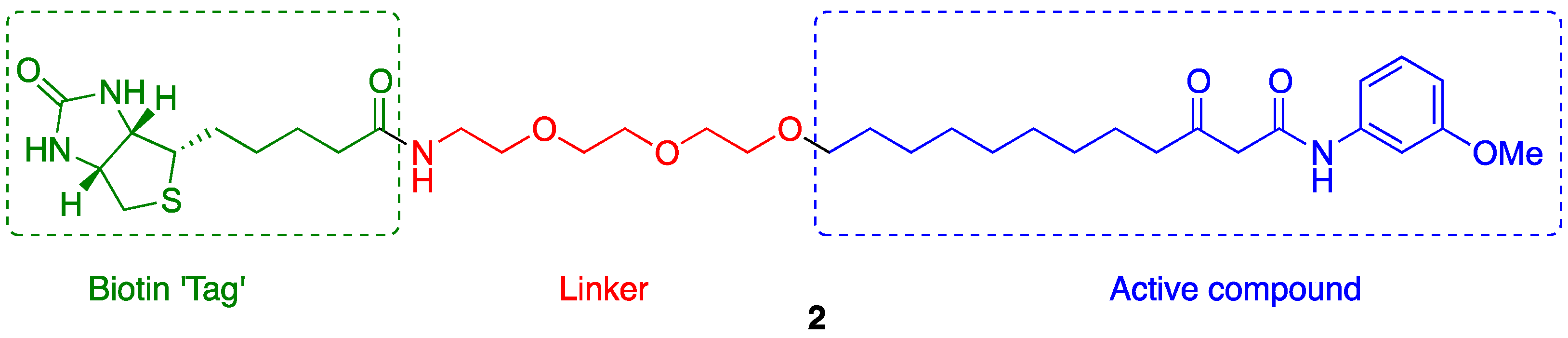
2.1. Probe Design
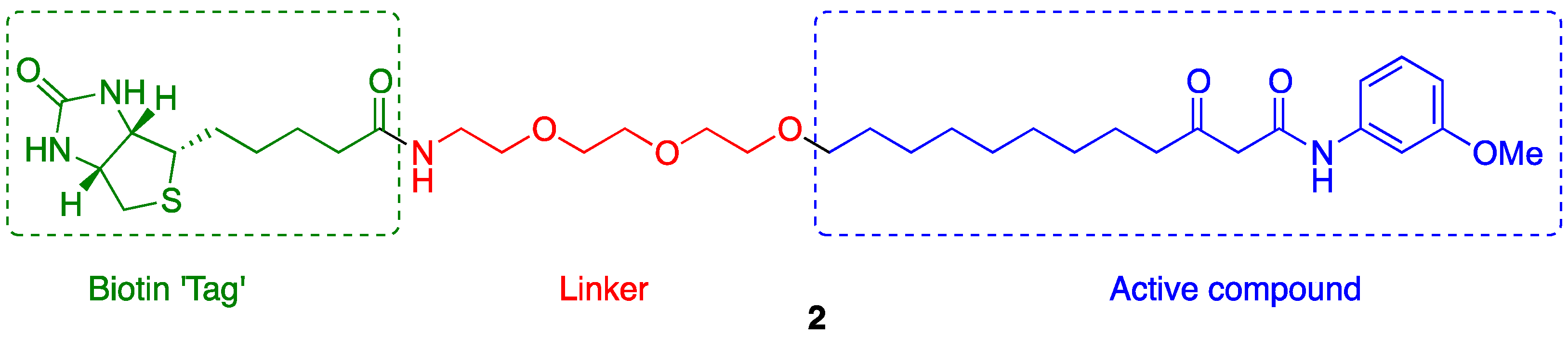
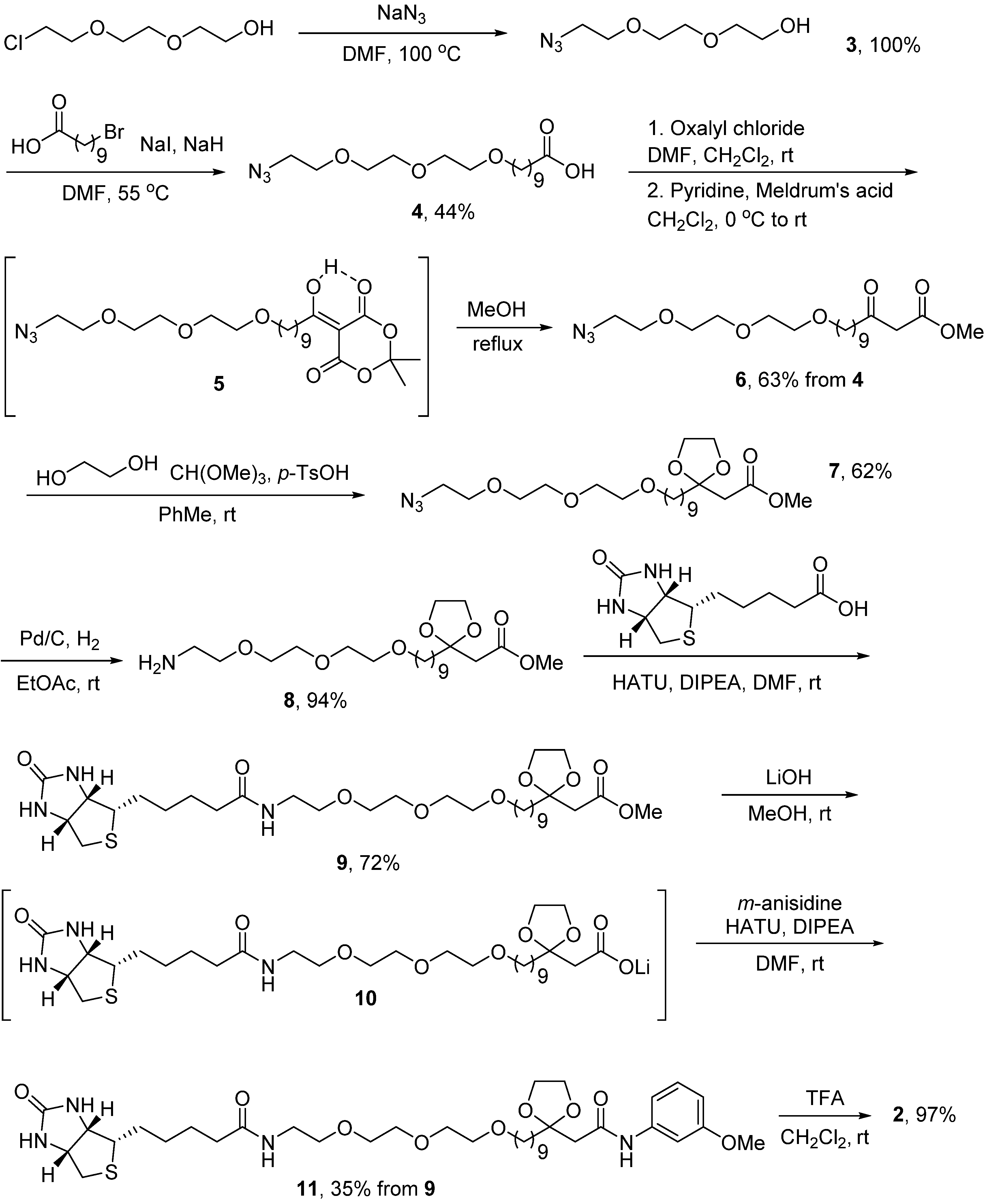
2.2. Probe Synthesis
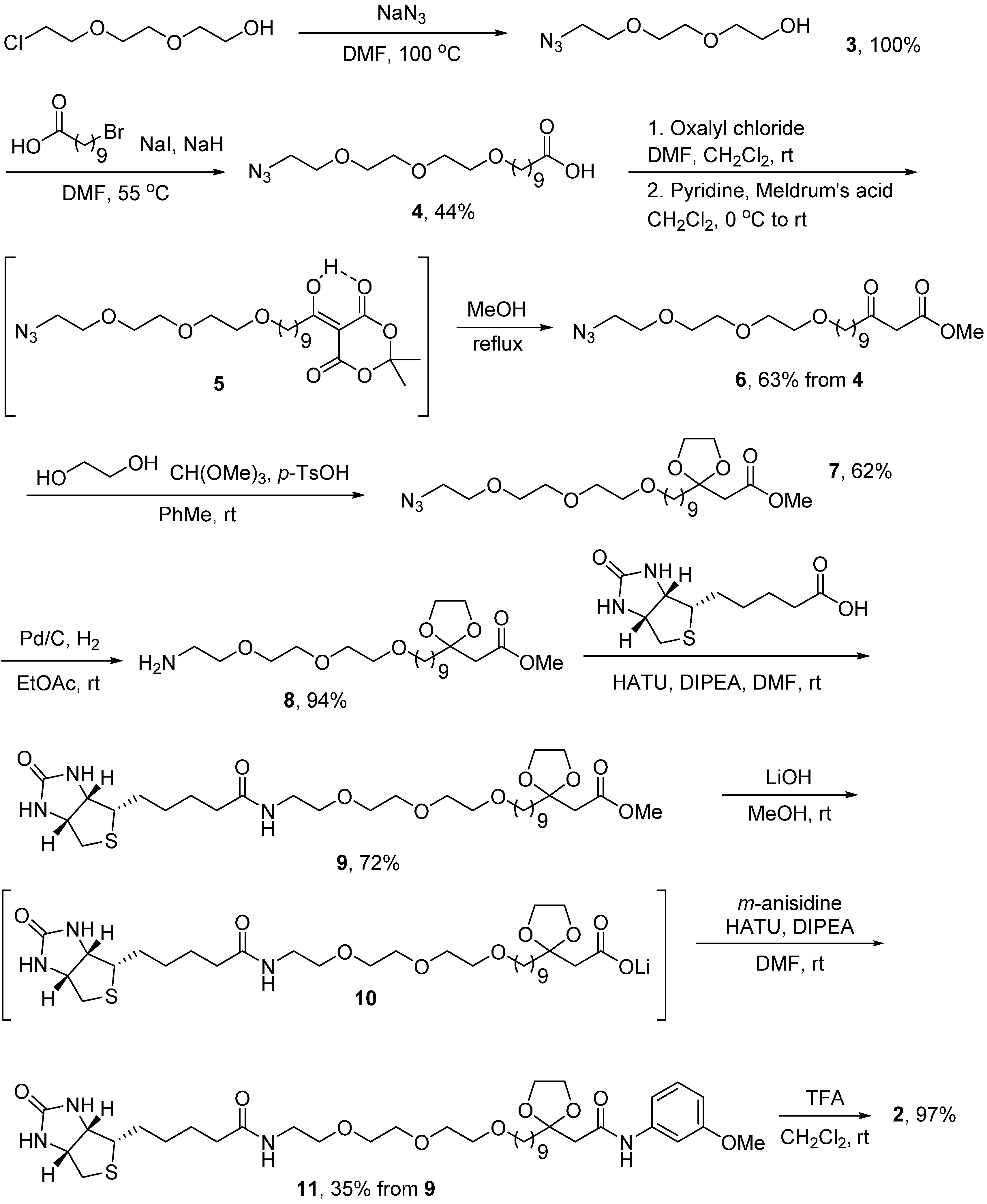
3. Experimental
3.1. General
3.2. Experimental Procedures
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Morkunas, B.; Galloway, W.R.J.D.; Wright, M.; Ibbeson, B.M.; Hodgkinson, J.T.; O'Connell, K.M.; Bartolucci, N.; Valle, M.D.; Welch, M.; Spring, D.R. Inhibition of the production of the Pseudomonas aeruginosa virulence factor pyocyanin in wild-type cells by quorum sensing autoinducer-mimics. Org. Biomol. Chem. 2012, 10, 8452–8464. [Google Scholar] [CrossRef]
- Mesaros, N.; Nordmann, P.; Plesiat, P.; Roussel-Delvallez, M.; Van Eldere, J.; Glupczynski, Y.; van Laethem, Y.; Jacobs, F.; Lebecque, P.; Malfroot, A.; et al. Pseudomonas aeruginosa: Resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infect. 2007, 13, 560–578. [Google Scholar] [CrossRef]
- Antunes, L.C.M.; Ferreira, R.B.R.; Buckner, M.M.C.; Finlay, B.B. Quorum sensing in bacterial virulence. Microbiology 2010, 156, 2271–2282. [Google Scholar] [CrossRef]
- Zaborina, O.; Kohler, J.E.; Wang, Y.; Bethel, C.; Shevchenko, O.; Wu, L.; Turner, J.R.; Alverdy, J.C. Identification of multi-drug resistant Pseudomonas aeruginosa clinical isolates that are highly disruptive to the intestinal epithelial barrier. Ann. Clin. Microbiol. Antimicrob. 2006, 5, 14. [Google Scholar] [CrossRef]
- Kipnis, E.; Sawa, T.; Wiener-Kronish, J. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Med. Maladies Infect. 2006, 36, 78–91. [Google Scholar] [CrossRef]
- Strateva, T.; Mitov, I. Contribution of an arsenal of virulence factors to pathogenesis of Pseudomonas aeruginosa infections. Ann. Microbiol. 2011, 61, 717–732. [Google Scholar] [CrossRef]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef]
- Barczak, A.K.; Hung, D.T. Productive steps toward an antimicrobial targeting virulence. Curr. Opin. Microbiol. 2009, 12, 490–496. [Google Scholar] [CrossRef]
- Galloway, W.R.J.D.; Hodgkinson, J.T.; Bovvden, S.; Welch, M.; Spring, D.R. Applications of small molecule activators and inhibitors of quorum sensing in Gram-negative bacteria. Trends Microbiol. 2012, 20, 449–458. [Google Scholar] [CrossRef]
- Deep, A.; Chaudhary, U.; Gupta, V. Quorum sensing and Bacterial Pathogenicity: From Molecules to Disease. J. Lab. Physicians 2011, 3, 4–11. [Google Scholar] [CrossRef]
- Whitehead, N.A.; Barnard, A.M.L.; Slater, H.; Simpson, N.J.L.; Salmond, G.P.C. Quorum-sensing in gram-negative bacteria. FEMS Microbiol. Rev. 2001, 25, 365–404. [Google Scholar] [CrossRef]
- O'Malley, Y.Q.; Reszka, K.J.; Spitz, D.R.; Denning, G.M.; Britigan, B.E. Pseudomonas aeruginosa pyocyanin directly oxidizes glutathione and decreases its levels in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Phys. 2004, 287, L94–L103. [Google Scholar] [CrossRef]
- Lau, G.W.; Ran, H.M.; Kong, F.S.; Hassett, D.J.; Mavrodi, D. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect. Immun. 2004, 72, 4275–4278. [Google Scholar] [CrossRef]
- Lau, G.W.; Hassett, D.J.; Ran, H.; Kong, F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol. Med. 2004, 10, 599–606. [Google Scholar] [CrossRef]
- Garner, A.L.; Struss, A.K.; Fullagar, J.L.; Argawal, A.; Moreno, A.Y.; Cohen, S.M.; Janda, K.D. 3-Hydroxy-1-alkyl-2-methylpyridine-4(1H)-thiones: Inhibition of the Pseudomonas aeruginosa Virulence Factor LasB. ACS Med. Chem. Lett. 2012, 3, 668–672. [Google Scholar] [CrossRef]
- Hodgkinson, J.T.; Galloway, W.R.J.D.; Wright, M.; Mati, I.K.; Nicholson, R.L.; Welch, M.; Spring, D.R. Design, synthesis and biological evaluation of non-natural modulators of quorum sensing in Pseudomonas aeruginosa. Org. Biomol. Chem. 2012, 10, 6032–6044. [Google Scholar] [CrossRef]
- Chong, Y.M.; Yin, W.F.; Ho, C.Y.; Mustafa, M.R.; Hadi, A.H.; Awang, K.; Narrima, P.; Koh, C.L.; Appleton, D.R.; Chan, K.G. Malabaricone C from Myristica cinnamomea exhibits anti-quorum sensing activity. J. Nat. Prod. 2011, 74, 2261–2264. [Google Scholar] [CrossRef]
- Krishnan, T.; Yin, W.F.; Chan, K.G. Inhibition of quorum sensing-controlled virulence factor production in Pseudomonas aeruginosa PAO1 by Ayurveda spice clove (Syzygium Aromaticum) Bud Extract. Sensors 2012, 12, 4016–4030. [Google Scholar] [CrossRef]
- Tan, L.Y.; Yin, W.F.; Chan, K.G. Silencing quorum sensing through extracts of melicope lunu-ankenda. Sensors 2012, 12, 4339–4351. [Google Scholar] [CrossRef]
- Geske, G.D.; O'Neill, J.C.; Blackwell, H.E. Expanding dialogues: From natural autoinducers to non-natural analogues that modulate quorum sensing in Gram-negative bacteria. Chem. Soc. Rev. 2008, 37, 1432–1447. [Google Scholar] [CrossRef]
- Dekimpe, V.; Deziel, E. Revisiting the quorum-sensing hierarchy in Pseudomonas aeruginosa: The transcriptional regulator RhlR regulates LasR-specific factors. Microbiology 2009, 155, 712–723. [Google Scholar] [CrossRef]
- Pukatzki, S.; Kessin, R.H.; Mekalanos, J.J. The human pathogen Pseudomonas aeruginosa utilizes conserved virulence pathways to infect the social amoeba Dictyostelium discoideum. Proc. Natl. Acad. Sci. USA 2002, 99, 3159–3164. [Google Scholar] [CrossRef]
- Galloway, W.R.J.D.; Hodgkinson, J.T.; Bowden, S.D.; Welch, M.; Spring, D.R. Quorum sensing in gram-negative bacteria: Small-molecule modulation of AHL and Al-2 quorum sensing pathways. Chem. Rev. 2011, 111, 28–67. [Google Scholar] [CrossRef]
- Stevens, A.M.; Queneau, Y.; Soulere, L.; von Bodman, S.; Doutheau, A. Mechanisms and synthetic modulators of AHL-dependent gene regulation. Chem. Rev. 2011, 111, 4–27. [Google Scholar] [CrossRef]
- Ng, W.L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef]
- McInnis, C.E.; Blackwell, H.E. Design, synthesis, and biological evaluation of abiotic, non-lactone modulators of LuxR-type quorum sensing. Bioorg. Med. Chem. 2011, 19, 4812–4819. [Google Scholar] [CrossRef]
- Williams, P.; Winzer, K.; Chan, W.C.; Camara, M. Look who’s talking: Communication and quorum sensing in the bacterial world. Philos. Trans. R. Soc. B Biol. Sci. 2007, 362, 1119–1134. [Google Scholar] [CrossRef]
- Hodgkinson, J.T.; Galloway, W.R.J.D.; Saraf, S.; Baxendale, I.R.; Ley, S.V.; Ladlow, M.; Welch, M.; Spring, D.R. Microwave and flow syntheses of Pseudomonas quinolone signal (PQS) and analogues. Org. Biomol. Chem. 2011, 9, 57–61. [Google Scholar] [CrossRef]
- Taha, M.O.; Al-Bakri, A.G.; Zalloum, W.A. Discovery of potent inhibitors of pseudomonal quorum sensing via pharmacophore modeling and in silico screening. Bioorg. Med. Chem. Lett. 2006, 16, 5902–5906. [Google Scholar] [CrossRef]
- Smith, K.M.; Bu, Y.G.; Suga, H. Library screening for synthetic agonists and antagonists of a Pseudomonas aeruginosa autoinducer. Chem. Biol. 2003, 10, 563–571. [Google Scholar] [CrossRef]
- Ishida, T.; Ikeda, T.; Takiguchi, N.; Kuroda, A.; Ohtake, H.; Kato, J. Inhibition of quorum sensing in Pseudomonas aeruginosa by N-acyl cyclopentylamides. Appl. Environ. Microbiol. 2007, 73, 3183–3188. [Google Scholar] [CrossRef]
- Popat, R.; Crusz, S.A.; Diggle, S.P. The social behaviours of bacterial pathogens. Br. Med. Bull. 2008, 87, 63–75. [Google Scholar] [CrossRef]
- Skindersoe, M.E.; Alhede, M.; Phipps, R.; Yang, L.; Jensen, P.O.; Rasmussen, T.B.; Bjarnsholt, T.; Tolker-Nielsen, T.; Hoiby, N.; Givskov, M. Effects of antibiotics on quorum sensing in pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 3648–3663. [Google Scholar] [CrossRef]
- Brint, J.M.; Ohman, D.E. Synthesis of multiple exoproducts in Pseudomonas-Aeruginosa is under the control of RhlR-RhlI, another set of regulators in strain pao1 with homology to the autoinducer-responsive LuxR-LuxI family. J. Bacteriol. 1995, 177, 7155–7163. [Google Scholar]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Ed. 2013, 52, 2744–2792. [Google Scholar] [CrossRef]
- Praneenararat, T.; Palmer, A.G.; Blackwell, H.E. Chemical methods to interrogate bacterial quorum sensing pathways. Org. Biomol. Chem. 2012, 10, 8189–8199. [Google Scholar] [CrossRef]
- Leslie, B.J.; Hergenrother, P.J. Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem. Soc. Rev. 2008, 37, 1347–1360. [Google Scholar] [CrossRef]
- Matsuyama, A.; Yashiroda, Y.; Yoshida, M. Chemical Proteomics: A Global Study of Protein-Small Molecule Interactions. In Chemical Genomics; Fu, H., Ed.; Cambridge University Press: Cambridge, UK, 2012; Chapter 3; p. 26. [Google Scholar]
- Praneenararat, T.; Beary, T.M.J.; Breitbach, A.S.; Blackwell, H.E. Synthesis and application of an N-acylated L-homoserine lactone derivatized affinity matrix for thte isolation of quourm sensing signal receptors. Bioorg. Med. Chem. Lett. 2011, 5054–5057. [Google Scholar]
- Seabra, R.; Brown, A.; Hooi, D.; Kerkhoff, C.; Chhabra, S.R.; Harty, C.; Williams, P.; Pritchard, D.I. A eukaryotic binding protein for the immune modulatory bacterial quorum sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone. Calcium Bind. Proteins 2008, 3, 31–37. [Google Scholar]
- Karlsson, T.; Turkina, M.V.; Yakymenko, O.; Magnusson, K.; Vikström, E. The Pseudomona aeruginosa N-acylhomoserine lactone quorum sensing molecules target IQGAP1 and modulate epithelial cell migrations. PLoS Pathog. 2012, 8, e1002953. [Google Scholar] [CrossRef]
- Spandl, R.J.; Nicholson, R.L.; Marsden, D.M.; Hodgkinson, J.T.; Su, X.B.; Thomas, G.L.; Salmond, G.P.C.; Welch, M.; Spring, D.R. Synthesis of a biotin-labeled quorum-sensing molecule: Towards a general method for target identification. Synlett 2008, 2008, 2122–2126. [Google Scholar] [CrossRef]
- Most successful applications of affinity probes involve active parent compounds with nanomolar activities. The active compound 1 can block pyocyanin production by approximately 75% at mid-micromolar levels (approximately 50 μM, see reference 1). This was also the concentration at which this compound was tested in a LasR ligand binding domain production assay, which provided indirect evidence that 1 can directly bind to LasR (see reference 16). Given this activity level, the identification of targets for compound 1 using affinity probe 2 is likely to be quite a challenging (though not implausible) endeavor.
- Sample Availability: Samples of the compounds 1–11 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baker, Y.R.; Galloway, W.R.J.D.; Hodgkinson, J.T.; Spring, D.R. Design and Synthesis of a Biotinylated Chemical Probe for Detecting the Molecular Targets of an Inhibitor of the Production of the Pseudomonas aeruginosa Virulence Factor Pyocyanin. Molecules 2013, 18, 11783-11796. https://doi.org/10.3390/molecules181011783
Baker YR, Galloway WRJD, Hodgkinson JT, Spring DR. Design and Synthesis of a Biotinylated Chemical Probe for Detecting the Molecular Targets of an Inhibitor of the Production of the Pseudomonas aeruginosa Virulence Factor Pyocyanin. Molecules. 2013; 18(10):11783-11796. https://doi.org/10.3390/molecules181011783
Chicago/Turabian StyleBaker, Ysobel R., Warren R. J. D. Galloway, James T. Hodgkinson, and David R. Spring. 2013. "Design and Synthesis of a Biotinylated Chemical Probe for Detecting the Molecular Targets of an Inhibitor of the Production of the Pseudomonas aeruginosa Virulence Factor Pyocyanin" Molecules 18, no. 10: 11783-11796. https://doi.org/10.3390/molecules181011783