3.1. General
All reactions were performed under argon with magnetic stirring unless otherwise specified. Dry solvents were used in all experiments. Thin layer chromatography was performed on E. Merck pre-coated 60 F254 plates and compounds were observed by UV or by charring the plates with an acidic anisaldehyde system. Flash chromatography was performed on an Armen SpotFlash with silica gel 60 (particle size 15–40 mm). Optical rotations were measured on a Perkin-Elmer 341 polarimeter at 20 °C. Electrospray ionization mass spectrometry experiments (MS and HRMS) were obtained on a hybrid tandem quadrupole/time-of-flight (Q-TOF) instrument, equipped with a pneumatically assisted electrospray (Z-spray) ion source (Micromass, Manchester, UK) operated in positive mode (EV = 30V, 80 °C, injection flow 5 mL/min). NMR spectra were recorded on Bruker 600, 500 or 250 MHz spectrometer. Chemical shifts were measured in δ (ppm) and coupling constants J in Hz (solvent peak reference: δ = 7.27 for 1H, 77.0 for 13C). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) or br (broad). Chemical shifts (δ) reported are referred to internal tetramethylsilane. FT-IR spectra were recorded on Nicolet Avatar 320 FT-IR as films. Elemental analyses were performed with a Thermo Flash EA 1112 Series.
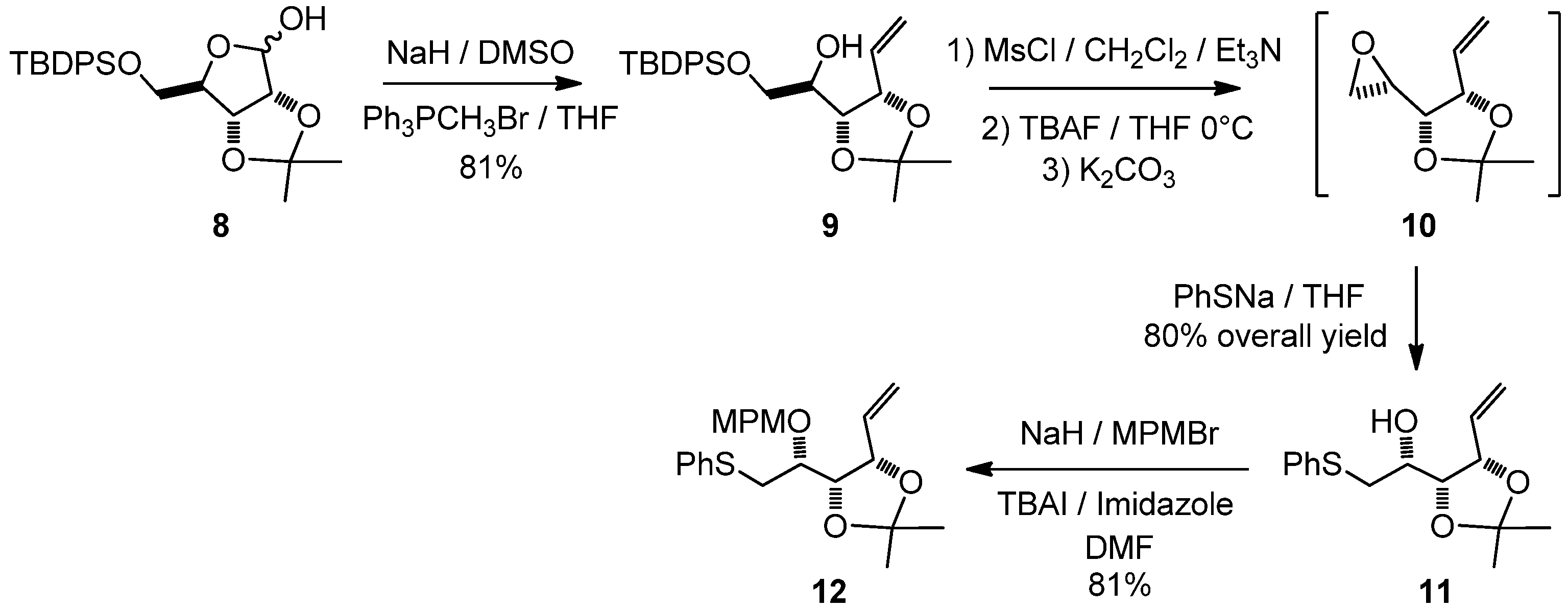
1,2-Dideoxy-6-O-(tert-butyldiphenylsilyloxy)-3,4-di-O-isopropylidene-D-ribo-hex-1-enitol (9)
A sodium hydride suspension in mineral oil (1.16 g, 28.9 mmol) was washed three times with n-hexane and then suspended in DMSO (15.9 mL). After heating to 68 °C for 45 min, the grey-green mixture was transferred via syringe to a suspension of methyltriphenylphosphonium bromide (10.33 g, 28.9 mmol, co-evaporated with toluene before use) in dry THF (110 mL). After stirring for 10 min, a solution of lactol 8 (3 g, 7.23 mmol) in dry THF (50 mL) was transferred via a cannula. The yellow mixture was stirred at room temperature for 16 h and then poured into ice water (200 mL). After extraction with diethyl ether (3 × 500 mL), the combined organic phases were washed with a saturated NaCl solution (50 mL). The organic phase was dried with MgSO4, and after filtration and evaporation to dryness, the residue was purified by column chromatography to give 9 (2.17 g, 5.1 mmol, 81%) as a slightly yellow oil. [α]20D = −0.4° (C = 1.04, CHCl3); 1H-NMR (CDCl3): δ 7.66–7.77 (m, 4 H, Ar), 7.34–7.51 (m, 6 H, Ar), 6.04 (ddd, 1 H, J2,1trans 17.1 Hz, J2,1cis 10.4 Hz, J2,3 6.7 Hz, H-2), 5.44 (d, 1 H, J1trans,2 17.1 Hz, H-1trans), 5.30 (d, 1 H, J1cis,2 10.4 Hz, H-1cis), 4.73 (t, 1 H, J3,2 6.7 Hz, J3,4 6.7 Hz, H-3), 4.17 (dt, 1 H, J4,5 13.7 Hz, J4,3 6.7 Hz, H-4), 3.87 (m, 2 H, H-6), 3.67-3.80 (m, 1 H, H-5), 2.65 (br s, 1 H, OH), 1.41 (s, 3 H, 1 CH3), 1.37 (s, 3 H, 1 CH3), 1.10 (s, 9 H, Si-(CH3)3); 13C-NMR (CDCl3): δ 136.1 (Cortho or Cmeta), 136.0 (Cortho or Cmeta), 134.5 (C-2), 133.5 (CqAr), 133.5 (CqAr), 130.3 (Cpara), 128.3 (CAr), 128.2 (CAr), 118.1 (C-1), 109.2 (Cq isopropylidene), 79.2 (C-3), 77.9 (C-4), 70.3 (C-5), 65.7 (C-6), 28.2 (CH3 isopropylidene), 27.3 (C(CH3)3), 25.9 (CH3 isopropylidene), 19.8 (C(CH3)3); IR (neat) vmax: 3564, 3481, 2934, 2859, 1429, 1218, 1114, 1059, 704 cm–1; HRMS (m/z, ESI) calculated for C25H34O4NaSi: (M+Na) = 449.2124 (calculated), 449.2120 (found).
1,2-Dideoxy-3,4-di-O-isopropylidene-6-S-phenylthio-L-lyxo-hex-1-enitol (11)
Triethylamine (4.8 mL, 34.5 mmol) followed by methanesulfonyl chloride (1.16 mL, 15 mmol) were added drop-wise to a solution of compound 9 (4.89 g, 11.5 mmol) in methylene chloride (70 mL) at 0 °C. After stirring for 16 h at room temperature, the mixture was diluted with diethyl ether (300 mL), washed with a saturated ammonium chloride solution (50 mL), and then with a saturated sodium chloride solution (50 mL). The organic phase was dried with MgSO4, filtered and evaporated to dryness. Dry THF (110 mL) was added and reaction mixture was cooled to 0 °C. A tetra-n-butyl ammonium fluoride solution (1 M in THF; 23 mL) was added drop-wise, and the reaction mixture was stirred at room temperature for 3 h. After addition of K2CO3 (2.8 g, 20 mmol) and stirring for 16 h, the reaction mixture was diluted with water (25 mL) and extracted with diethyl ether (3 × 300 mL). The combined organic phases were dried with MgSO4, filtered and evaporated to dryness. The crude epoxide 10 was used in the next step without further purification. Dry THF (50 mL) was then added and the reaction mixture was cooled to 0 °C, and a THF solution of sodium thiophenoxide was added [prepared by addition of thiophenol (2.94 mL, 28.8 mmol) to a suspension of sodium (1.15 g, 28.8 mmol) in dry THF (50 mL)]. The reaction was stirred at room temperature for 16 h. The crude mixture was then slowly quenched with methanol (5 mL), a half-saturated ammonium chloride solution (30 mL) was added and the aqueous layer was extracted with diethyl ether (3 × 300 mL). The combined organic phases were dried with MgSO4, filtered and evaporated, and the residue purified by column chromatography to give 11 (2.56 g, 9.1 mmol, 80%) as a colorless oil. [α]20D = +27.3° (C = 1.04, CHCl3); 1H-NMR (CDCl3): δ 7.13–7.43 (m, 5 H, HAr), 5.95 (ddd, 1 H, J2,1trans 17.4 Hz, J2,1cis 10.1 Hz, J2,3 8.1 Hz, H-2), 5.33 (d, 1 H, H-1trans), 5.24 (d, 1 H, H-1cis), 4.59 (t, 1 H, J3,4 7.5 Hz, H-3), 4.27 (dd, 1 H, J4,5 3.6 Hz, H-4), 3.68 (td, 1 H, J5,6 6.5 Hz, H-5), 3.05 (dd, 2 H, H-6), 2.44 (br s, 1 H, OH), 1.52 (s, 3 H, 1 CH3), 1.38 (s, 3 H, 1 CH3); 13C-NMR (CDCl3): δ 135.9 (CqAr), 134.2 (C-2), 130.2 (Cortho or Cmeta), 129.4 (Cortho or Cmeta), 126.9 (Cpara), 120.3 (C-1), 109.3 (Cq isopropylidene), 79.5 (C-3 or C-4), 78.9 (C-3 or C-4), 68.8 (C-5), 38.3 (C-6), 27.5 (CH3 isopropylidene), 25.3 (CH3 isopropylidene); IR (neat) vmax: 3480, 2988, 2935, 1585, 1482, 1458, 1439, 1380, 1215, 1057, 741, 693 cm–1; HRMS (m/z, ESI) calculated for C15H20O3NaS: (M+Na) = 303.1031 (calculated), 303.1038 (found).
1,2-Dideoxy-3,4-di-O-isopropylidene-5-O-(4-methoxybenzyl)-6-S-phenylthio-L-lyxo-hex-1-enitol (12)
A solution of 11 (2.38 g, 8.49 mmol) in a mixture of THF/DMF (18 mL/4.5 mL) was added drop-wise to a stirring suspension of sodium hydride (560 mg, 17 mmol) in a mixture of THF/DMF (18 mL/4.5 mL) at 0 °C. After addition of tetra-n-butylammonium iodide (314 mg, 0.85 mmol) and imidazole (58 mg, 0.85 mmol), p-methoxybenzyl bromide (2.48 mL, 17 mmol) was added drop-wise and the yellow suspension was stirred at room temperature for 16 h. The reaction was quenched by the addition of methanol (5 mL) at 0 °C and after addition of water (30 mL), the reaction mixture was extracted with diethyl ether (3 × 300 mL). The combined organic phases were dried with MgSO4, filtered and evaporated, and the residue purified by column chromatography to give 12 (2.76 g, 6.89 mmol, 81%) as a colorless oil. [α]20D = −2.8° (C = 1.02, CH3OH); 1H-NMR (CDCl3): δ 7.11–7.50 (m, 7 H, HAr), 6.77–6.95 (m, 2 H, HAr), 5.84 (ddd, 1 H, J2,1trans 17.2 Hz, J2,1cis 10.2 Hz, J2,3 8.3 Hz, H-2), 5.26 (d, 1 H, H-1trans), 5.16 (d, 1 H, H-1cis), 4.59 (s, 2 H, CH2Ph), 4.50 (dd, 1 H, J3,4 6.1 Hz, H-3), 4.39 (t, 1 H, J4,5 6.1 Hz, H-4), 3.80 (s, 3 H, OMe), 3.55 (dd, 1 H, J5,6 11.7 Hz, H-5), 3.11 (dd, 2 H, H-6), 1.52 (s, 3 H, 1 CH3), 1.37 (s, 3 H, 1 CH3); 13C-NMR (CDCl3): δ 159.5 (CqArOMe), 136.7 (CqArSPh), 134.8 (C-2), 130.9 (CqArCH2O), 130.3 (CAr), 129.8 (CAr), 129.3 (CAr), 126.7 (CAr/paraS), 119.6 (C-1), 114.1 (CAr/orthoOMe), 109.7 (Cq isopropylidene), 80.0 (C-4), 79.5 (C-3), 77.0 (C-5), 72.8 (CH2Ph), 55.7 (OMe), 35.5 (C-6), 28.0 (CH3 isopropylidene), 26.0 (CH3 isopropylidene); IR (neat) vmax: 2986, 2935, 2837, 1613, 1614, 1248, 1038, 742, 693 cm–1; HRMS (m/z, ESI) calculated for C23H28O4NaS: (M+Na) = 423.1606 (calculated), 423.1601 (found).
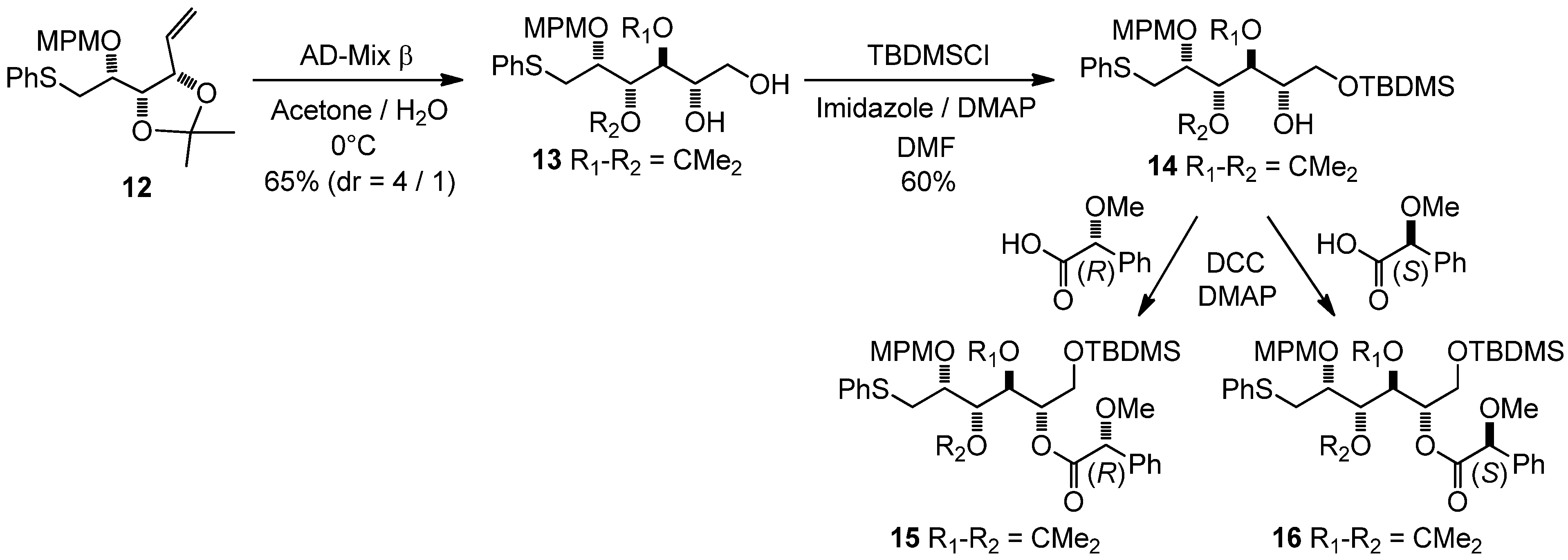
3,4-Di-O-isopropylidene-2-O-(4-methoxybenzyl)-1-S-phenylthio-L-altritol (13)
A solution of 12 (654 mg, 1.63 mmol) in a mixture of acetone/water (4 mL 1:1) was added to AD-Mix b (3 g) in a mixture of acetone/water (16 mL 1:1) at 0 °C. After stirring for 18 h, sodium sulfite (3.34 g) was added. After dilution with water (2 mL), the reaction was extracted with methylene chloride (3 × 100 mL). The combined organic phases were dried with MgSO4, filtered and evaporated, and the residue purified by column chromatography to give 13 (368 mg, 52%) as the major diastereoisomer, and 3,4-di-O-isopropylidene-2-O-(4-methoxybenzyl)-1-S-phenylthio-D-galactitol (92 mg, 13%) as the minor one, both as colorless oils (diastereoisomeric ratio 4/1), with 11% of recovered starting material. The same reaction with AD-Mix α gave 13 (336 mg, 47.5%) as the major diastereoisomer, and 3,4-di-O-isopropylidene-2-O-(4-methoxybenzyl)-1-S-phenylthio-D-galactitol (46 mg, 6.5%) as the minor diastereoisomer, (diastereoisomeric ratio 7.3/1), with 20% of recovered starting material.
Major diol (13): [α]20D = −0.4° (C = 0.975, CH3OH); 1H-NMR (CDCl3): δ 7.10–7.46 (m, 7 H, HAr), 6.84 (d, 2 H, Jortho,meta/OMe 8.6 Hz, HAr), 4.70 (d, 1 H, JCHPh,CPh 10.9 Hz, CH2Ph), 4.52 (d, 1 H, CH2Ph), 4.41 (dd, 1 H, J3,4 6.1 Hz, J3,2 4.3 Hz, H-3), 4.09 (dd, 1 H, J4,5 9.3 Hz, H-4), 3.93–4.02 (m, 1 H, H-2), 3.84–3.93 (m, 1 H, H-5), 3.73–3.82 (m, 4 H, H-6a, OMe), 3.61 (dd, 1 H, J6b,6a 11.2 Hz, J6b,5 5.6 Hz, H-6b), 3.42–3.56 (br s, 1 H, OH), 3.46 (dd, 1 H, J1a,2 5.0 Hz, J1a,1b 13.7 Hz, H-1a), 3.25 (dd, 1 H, J1b,2 7.2 Hz, H-1b), 2.22 (br s, 1 H, OH), 1.46 (s, 3 H, 1 CH3), 1.31 (s, 3 H, 1 CH3); 13C-NMR (CDCl3): δ 159.9 (CqArOMe), 136.7 (CqArSPh), 130.1 (CqArCH2O), 130.1 (CAr), 129.9 (CAr), 129.5 (CAr), 126.7 (CAr/paraS), 114.3 (CAr/orthoOMe), 109.2 (Cq isopropylidene), 78.0 (C-2), 77.8 (C-4), 77.6 (C-3), 73.3 (CH2Ph), 70.0 (C-5), 65.1 (C-6), 55.7 (OMe), 35.3 (C-1), 27.4 (CH3 isopropylidene), 25.5 (CH3 isopropylidene); IR (neat) vmax: 3422, 2987, 2933, 1612, 1514, 1249, 1070, 1035, 744 cm–1; HRMS (m/z, ESI) calculated for C23H30O6NaS: (M+Na) = 457.1661 (calculated), 457.1649 (found).
Minor diol (3,4-di-O-isopropylidene-2-O-(4-methoxybenzyl)-1-S-phenylthio-D-galactitol): 1H-NMR (CDCl3): δ 7.23-7.45 (m, 5 H, HAr), 7.19 (d, 2 H, Jortho,meta/OMe 8.6 Hz, H ortho,meta/OMe), 6.84 (m, 2 H, H meta,ortho/OMe), 4.65 (d, 1 H, JCHPh,CPh 11.0 Hz, CH2Ph), 4.52 1 H, (dd, J3,4 6.6 Hz, J3,2 2.0 Hz, H-3), 4.40 (d, 1 H, CH2Ph), 4.02 (d, 1 H, H-4), 3,80 (s, 3 H, OMe), 3.68–3.76 (m, 1 H, H-2 or H-5), 3.61–3.68 (m, 1 H, H-2 or H-5), 3.56 (s, 1 H, OH), 3.50 (s, 2 H, H-6), 3.35 (dd, 1 H, J1a,2 4.1 Hz, J1a,1b 13.7 Hz, H-1a), 3.22 (dd, 1 H, J1a,2 8.9 Hz, H-1b), 1.51 (s, 3 H, 1 CH3), 1.27 (s, 3 H, 1 CH3).
6-O-(tert-Butyldimethylsilyloxy)-3,4-di-O-isopropylidene-2-O-(4-methoxybenzyl)-1-S-phenylthio-L-altritol (14)
Imidazole (28 mg, 0.4 mmol), TBDMSCl (67 mg, 0.44 mmol) and DMAP (10 mg, 80 mmol) were added to a solution of 13 (175 mg, 0.4 mmol) in dry DMF (4 mL) at 0 °C. After warming to room temperature and stirring for 6 h, the reaction mixture was diluted with CH2Cl2 (50 mL), washed with water (5 mL), and then with saturated NH4Cl (5 mL). The organic phase was dried with MgSO4, filtered and evaporated, and the residue purified by column chromatography to give 14 (130 mg, 60%) as a colorless oil. The crude alcohol 14 was used without any further purification.
6-O-(tert-Butyldimethylsilyloxy)-3,4-di-O-isopropylidene-2-O-(4-methoxybenzyl)-5-O-(R)-2-methoxy-phenylacetyl-1-S-phenylthio-L-altritol (15) and 6-O-(tert-Butyldimethylsilyloxy)-3,4-di-O-isopropyl-idene-2-O-(4-methoxybenzyl)-5-O-(S)-2-methoxyphenylacetyl-1-S-phenylthio-L-altritol (16)
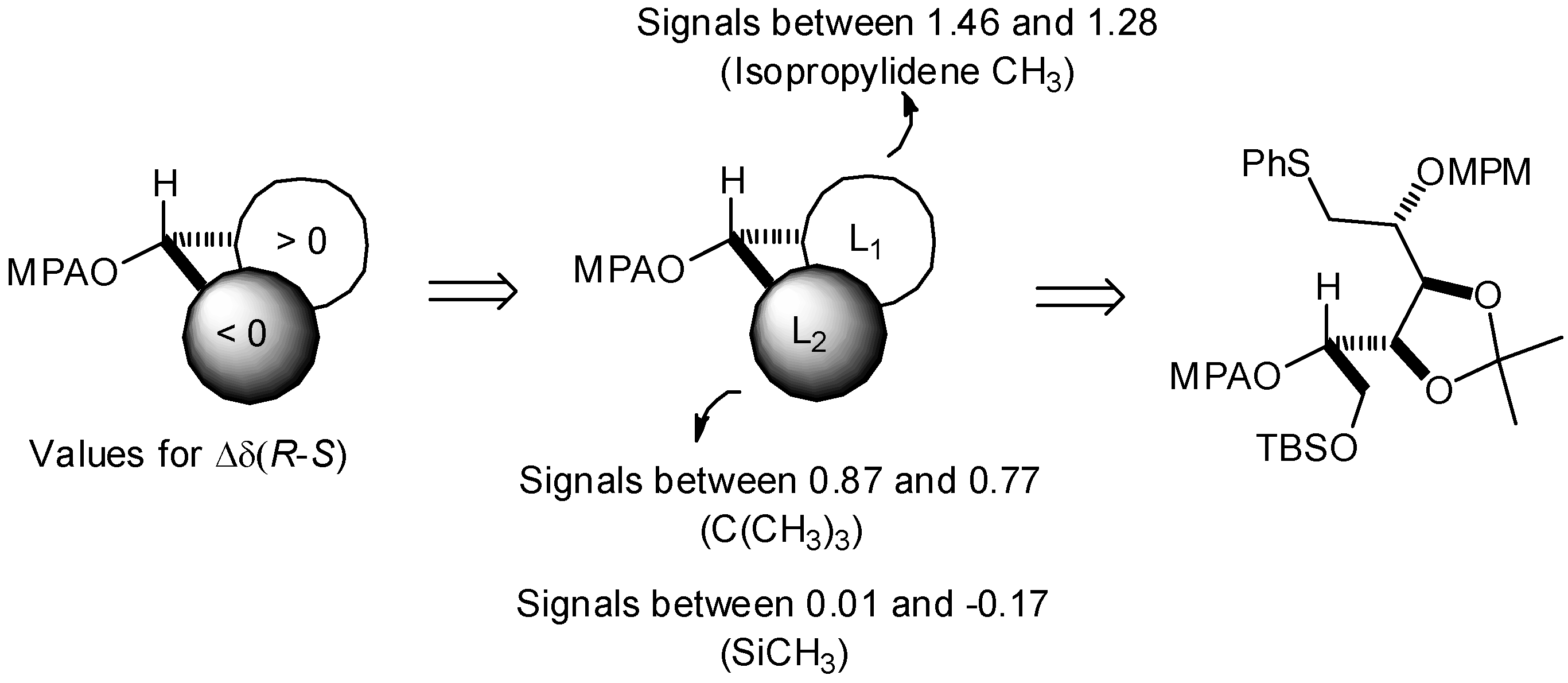
DMAP (3 mg, 20 mmol), (−)-R-MPA (80 mg, 0.48 mmol) and DCC (99 mg, 0.48 mmol) were added to a solution of 14 (65 mg, 120 mmol) in dry CH2Cl2 (5 mL) at 0 °C. After stirring for 1 h, the reaction mixture was diluted with CH2Cl2 (30 mL), washed with saturated NH4Cl (5 mL), and then with brine (5 mL). The organic phase was dried with MgSO4, filtered and evaporated. The residue was purified by column chromatography to give 15 as a colorless oil. 1H-NMR (CDCl3): δ 7.13–7.46 (m, 12 H, HAr), 6.85 (d, 2 H, Jortho,meta/OMe 8.6 Hz, HAr), 5.17 (m, 1 H, J5,4 8.0 Hz H-5), 4.63 (s, 3 H, CHOMe), 4.58 (d, 1 H, JCHPh,CPh 11.1 Hz, CH2Ph), 4.50 (d, 1 H, CH2Ph), 4.47 (m, 2 H, H-3, H-4), 3.80 (s, 3 H, OMe), 3.73 (m, 2 H, 2 H-6), 3.44 (m, 1 H, H-2), 3.29 (m, 3 H, CHOMe), 3.23 (m, 2 H, 2 H-1), 1.46 (s, 3 H, 1 CH3), 1.34 (s, 3 H, 1 CH3), 0.77 (s, 9 H, 1 C(CH3)3), −0.11 (s, 3 H, SiCH3); −0.17 (2s, 2 × 3 H, 2 SiCH3). The same procedure with (+) S-MPA gave 16 as a colorless oil. 1H-NMR (CDCl3): δ 7.11–7.47 (m, 12 H, HAr), 6.82 (d, 2 H, Jortho,meta/OMe 8.6 Hz, HAr), 5.20 (m, 1 H, J5,4 7.2 Hz H-5), 4.69 (s, 3 H, CHOMe), 4.52 (d, 1 H, JCHPh,CPh 11.0 Hz, CH2Ph), 4.37 (d, 1 H, CH2Ph), 4.34 (dd, 1 H, J4,3 6.2 Hz, H-4), 4.03 (dd, 1 H, J3,2 1.2 Hz, H-3), 3.91 (dd, 1 H, J6a,6b 11.5 Hz, J6a,5 1.5 Hz, H-6a), 3.78 (s, 3 H, OMe), 3.76 (dd, 1 H, J6b,5 4.4 Hz, H-6b), 3.35 (m, 3 H, CHOMe), 3.12 (m, 1 H, H-2), 3.00 (dd, 1 H, J1a,2 4.5 Hz, J1a,1b 12.7 Hz, H-1a), 2.94 (dd, 1 H, H-1b), 1.41 (s, 3 H, 1 CH3), 1.28 (s, 3 H, 1 CH3), 0.87 (s, 9 H, 1 C(CH3)3), 0.01 (2s, 2 × 3 H, 2 SiCH3).
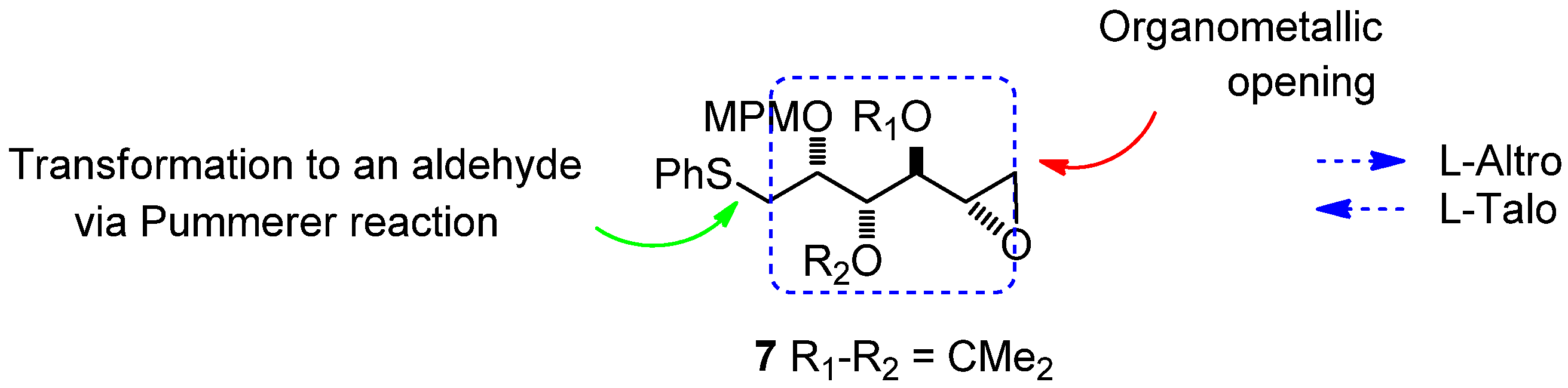
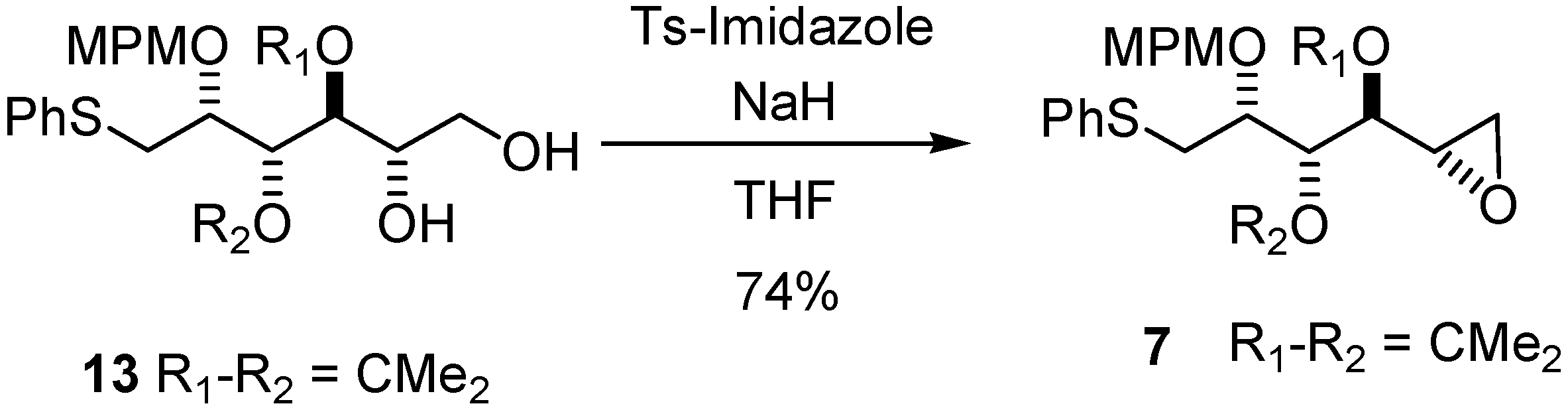
1,2-Anhydro-3,4-di-O-isopropylidene-2-O-(4-methoxybenzyl)-1-S-phenylthio-L-altritol (7)
Sodium hydride 60% suspension in mineral oil (56 mg, 1.7 mmol) was added to a solution of 13 (100 mg, 0.23 mmol) in dry THF (2.4 mL) at 0 °C After 15 min at 0 °C tosylimidazole (104 mg, 468 mmol) was then added. The reaction mixture was stirred for 16 h. After addition of diethylether (20 mL), washing with saturated sodium bicarbonate (5 mL), saturated ammonium chloride (5 mL) and brine (5 mL), the combined organic phases were dried with MgSO4, filtered and evaporated, and the residue purified by column chromatography to give 7 (71 mg, 74%) as a slightly yellow oil. [α]20D = −16.4° (C = 0.22, CHCl3); 1H-NMR (CDCl3): δ 7.43 (d, 2 H, Jmeta,ortho 7.7 Hz, HAr), 7. 3 (m, 4 H, HAr), 7.21 (m, 1 H, HAr), 6.87 (d, 2 H, Jortho 8.5 Hz, HAr), 4.67 (d, 1 H, Jgem 11.0 Hz, CH2Ph), 4.64 (d, 1 H, Jgem 11.0 Hz, CH2Ph), 4.52 (dd, 1 H, J3,4 5.7 Hz, J3,2 5.6 Hz, H-3), 3.96 (ddd, 1 H, J2,1 5.6 Hz, H-2), 3.82 (s, 3 H, OMe), 3.59 (dd, 1 H, J4,5 8.1 Hz, H-4), 3.35 (dd, 1 H, Jgem 13.6 Hz, H-1a), 3.25 (dd, 1 H, H-1b), 3.12 (ddd, J5,6a 4.9 Hz, J5,6b 2.5 Hz, 1 H, H-5), 2.83 (dd, 1 H, H-6a), 2.62 (dd, 1 H, H-6b), 1.53 (s, 3 H, 1 CH3), 1.36 (s, 3 H, 1 CH3); 13C-NMR (CDCl3): δ 159.2 (CqArOMe), 136.3 (CqArSPh), 130.4 (CqArCH2O), 129.6 (2CAr), 128.9 (CAr), 126.2 (CAr/paraS), 113.7 (CAr/orthoOMe), 109.2 (Cq isopropylidene), 78.8 (C-4), 78.7 (C-3), 76.3 (C-2), 72.3 (CH2Ph), s55.3 (OMe), 49.4 (C-5), 46.1 (C-6), 35.1 (C-1), 27.6 (CH3 isopropylidene), 25.3 (CH3 isopropylidene); IR (neat) vmax: 3057, 2989, 2933, 1653, 1615, 1559, 1540, 1515, 1248, 1086, 1038, 741 cm–1; HRMS (m/z, ESI) calculated for C23H28O5NaS: (M+Na) = 439.1555 (calculated), 439.1557 (found).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















