Synthesis of 9,9′-[1,2-Ethanediylbis(oxymethylene)]bis-2-amino-1,9-dihydro-6H-purin-6-one, an Impurity of Acyclovir

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
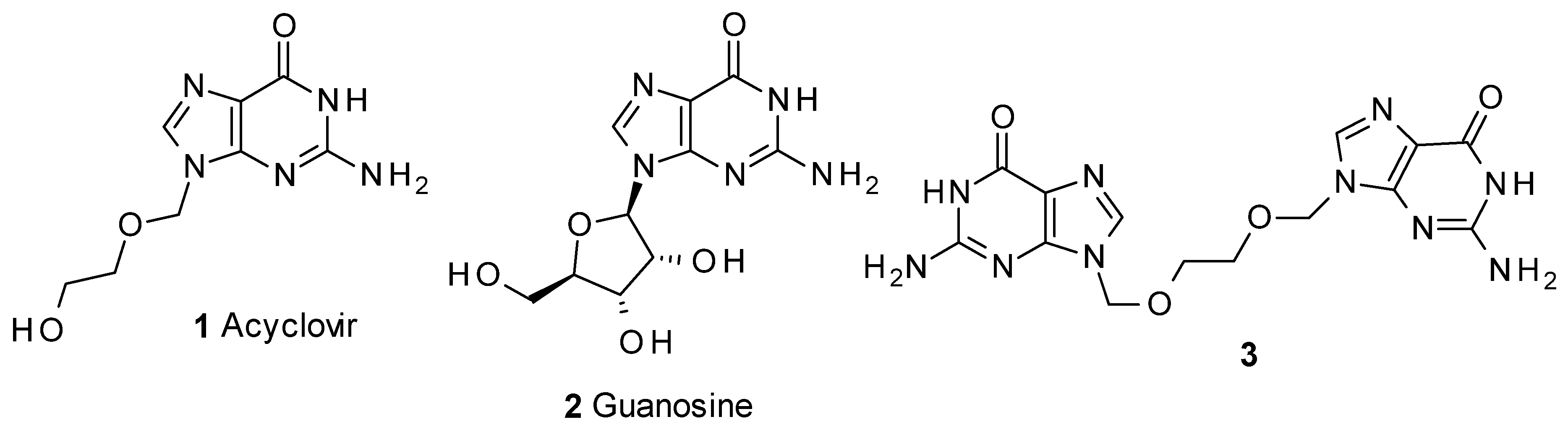
:1. Introduction
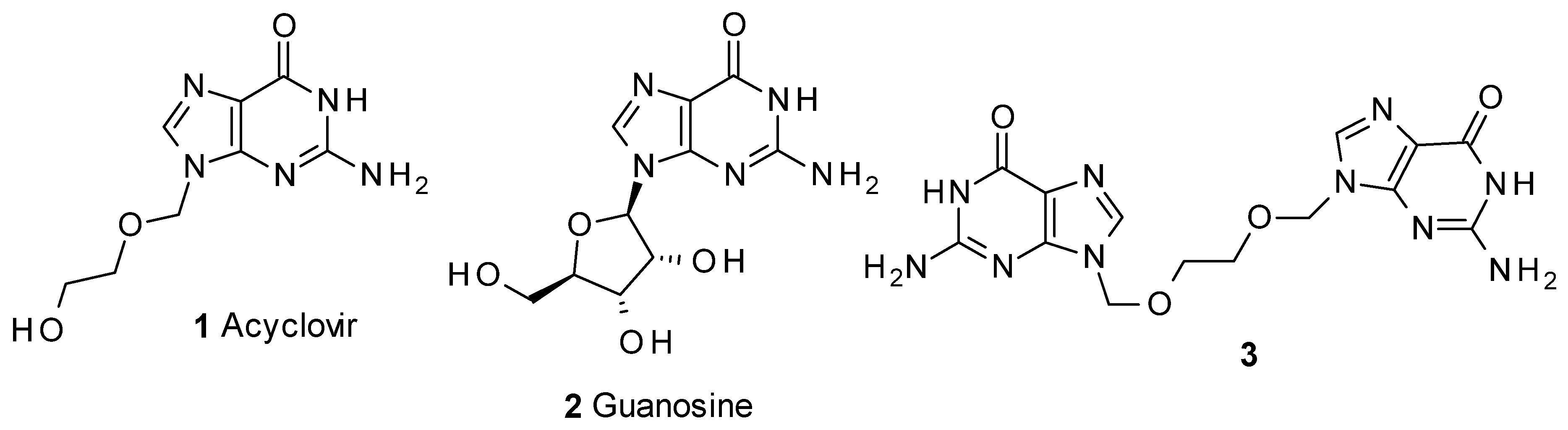

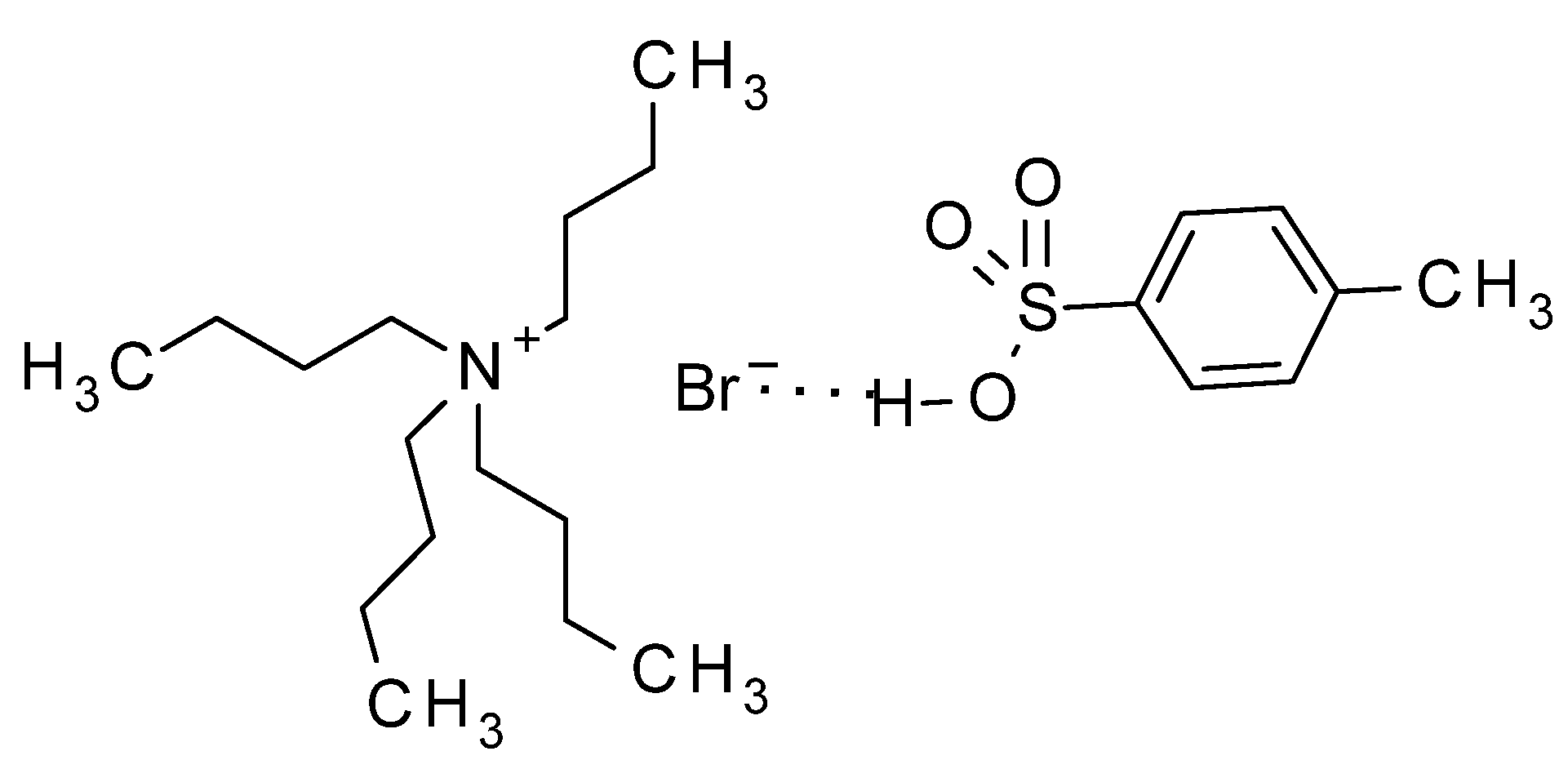
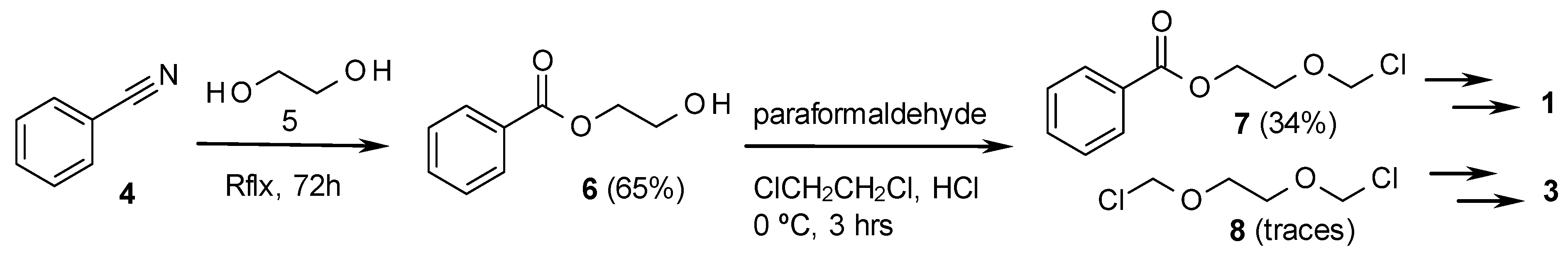
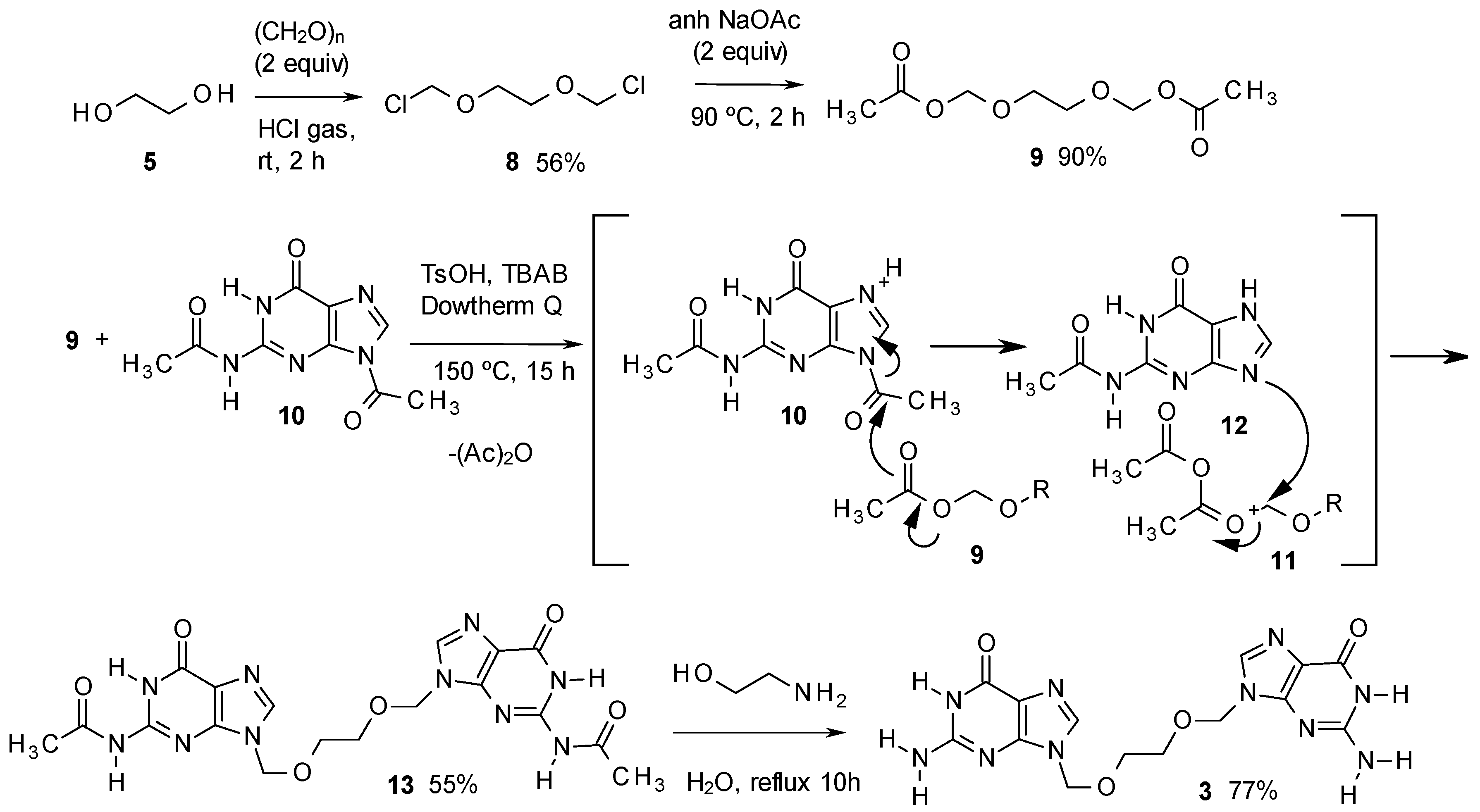
2. Results and Discussion
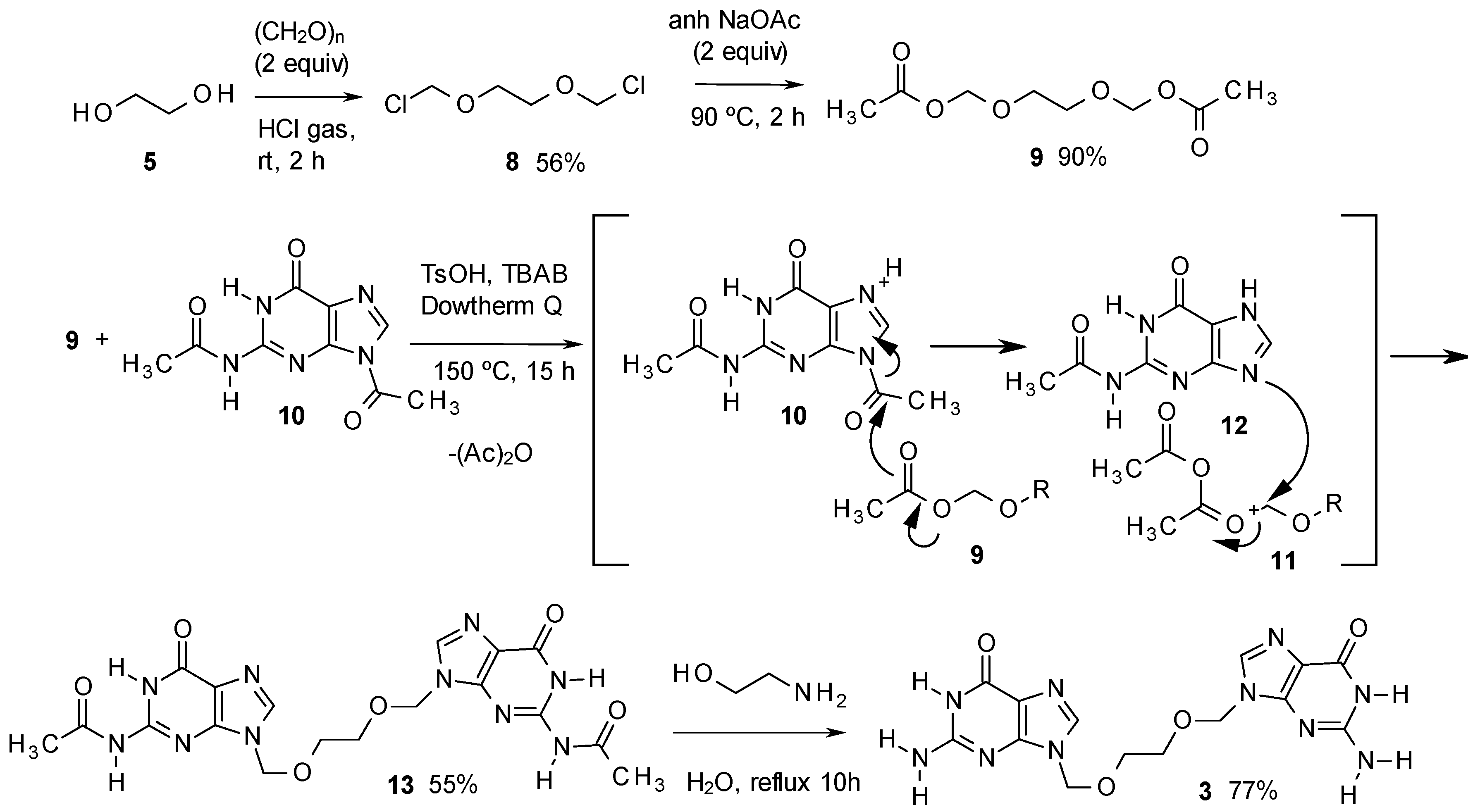
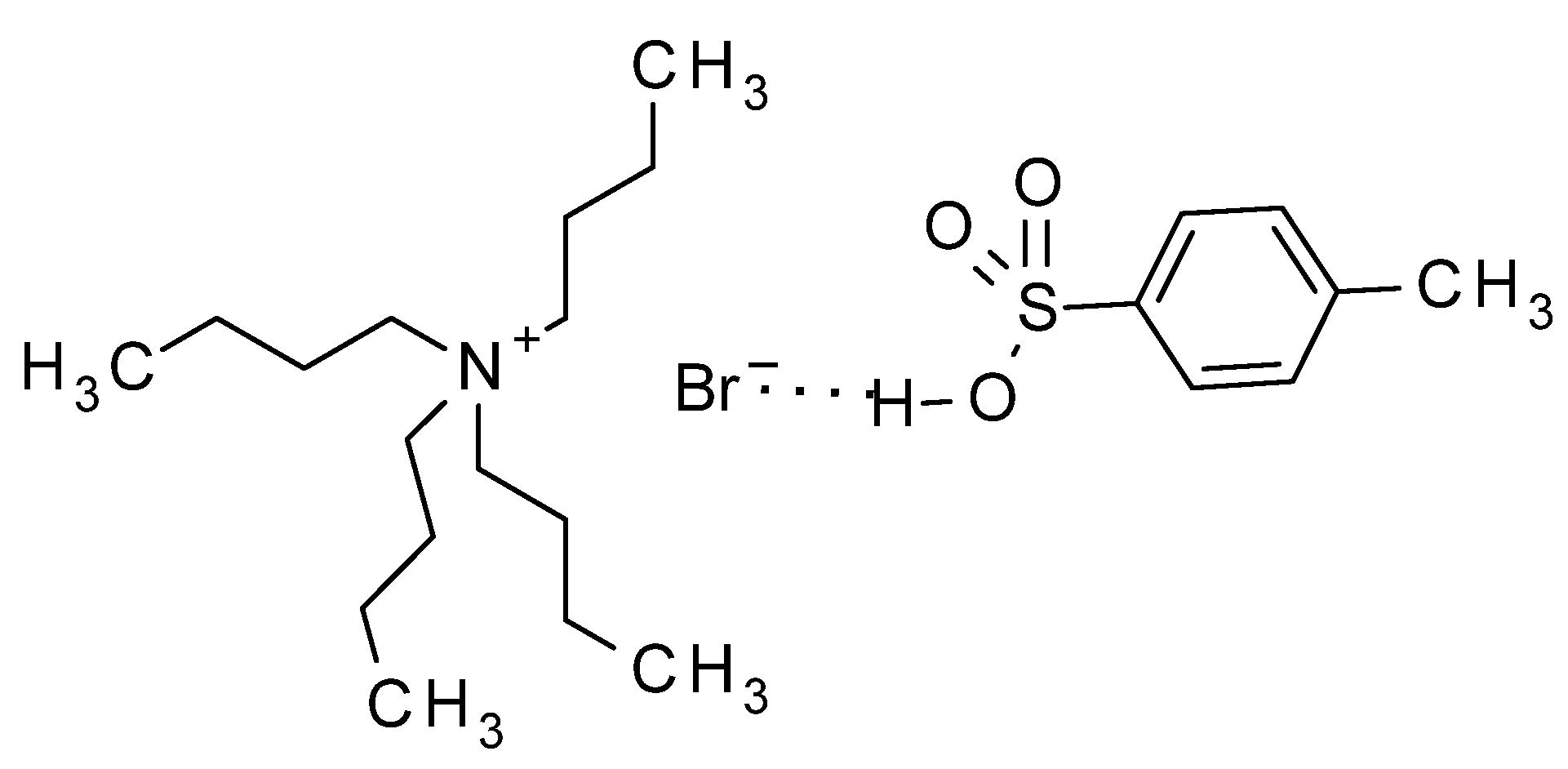
3. Experimental
General
4. Conclusions
Acknowledgments
References
- International Conference on Harmonization (ICH) Guidelines, Q3A (R) Impurities in New Drug Substances, February 2002.
- Gorog, S. The role of impurity profiling in drug research, development and production. Progr. Pharm. Biomed. Anal. 2000, 4, 38–47. [Google Scholar] [CrossRef]
- Gorog, S. Identification, structure elucidation and determination of related organic impurities 2.1. Strategies in impurity profiling. Progr. Pharm. Biomed. Anal. 2000, 4, 67–83. [Google Scholar] [CrossRef]
- Biba, M.; Welch, C.J.; da Silva, J.O.; Mathre, D.J. Impurity isolation in support of pharmaceutical process research. Am. Pharm. Rev. 2005, 8, 68–72. [Google Scholar]
- Grekas, N. Organic impurities in chemical drug substances. Pharm. Tech. Eur. 2005, 24–32. [Google Scholar]
- Glazkov, I.N.; Bochkareva, N.L.; Revel’skii, A. Determination of organic impurities in pharmaceutical preparations. J. Anal. Chem. 2005, 60, 107–118. [Google Scholar]
- Schaeffer, H.J.; Beauchamp, L.; De Miranda, P.; Elion, G.B.; Bauer, D.J.; Collins, P. 9-(2-Hydroxyethoxymethyl)guanine activity against viruses of the herpes group. Nature 1978, 272, 583–585. [Google Scholar]
- De Clercq, E.; Field, H.J. Antiviral prodrugs—The development of successful prodrug strategies for antiviral chemotherapy. Br. J. Pharmacol. 2006, 147, 1–11. [Google Scholar] [CrossRef]
- Schaeffer, H.J. Substituted purines. DE 2539963, 1976. [Google Scholar]
- Schaeffer, H.J. Compositions for treating viral infections and guanine acyclic nucleosides. U.S. Patent 4199574, 1980. [Google Scholar]
- Matsumoto, H.; Kaneko, C.; Yamada, K.; Takeuchi, T.; Mori, T.; Mizuno, Y. A convenient synthesis of 9-[(2-hydroxyethoxy)methyl]guanine (acyclovir) and related compounds. Chem. Pharm. Bull. 1988, 36, 1153–1157. [Google Scholar] [CrossRef]
- Schaeffer, H.J. Esters of 9-(2-hydroxyethoxymethyl)guanine and 2-amino-9-(2-hydroxyethoxy-methyl)adenine and pharmaceutical compositions containing them. GB 1567671, 1976. [Google Scholar]
- Schloemer, G.C.; Han, Y.K.; Harrington, P.J. Preparation of acyclovir using 1,3 dioxolane. US 5567816, 1996. [Google Scholar]
- Hansen, H.; Scheuplein, S.W.; Stuhltraeger, K.; Bauer, K. Improved procedure for the preparation of acyclovir. DE 19536164, 1996. [Google Scholar]
- Gao, H.; Mitra, A.K. Synthesis of acyclovir, ganciclovir and their prodrugs: A review. Synthesis 2000, 329–351. [Google Scholar]
- Ashton, D.S.; Beddell, C.; Ray, A.D.; Valko, K. Quantitative structure-retention relationships of acyclovir esters using immobilized albumin high-performance liquid chromatography and reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1995, 707, 367–372. [Google Scholar]
- Martin, J.C.; Dvorak, C.A.; Smee, D.F.; Matthews, T.R.; Verheyden, J.P.H. 9-(1,3-Dihydroxy-2-propoxymethyl)guanine: A new potent and selective antiherpes agent. J. Med. Chem. 1983, 26, 759–761. [Google Scholar] [CrossRef]
- Robinson, B. Non-catalytic alcoholysis of nitriles. J. Chem. Soc. 1963, 2417–2419. [Google Scholar] [CrossRef]
- Senkus, M. Reaction of some cyclic acetals with acid anhydrides. J. Am. Chem. Soc. 1946, 68, 734–736. [Google Scholar] [CrossRef]
- Zaragoza Dörwald, F. Side Reactions in Organic Synthesis; John Wiley & Sons: Weinheim, Germany, 2005; p. 336. [Google Scholar]
- European Pharmacopeia 6.0. Council of Europe: Strasbourg, France, 2007; Volume 2, p. 1107.
- Yang, G.Y.; Oh, K.-A.; Park, N.-J.; Jung, Y.-S. New oxime reactivators connected with CH2O(CH2)nOCH2 linker and their reactivation potency for organophosphorus agents-inhibited acetylcholinesterase. Bioorg. Med. Chem. 2007, 15, 7704–7710. [Google Scholar]
- Landini, D.; Montanari, F.; Rolla, F. Conversion of primary alcohols to alkyl chlorides using aqueous hydrochloric acid in the presence of phase-transfer catalysts. Synthesis 1974, 37–38. [Google Scholar]
- Landini, D.; Montanari, F.; Rolla, F. Cleavage of dialkyl and aryl alkyl esters with hydrobromic acid in the presence of phase-transfer catalysts. Synthesis 1978, 771–773. [Google Scholar]
- Landini, D.; Rolla, F. Addition of hydrohalogenic acids to alkenes in aqueous-organic, two-phase systems in the presence of catalytic amounts of onium salts. J. Org. Chem. 1980, 45, 3527–3529. [Google Scholar] [CrossRef]
- Goverdhan, L.K.; Irvinder, K.; Monica, B.; Jasvinder, S.; Jasamrit, K. Functional Group Transformations of Diols, Cyclic Ethers, and Lactones Using Aqueous Hydrobromic Acid and Phase Transfer Catalyst under Microwave Irradiation. Org. Proc. Res. Dev. 2003, 7, 339–340. [Google Scholar] [CrossRef]
- Radl, S.; Zikan, V. A new method of preparation of the antiviral agent acyclovir. Cesk. Farm. 1987, 36, 58–60. [Google Scholar]
- Saleh, A.; D’Angelo, J.G.; Morton, M.D.; Quinn, J.; Redden, K.; Mielguz, R.W. Pavlik, C.; Smith, M.B. Synthesis of 3-Guaninyl- and 3-Adeninyl-5-hydroxymethyl-2-pyrrolidinone Nucleosides. J. Org. Chem. 2011, 76, 5574–5583. [Google Scholar]
- Schaeffer, H.J. Compositions for treating viral infections and guanine acyclic nucleosides. U.S. Patent 4146715.
- Madre, M.A.; Zhuk, R.A.; Lidak, M.J. Analogs of purine nucleosides. I. Methods for synthesis of 9-(2-hydroxyethoxymethyl)guanine-acycloguanosine. Khim.-Farm. Zh. 1985, 19, 1371–1375. [Google Scholar]
- It is important to obtain 11 pure, because 3 is more difficult to purify. Compound 10 can be separated from 11 by addition of DMF (3 mL) to a sample of 1 g of the product and heating the mixture to 100 °C for 30 s. In this way, up to 100 mg of 6 can be dissolved and remains in solution even at room temperature, while 11 is insoluble. Thus, the mixture can be filtered, the solid recovered and dried.
- Sample Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suárez, R.M.; Matía, M.P.; Novella, J.L.; Molina, A.; Cosme, A.; Vaquero, J.J.; Alvarez-Builla, J. Synthesis of 9,9′-[1,2-Ethanediylbis(oxymethylene)]bis-2-amino-1,9-dihydro-6H-purin-6-one, an Impurity of Acyclovir. Molecules 2012, 17, 8735-8741. https://doi.org/10.3390/molecules17088735
Suárez RM, Matía MP, Novella JL, Molina A, Cosme A, Vaquero JJ, Alvarez-Builla J. Synthesis of 9,9′-[1,2-Ethanediylbis(oxymethylene)]bis-2-amino-1,9-dihydro-6H-purin-6-one, an Impurity of Acyclovir. Molecules. 2012; 17(8):8735-8741. https://doi.org/10.3390/molecules17088735
Chicago/Turabian StyleSuárez, Rosa M., Maria Paz Matía, José Luis Novella, Andres Molina, Antonio Cosme, Juan José Vaquero, and Julio Alvarez-Builla. 2012. "Synthesis of 9,9′-[1,2-Ethanediylbis(oxymethylene)]bis-2-amino-1,9-dihydro-6H-purin-6-one, an Impurity of Acyclovir" Molecules 17, no. 8: 8735-8741. https://doi.org/10.3390/molecules17088735