Reactivity of Ruthenium Vinylidene Complexes Containing Indenyl/dppe Ligands and Unsaturated Bonds at Cd with Trimethylsilyl Azide

Abstract
:1. Introduction
2. Results and Discussion
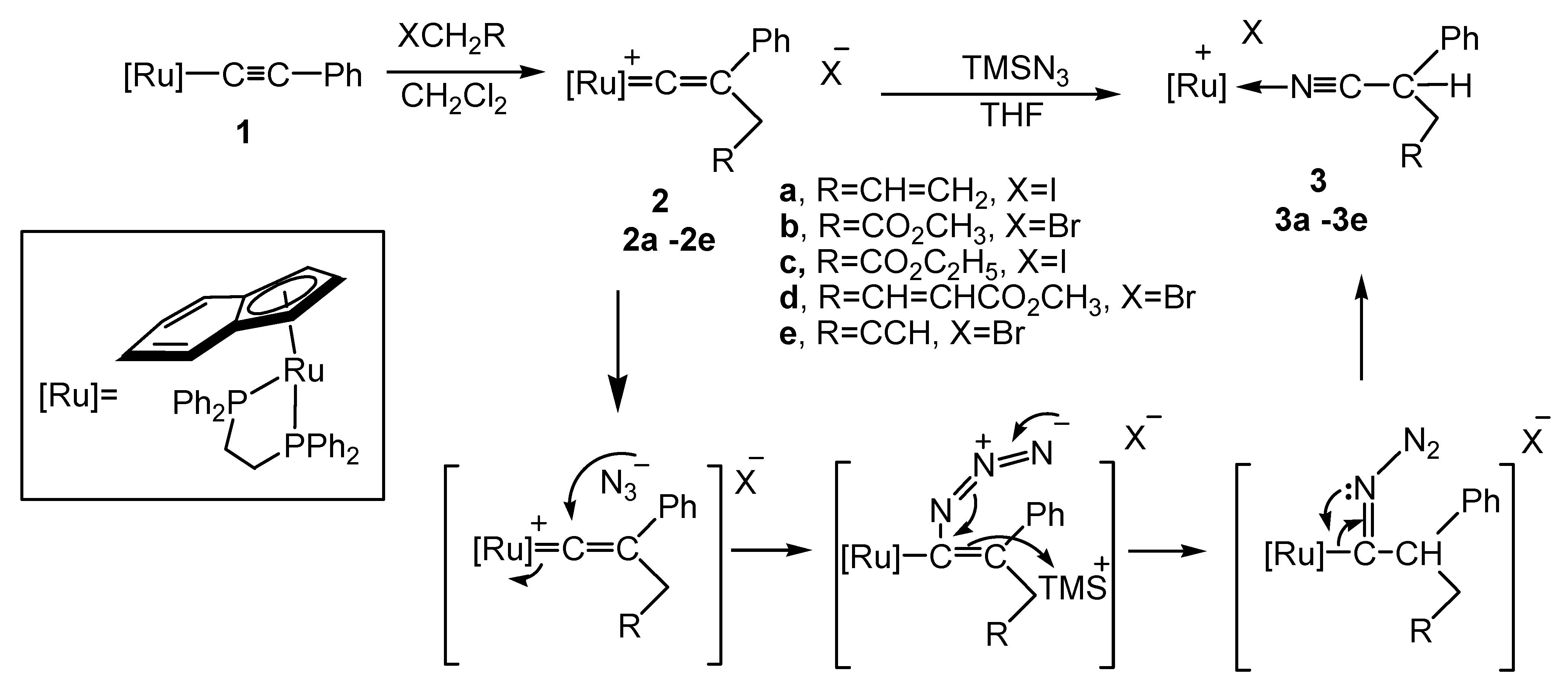
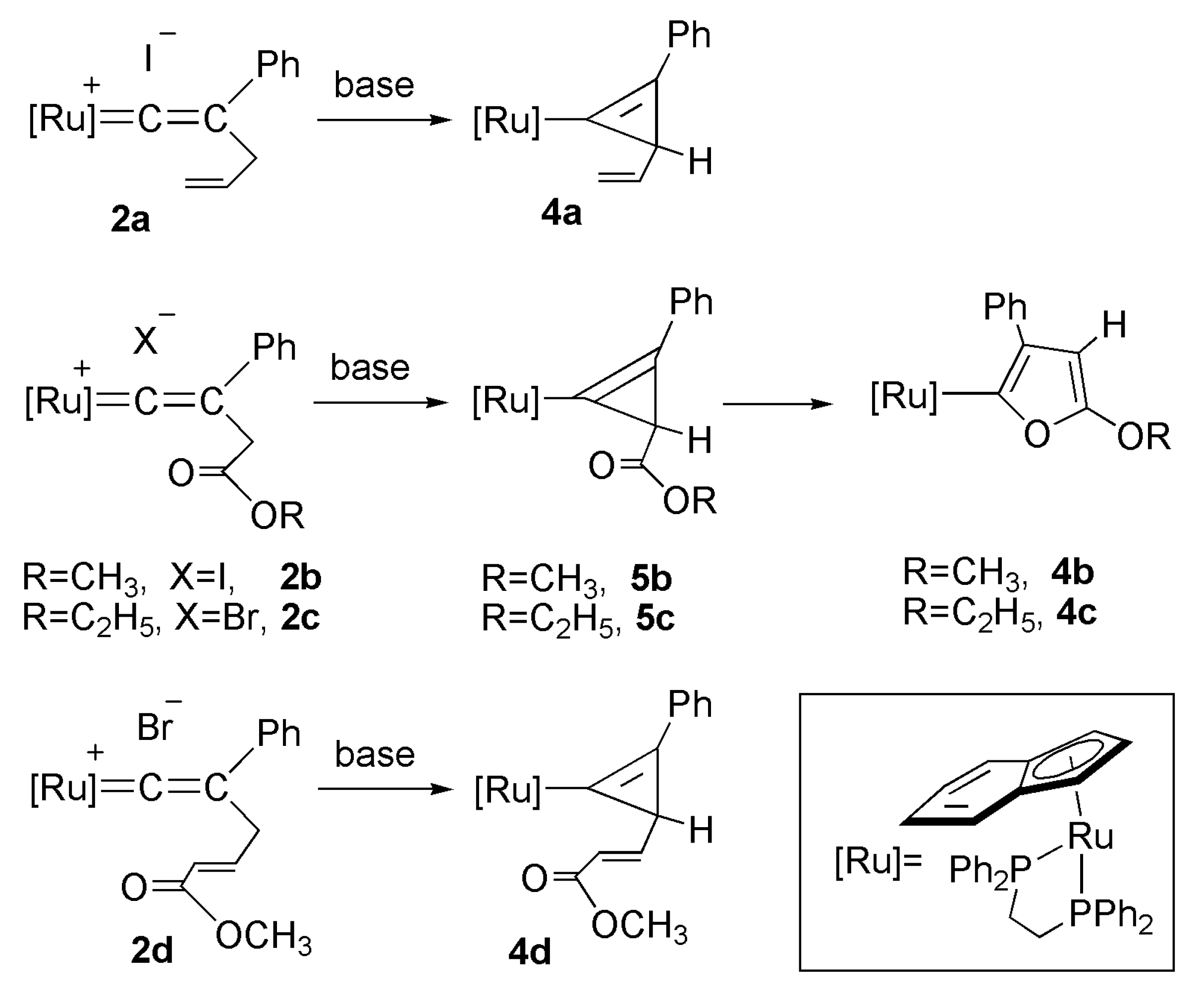
2.1. Preparation of Cationic Ruthenium Vinylidene Complexes 2a–e

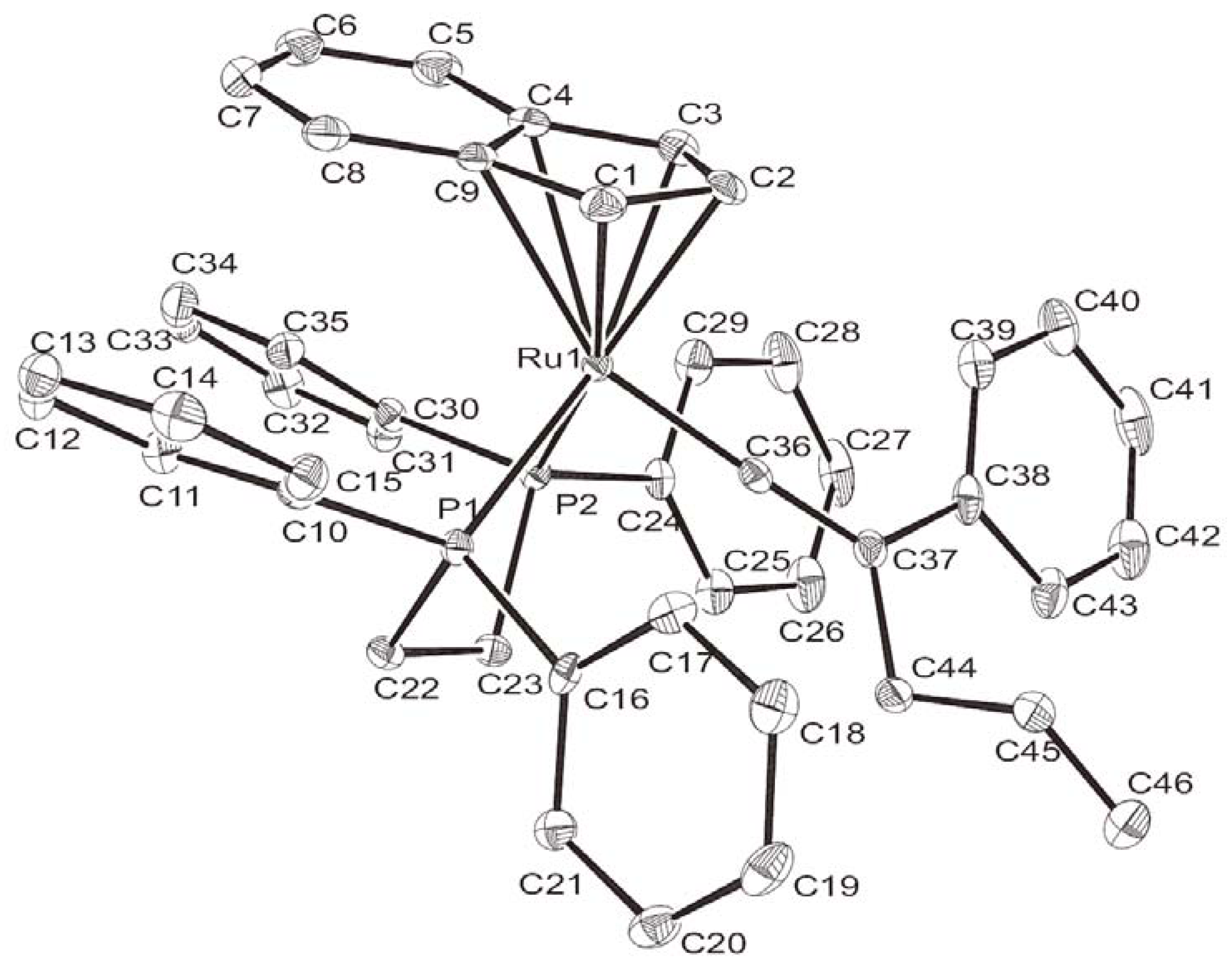

{kind=link}
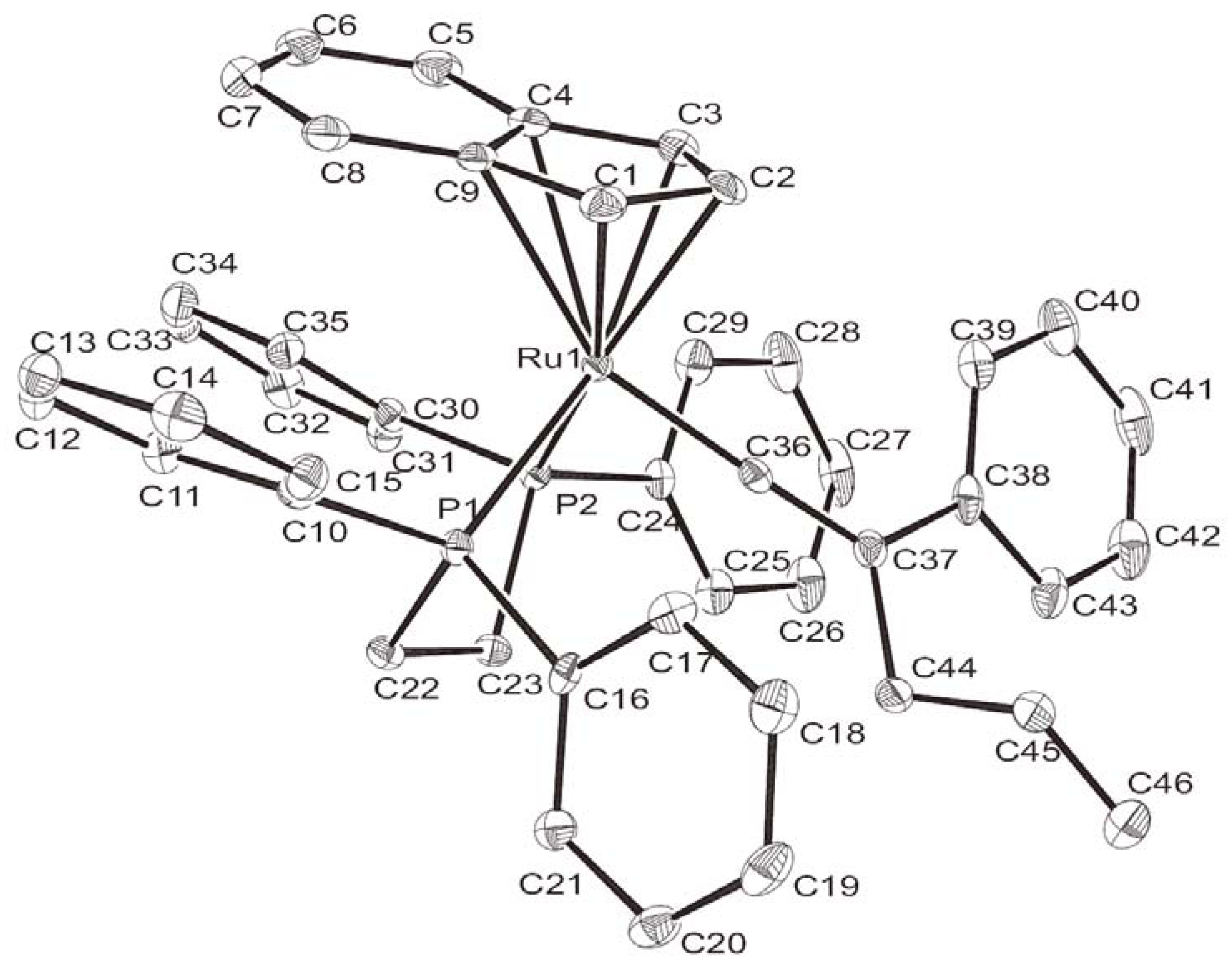
{kind=link}
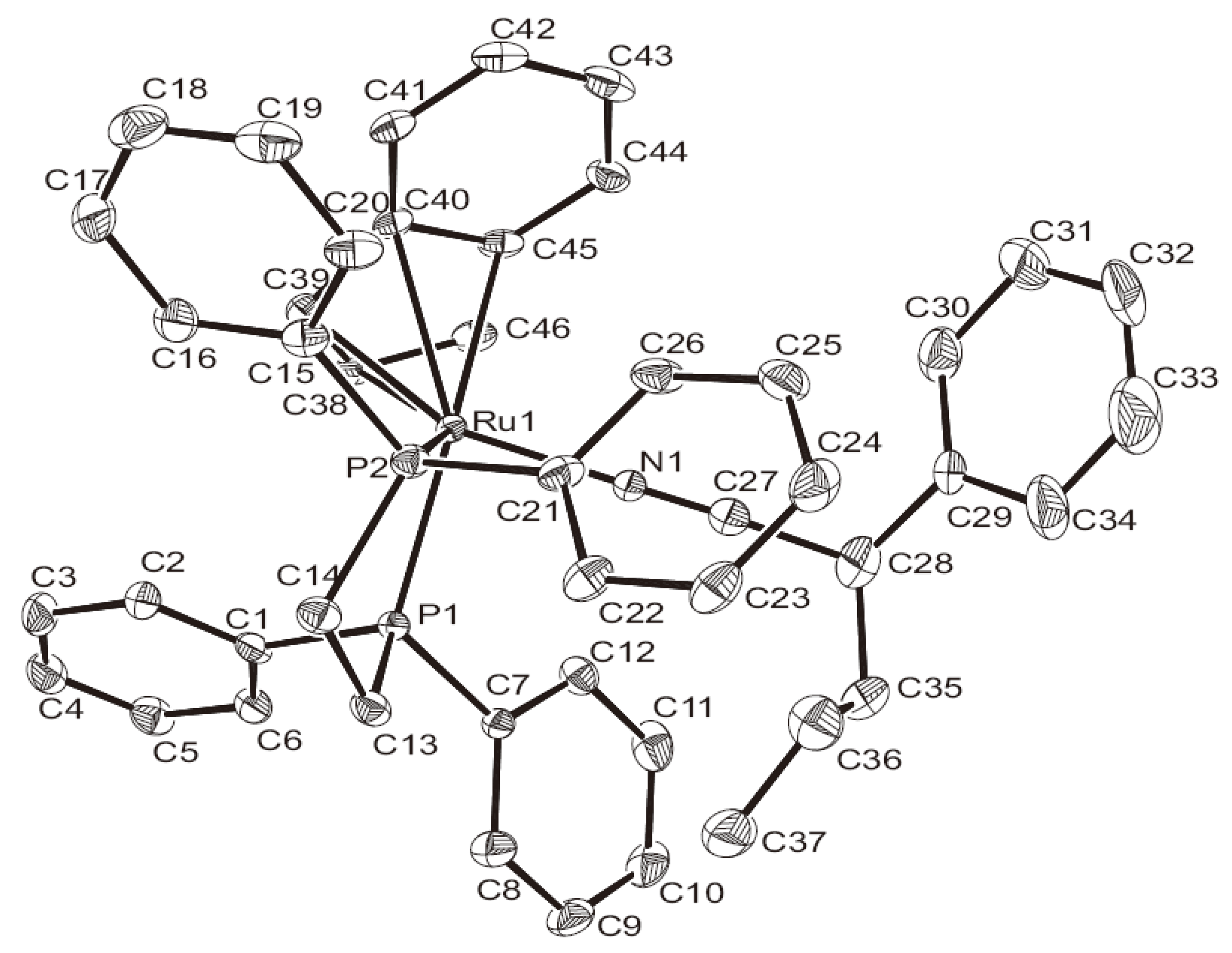
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
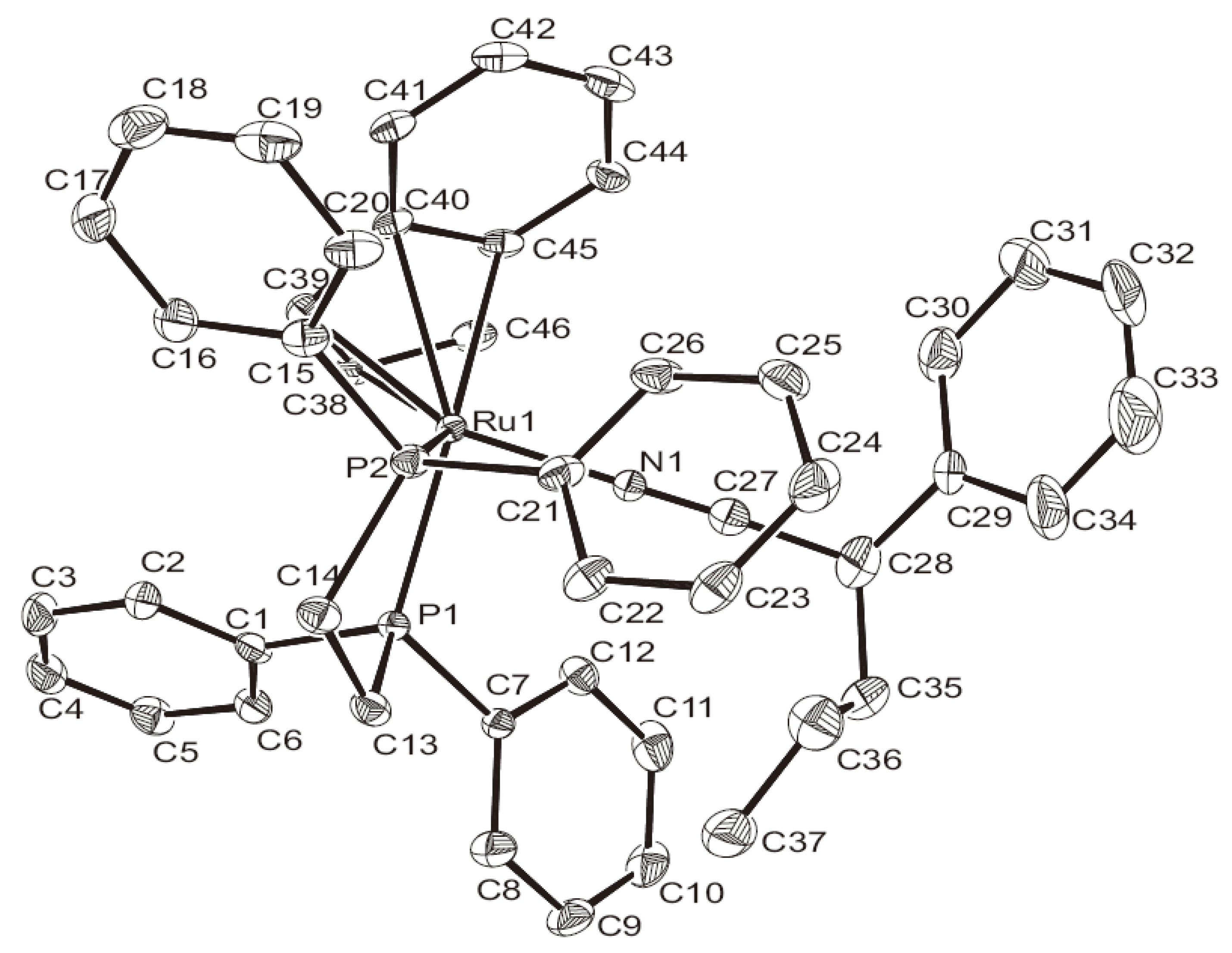
{kind=link}
| C(36)–Ru(1) | 1.841(4) | C(36)–C(37) | 1.326(6) |
| C(37)–C(44) | 1.538(6) | C(44)–C(45) | 1.515(6) |
| P(1)–Ru(1) | 2.3380(10) | C(45)–C(46) | 1.321(6) |
| P(2)–Ru(1) | 2.3104(10) | P(1)–Ru(1)–P(2) | 83.29(3) |
| C(37)–C(36)–Ru(1) | 177.2(4) | C(36)–C(37)–C(44) | 119.1(4) |
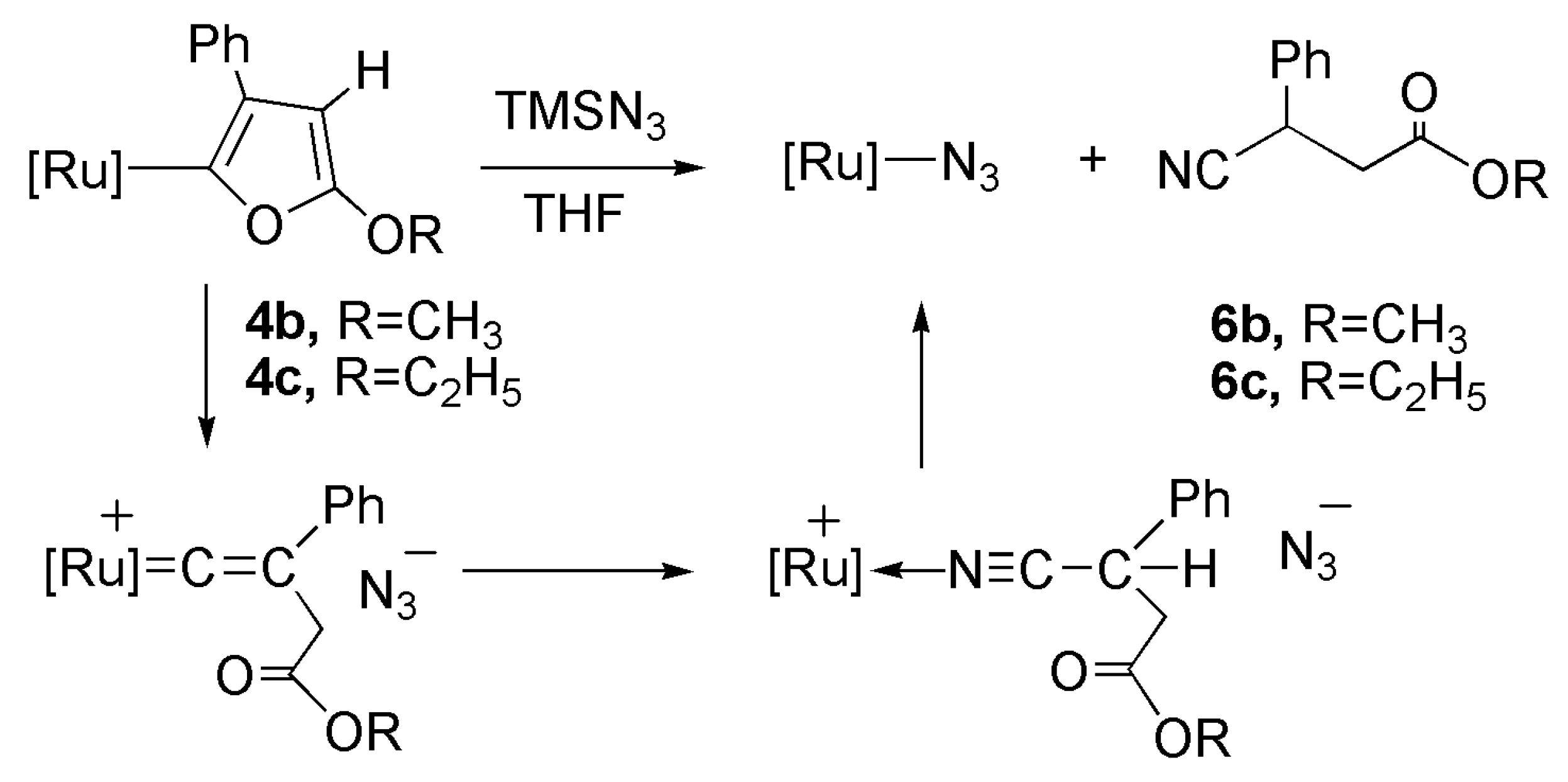
2.2. Reactivity of the Vinylidene Complexes with TMSN3
| N(1)–Ru(1) | 2.031(4) | C(27)–N(1) | 1.130(7) |
| C(27)–C(28) | 1.494(9) | C(28)–C(35) | 1.515(10) |
| P(1)–Ru(1) | 2.2637(13) | P(2)–Ru(1) | 2.2989(15) |
| C(27)–N(1)–Ru(1) | 174.1(6) | N(1)–C(27)–C(28) | 176.8(8) |
| P(1)-Ru(1)-P(2) | 84.34(5) | N(1)-Ru(1)-P(1) | 88.88(12) |
| N(1)-Ru(1)-P(2) | 92.02(15) |
| 2a | 3a | |
|---|---|---|
| Empirical formula | C46H41IP2Ru | C46H42INP2Ru |
| Temperature | 200(2) K | 200(2) K |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | P n a 21 | P 21/n |
| a, Å | 22.1483(8) | 10.4855(3) |
| b, Å | 11.8391(5) | 19.1064(6) |
| c, Å | 14.4101(6) | 19.8563(6) |
| α, deg | 90 | 90 |
| β, deg | 90 | 92.248(2) |
| γ, deg | 90 | 90 |
| Volume, Å3 | 3778.6(3) | 3975.0(2) |
| Z | 4 | 4 |
| Crystal size, mm3 | 0.27 × 0.14 × 0.11 | 0.28 × 0.24 × 0.08 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Flack parameters | −0.013(14) | |
| Final R indices [I > 2sigma(I)] | R1 = 0.0240, wR2 = 0.0548 | R1= 0.0465, wR2 = 0.1133 |
| R indices (all data) | R1 = 0.0283, wR2 = 0.0662 | R1 = 0.0682, wR2 = 0.1318 |
| 0.311 and −0.323 e | 1.323 and −0.897 e | |
| Largest diff. peak and hole, Å−3 | 776702 | 776705 |
| CCDC number |
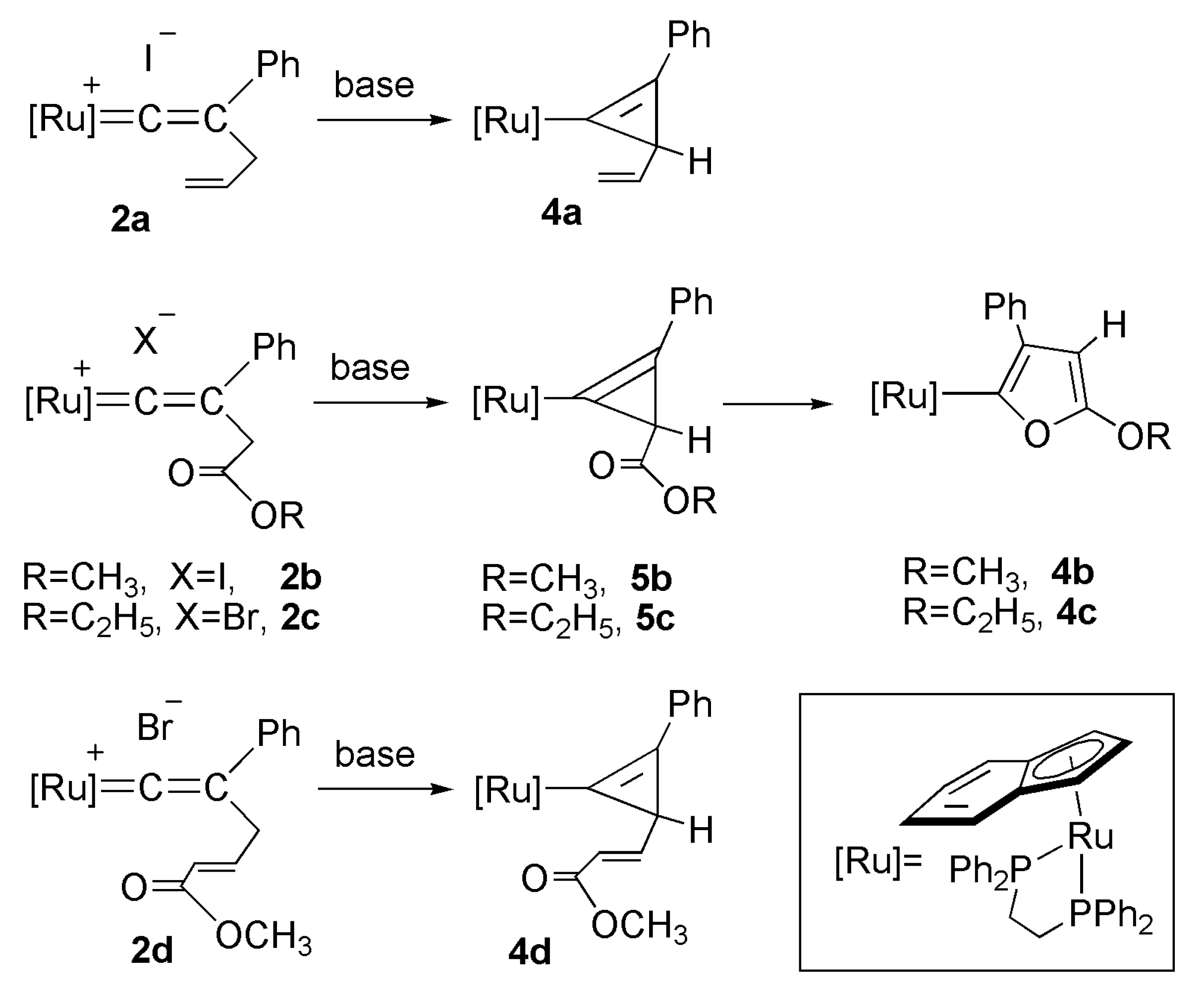
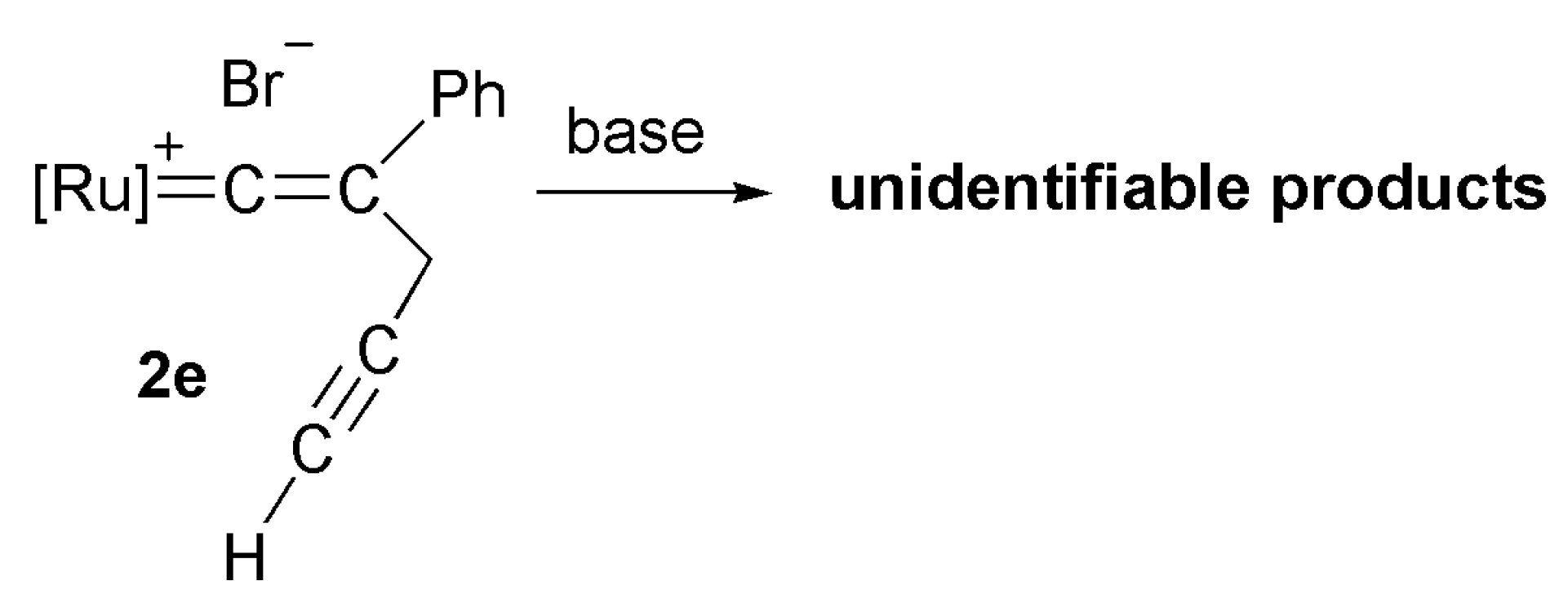
2.3. Deprotonation Reaction of the Vinylidene Complexes

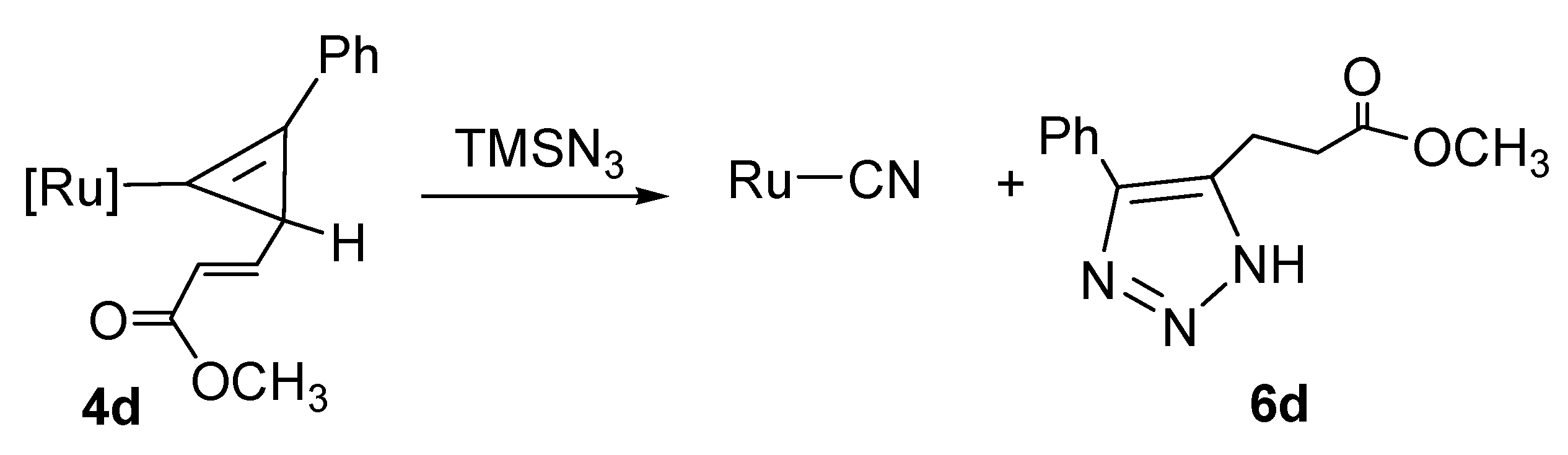
2.4. Reaction of Cyclopropenyl Complexes with TMSN3

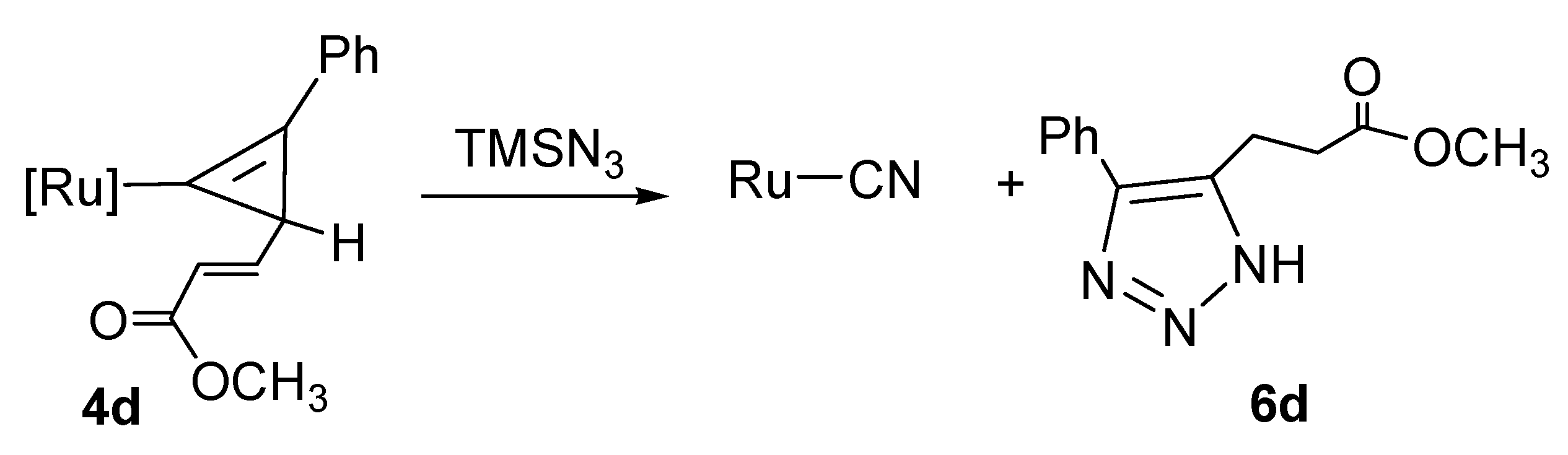
2.5. Reaction of Furyl Complexes with TMSN3

3. Experimental
3.1. General
3.2. Synthesis of [(η5-C9H7)(dppe)Ru=C=C(Ph)CH2CH=CH2][I] (2a)
3.3. Synthesis of [(η5-C9H7)(dppe)Ru=C=C(Ph)CH2CO2CH3][Br] (2b)
3.4. Synthesis of [(η5-C9H7)(dppe)Ru=C=C(Ph)CH2CO2C2H5][I] (2c)
3.5. Synthesis of [(η5-C9H7)(dppe)Ru=C=C(Ph)CH2CH=CHCO2CH3][Br] (2d)
3.6. Synthesis of [(η5-C9H7)(dppe)Ru=C=C(Ph)CH2C≡CH][Br] (2e)
3.7. Synthesis of the N-Coordinated Complexes [(η5-C9H7)(dppe)Ru–NCCH(Ph)CH2CH=CH2][I] (3a)
3.8. Synthesis of the N-Coordinated Complexes [(η5-C9H7)(dppe)Ru–NCCH(Ph)CH2CO2CH3][Br] (3b)
3.9. Synthesis of the N-Coordinated Complexes [(η5-C9H7)(dppe)Ru–NCCH(Ph)CH2CO2C2H5][I] (3c)
3.10. Synthesis of the N-Coordinated Complexes [(η5-C9H7)(dppe)Ru–NCCH(Ph)CH2CH=CH CO2CH3] [Br] (3d)
3.11. Synthesis of the N-Coordinated Complexes [(η5-C9H7)(dppe)Ru–NCCH(Ph)CH2C≡CH][Br](3e)
3.12. Synthesis of Cyclopropenylruthenium Complex (η5-C9H7)(dppe)Ru–C=C(Ph)CH-CH=CH2 (4a )
3.13. Synthesis of Furylruthenium Complex (η5-C9H7)(dppe)Ru–C=C(Ph)CH=C(OCH3)O (4b)
3.14. Synthesis of Furylruthenium Complex (η5-C9H7)(dppe)Ru–C=C(Ph)CH=C(OC2H5)O (4c)
3.15. Synthesis of Cyclopropenylruthenium Complex (η5-C9H7)(dppe)Ru–C=C(Ph)CHCH=CHC(O) OCH3 (4d)
3.16. Reaction of 4a with TMSN3
3.17. Reaction of 4b with TMSN3
3.18. Reaction of 4c with TMSN3
3.19. Reaction of 4d with TMSN3
3.20. X-ray Analysis of 2a and 3a
4. Conclusions
Acknowledgments
References
- Bruce, M.I. Organometallic chemistry of vinylidene and related unsaturated carbenes. Chem. Rev. 1991, 91, 197–257. [Google Scholar]
- Bruce, M.I. Transition Metal Complexes Containing Allenylidene, Cumulenylidene, and Related Ligands. Chem. Rev. 1998, 98, 2797–2858. [Google Scholar] [CrossRef]
- Trost, B.M.; McClory, A. Metal Vinylidenes as Catalytic Species in Organic Reactions. Chem. Asian J. 2008, 3, 164–194. [Google Scholar] [CrossRef]
- Davies, S.G.; McNally, J.P.; Smallridge, A.J. Chemistry of the Cyciopentadienyl Bisphosphine Ruthenium Auxiliary. Adv. Organomet. Chem. 1990, 30, 1–76. [Google Scholar]
- Cadierno, V.; Gamasa, M.P.; Gimeno, J. Synthesis and reactivity of α,β-unsaturated alkylidene and cumulenylidene Group 8 half-sandwich complexes. Coord. Chem. Rev. 2004, 248, 1627–1657. [Google Scholar]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom-Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Bruneau, C.; Dixneuf, P.H. Metal Vinylidenes in Catalysis. Accounts Chem. Res. 1999, 32, 311–323. [Google Scholar] [CrossRef]
- Trost, B.M.; Toste, F.D.; Pinkerton, A.B. Non-Metathesis Ruthenium-Catalyzed C-C Bond Formation. Chem. Rev. 2001, 101, 2067–2096. [Google Scholar]
- Bruce, M.I.; Swincer, A.G. Vinylidene and Propadienylidene (Allenylidene) Metal Complexes. Adv. Organomet. Chem. 1983, 22, 59–128. [Google Scholar]
- Rigaut, S.; Touchard, D.; Dixneuf, P.H. Ruthenium-allenylidene complexes and their specific behavior. Coord. Chem. Rev. 2004, 248, 1585–1601. [Google Scholar]
- Werner, H.; Weinhand, R.; Knaup, W. Vinylidene transition-metal complexes. 18. (Arene)osmium complexes containing alkynyl, vinyl, vinylidene, and thio- and selenoketene units as ligands: A series of organometallic compounds built up from 1-alkynes. Organometallics 1991, 10, 3967–3977. [Google Scholar]
- Rappert, T.O.; Mahr, N.; Wolf, J.; Werner, H. Vinylidene transition-metal complexes. Synthesis, reactivity, and structure of four-coordinate (vinylvinylidene)- and five- and six-coordinate enynyl(hydrido)rhodium complexes with [RhCl(PiPr3)2] as a molecular unit. Organometallics 1992, 11, 4156–4164. [Google Scholar]
- Puerta, M.C.; Valerga, P. Ruthenium and osmium vinylidene complexes and some related compounds. Coord. Chem. Rev. 1999, 193–195, 977–1025. [Google Scholar]
- Wakatsuki, Y. Mechanistic aspects regarding the formation of metal vinylidenes from alkynes and related reactions. J. Organomet. Chem. 2004, 689, 4092–4109. [Google Scholar] [CrossRef]
- Ghazala, S.I.; Paul, F.; Toupet, L.; Roisnel, T.; Hapiot, P.; Lapinte, C. Di-organoiron Mixed Valent Complexes Featuring “(η2-dppe)( η5-C5Me5)Fe” Endgroups: Smooth Class-III to Class-II Transition Induced by Successive Insertion of 1,4-Phenylene Units in a Butadiyne-Diyl Bridge. J. Am. Chem. Soc. 2006, 128, 2463–2476. [Google Scholar]
- Basallote, M.G.; Besora, M.; Duran, J.; Fernandez-Trujillo, M.J.; Lledos, A.; Manez, M.A.; Maseras, F. The Effect of the “Inert” Counteranions in the Deprotonation of the Dihydrogen Complex trans-[FeH(η2-H2)(dppe)2]+: Kinetic and Theoretical Studies. F. J. Am. Chem. Soc. 2004, 126, 2320–2321. [Google Scholar] [CrossRef]
- Narvor, N.L.; Toupet, L.; Lapinte, C. Elemental Carbon Chain Bridging Two Iron Centers: Syntheses and Spectroscopic Properties of [Cp*(dppe)Fe-C4-FeCp*(dppe)]n+·n[PF6]−. X-ray Crystal Structure of the Mixed Valence Complex (n = l). J. Am. Chem. Soc. 1995, 117, 7129–7138. [Google Scholar]
- Adams, R.D.; Davison, A.; Selegue, J.P. Cationic vinylidene complexes. Preparation and structural characterization of (h5-cyclopentadienyl)(2-methyl-4,5-bis(diphenylphosphino)-2-penten-3-yl)iron (II). A base-induced interligand reaction in a vinylidene complex. J. Am. Chem. Soc. 1979, 101, 7232–7238. [Google Scholar]
- Sung, H.-L.; Hsu, H.-L. The reactivity of TMSN3 with ruthenium cyclopropenyl complexes containing different ligands and different substituent at Cγ. J. Organomet. Chem. 2011, 696, 1280–1288. [Google Scholar]
- Tanase, T.; Mochizuki, H.; Sato, R.; Yamamoto, Y. Formation of nitriles by a novel metathesis reaction of ruthenium σ-acetylide complexes, (h5-indenyl)Ru(h1-C≡CR)(phosphine)2, with nitric oxide. J. Organomet. Chem. 1994, 466, 233–236. [Google Scholar]
- Hill, A.F.; Hulkes, A.G.; White, A.J.P.; Williams, D.J. Selenolatovinylidene Complexes: Metal-Mediated Alkynyl Selenoether Rearrangements. Organometallics 2000, 19, 371–373. [Google Scholar]
- Bruce, M.I.; Ellis, B.G.; Low, P.J.; Skelton, B.W.; White, A.H. Syntheses, Structures, and Spectro-electrochemistry of {Cp*(PP)Ru}CCCC{Ru(PP)Cp*} (PP = dppm, dppe) and Their Mono- and Dications. Organometallics 2003, 22, 3184–3198. [Google Scholar]
- Cadierno, V.; Gamasa, M.P.; Gimeno, J.; Perez-Carreno, E.; Garcia-Granda, S. A Novel Route to Functionalized Terminal Alkynes through η1-Vinylidene to η2-Alkyne Tautomerizations in Indenyl−Ruthenium(II) Monosubstituted Vinylidene Complexes: Synthetic and Theoretical Studies. Organometallics 1999, 18, 2821–2832. [Google Scholar]
- Cadierno, V.; Gamasa, M.P.; Gimeno, J.; Gonzalez-Bernardo, C. Reactivity of Indenyl-Ruthenium(II) Vinylidene Complexes: Selective Synthesis of Alkenyl-Phosphonio Derivatives via Nucleophilic Addition of Triphenylphosphine on Their h2-Alkyne Tautomers. Theoretical Study of the h1-Vinylidene-h2-Alkyne Tautomerization. Organometallics 2001, 20, 5177–5188. [Google Scholar]
- Gamasa, M.P.; Gimeno, J.; Martin-Vaca, M.B.; Borge, J.; Garcia-Granda, S.; Perez-Carreno, E. Synthesis and Characterization of (Alkyny1)-, (Vinylidene)-, and (Carbene)ruthenium Indenyl Complexes: X-ray Crystal Structure of [Ru(=C=CMe2)(h5-C9H7)( PPh3)2[ CF3SO3]·1/2CH2C12 and EHMO Calculation. Organometallics 1994, 13, 4045–4057. [Google Scholar] [CrossRef]
- Cadierno, V.; Gamasa, M.P.; Gimeno, J.; Borge, J.; Garcia-Granda, S. Selective Synthesis of Indenylruthenium(II) Vinylvinylidene Complexes via Unstable Allenylidene Intermediates: Unexpected Formation of Alkenyl-Phosphonio Complexes (E)-[Ru{C(H)=C(PPh3)R}(η5-C9H7)PPh3)2][PF6](R=1-Cyclohexenyl, 1-Cycloheptenyl) through Nucleophilic Addition of Triphenylphosphine on Vinylvinylidene Derivatives. Organometallics 1997, 16, 3178–3187. [Google Scholar] [CrossRef]
- Davison, A.; Solar, J.P. The reactivity of cyclopentadienylcarbonyliron acetylides with electrophiles: The isolation of a 1,3-dimetallo-stabilized cyclobutenium ion and a cationic “vinylidene”. J. Organomet. Chem. 1978, 155, C8–C12. [Google Scholar]
- Jolly, P.W.; Pettit, R. 1-(π-Cyclopentadienyliron dicarbonyl)propyne. J. Organomet. Chem. 1968, 12, 491–495. [Google Scholar]
- Bell, R.A.; Chisholm, M.H.; Couch, D.A.; Rankel, L.A. Reactions of alkynyl- and alkenylplatinum(II) compounds. 1. Formation of alkoxycarbene ligands within the coordination sphere of platinum. Inorg. Chem. 1977, 16, 677–686. [Google Scholar] [CrossRef]
- Bell, R.A.; Chisholm, M.H. Reactions of Furylruthenium Complexes with Oxygen and Trimethylsilyl Azide. Inorg. Chem. 1977, 16, 687–697. [Google Scholar]
- Gamasa, M.P.; Gimeno, J.; Lastra, E.; Lanfranchi, M.; Tiripicchio, A. Reactions of cationic vinylidene complexes [Fe{=C=C(R1)R2}(η5-C5H5)(dppm)]+[dppm=bis(diphenylphosphino)methane] with nucleophiles: Stereoselective synthesis and crystal structure of the alkenyl complex (E)-[Fe{C(H)=C(Me)Ph}(η5-C5H5)(dppm)]. J. Organomet. Chem. 1992, 430, C39–C43. [Google Scholar] [CrossRef]
- Kostic, N.M.; Fenske, R.F. Molecular orbital study of bonding, conformations, and reactivity of transition-metal complexes containing unsaturated organic ligands. Electrophilic and nucleophilic additions to acetylide, vinylidene, vinyl, and carbene ligands. Organometallics 1982, 1, 974–982. [Google Scholar]
- Werner, H.; Wolf, J.; Muller, G.; Kruger, C. Ambidentate Behavior of Mononuclear Vinylidenerhodium Complexes—Novel CC Coupling of a Methyl to a Vinylidene Group. Angew. Chem. Int. Ed. Engl. 1984, 23, 431–432. [Google Scholar] [CrossRef]
- Bruce, M.I.; Swincer, A.G. Cyclopentadienyl-ruthenium and -osmium chemistry. X. Reactions of vinylidene complexes with alcohols and water: Syntheses of alkoxy(alkyl)carbene, acyl and alkyl complexes. Aust. J. Chem. 1980, 33, 1471–1483. [Google Scholar]
- Barrett, A.G.M.; Carpenter, N.E.; Sabat, M.J. Isolation of [Cp(CO)(Ph3P)FeNCCH3]+BF4− from a vinylidene precursor; an organometallic Beckmann rearrangement. Organomet. Chem. 1988, 352, C8–C12. [Google Scholar]
- Chang, K.H.; Lin, Y.C. Reactions of ruthenium cyclopropenyl complexes with trimethylsilyl azide. Chem. Commun. 1998, 21, 1441–1442. [Google Scholar] [CrossRef]
- Chang, K.H.; Lin, Y.C.; Liu, Y.H.; Wang, Y. Reactions of ruthenium cyclopropenyl complexes with trimethylsilyl azide. J. Chem. Soc. Dalton Trans. 2001, 21, 3154–3159. [Google Scholar]
- Chang, C.W.; Lin, Y.C.; Lee, G.-H.; Huang, S.L.; Wang, Y. Reactions of Ruthenium Acetylide Complexes with Isothiocyanate. Organometallics 1998, 17, 2534–2542. [Google Scholar] [CrossRef]
- Kawata, Y.; Sato, M. Double Deprotonation of a Cationic Ruthenium(II) Terminal Vinylidene Complex and Molecular Structures of the Terminal Vinylidene Complex [(h5-C5Me5)(PPh3)2Ru(CCH2)]PF6 and the Acetylide Complex (h5-C5Me5)(PPh3)2Ru(CCSiMe3). Organometallics 1997, 16, 1093–1096. [Google Scholar] [CrossRef]
- Huisgen, R. Descript reactions of organic alkynes with azide. In 1,3-Dipolar Cycloadditions Chemistry; Padwa, A., Ed.; Wiley: New York, NY, USA, 1984; Volume 1, pp. 1–176. [Google Scholar]
- Maiorana, S.; Pocar, D.; Croce, P.D. Studies in the enamine field reactions of sulfonyl- and nitro-enamines with azides. Tetrahedron Lett. 1966, 7, 6043–6045. [Google Scholar] [CrossRef]
- Meek, J.S.; Fowler, J. Nucleophilic addition-elimination reactions of 1,2-bis(p-tolylsulfonyl)ethane. J. Org. Chem. 1968, 33, 985–991. [Google Scholar] [CrossRef]
- Gouault, N.; Cupif, J.F.; Sauleau, A.; David, M. Methyl-substituted-γ-butyrolactones: Solid-phase synthesis employing a cyclisation-cleavage strategy. Tetrahedron Lett. 2000, 41, 7293–7297. [Google Scholar]
- Rostovtsec, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Raghavendra, M.S.; Lam, Y. Regiospecific solid-phase synthesis of substituted 1,2,3-triazoles. Tetrahedron Lett. 2004, 45, 6129–6132. [Google Scholar] [CrossRef]
- Krasinski, A.; Fokin, V.V.; Sharpless, K.B. Direct Synthesis of 1,5-Disubstituted-4-magnesio-1,2,3-triazoles. Org. Lett. 2004, 6, 1237–1240. [Google Scholar] [CrossRef]
- Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Zunino, E.; Vaccaro, L. Synthesis of 4-Aryl-1H-1,2,3-triazoles through TBAF-Catalyzed [3+2] Cycloaddition of 2-Aryl-1-nitroethenes with TMSN3 under Solvent-Free Conditions. J. Org. Chem. 2005, 70, 6526–6529. [Google Scholar]
- Coats, S.J.; Link, J.S.; Gauthier, D.; Hlasta, D.J. Trimethylsilyl-Directed 1,3-Dipolar Cycloaddition Reactions in the Solid-Phase Synthesis of 1,2,3-Triazoles. Org. Lett. 2005, 7, 1469–1472. [Google Scholar] [CrossRef]
- Huang, C.C.; Lin, Y.C.; Huang, S.L.; Liu, Y.H.; Wang, Y. Synthesis of Dinuclear and Trinuclear Ruthenium Cyclopropenyl Complexes. Organometallics 2003, 22, 1512–1518. [Google Scholar]
- Chang, C.W.; Lin, Y.C.; Lee, G.H.; Wang, Y. Reactions of Ruthenium Cyclopropenyl Complexes Containing Pentamethylcyclopentadienyl Ligands. Organometallics 2000, 19, 3211–3219. [Google Scholar]
- Lo, Y.H.; Lin, Y.C.; Lee, G.H.; Wang, Y. Synthesis and Reactivity of the Ruthenium Cyclopropenyl Complex with a Tp Ligand. Organometallics 1999, 18, 982–988. [Google Scholar]
- Ting, P.C.; Lin, Y.C.; Lee, G.H.; Cheng, M.C.; Wang, Y. Cyclopropenation and Related Reactions of Ruthenium Vinylidene Complexes. J. Am. Chem. Soc. 1996, 118, 6433–6444. [Google Scholar] [CrossRef]
- Ting, P.C.; Lin, Y.C.; Cheng, M.C.; Wang, Y. Novel Method for the Preparation of Metal Cyclopropenyl Complexes from Vinylidene Complexes with an Electron-Withdrawing Substituent. Organometallics 1994, 13, 2150–2152. [Google Scholar]
- Yen, Y.S.; Lin, Y.C.; Liu, Y.H.; Wang, Y. Deprotonation of Iron Vinylidene Complexes Containing a dppe Ligand. Organometallics 2007, 26, 1250–1255. [Google Scholar]
- Alvarez, P.; Lastra, E.; Gimeno, J.; Bassetti, M.; Falvello, L.R. Formation of a Cyclobutylidene Ring: Intramolecular [2+2] Cycloaddition of Allyl and Vinylidene CC Bonds under Mild Conditions. J. Am. Chem. Soc. 2003, 125, 2386–2387. [Google Scholar]
- Chang, K.H.; Sung, H.L.; Lin, Y.C. Reactions of Furylruthenium Complexes with Oxygen and Trimethylsilyl Azide. Eur. J. Inorg. Chem. 2006, 3, 649–655. [Google Scholar]
- Lo, Y.H.; Lin, Y.C.; Huang, C.C. Synthesis and reactivity of ruthenium tetrazolate complexes containing a tris(pyrazolyl)borato (Tp) ligand. J. Organomet. Chem. 2008, 693, 117–127. [Google Scholar]
- Chang, K.H.; Lin, Y.C. Reactions of ruthenium cyclopropenyl complexes with trimethylsilyl azide. Chem. Commun. 1998, 14, 1441–1442. [Google Scholar] [CrossRef]
- Selnau, H.E.; Merola, J.S. Reactions of iridium complex [Ir(COD)(PMe3)3]Cl with benzene, pyridine, furan, and thiophene: Carbon-hydrogen cleavage vs. ring opening. Organometallics 1993, 12, 1583–1591. [Google Scholar]
- Sample Availability: Samples of complexes 2-5 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sung, H.-L.; Her, T.-M.; Su, W.-H.; Cheng, C.-P. Reactivity of Ruthenium Vinylidene Complexes Containing Indenyl/dppe Ligands and Unsaturated Bonds at Cd with Trimethylsilyl Azide. Molecules 2012, 17, 8533-8553. https://doi.org/10.3390/molecules17078533
Sung H-L, Her T-M, Su W-H, Cheng C-P. Reactivity of Ruthenium Vinylidene Complexes Containing Indenyl/dppe Ligands and Unsaturated Bonds at Cd with Trimethylsilyl Azide. Molecules. 2012; 17(7):8533-8553. https://doi.org/10.3390/molecules17078533
Chicago/Turabian StyleSung, Hui-Ling, Tze-Min Her, Wen-Hsien Su, and Chin-Pao Cheng. 2012. "Reactivity of Ruthenium Vinylidene Complexes Containing Indenyl/dppe Ligands and Unsaturated Bonds at Cd with Trimethylsilyl Azide" Molecules 17, no. 7: 8533-8553. https://doi.org/10.3390/molecules17078533
APA StyleSung, H.-L., Her, T.-M., Su, W.-H., & Cheng, C.-P. (2012). Reactivity of Ruthenium Vinylidene Complexes Containing Indenyl/dppe Ligands and Unsaturated Bonds at Cd with Trimethylsilyl Azide. Molecules, 17(7), 8533-8553. https://doi.org/10.3390/molecules17078533