Development of a Method for the Preparation of Ruthenium Indenylidene-Ether Olefin Metathesis Catalysts

Abstract
:1. Introduction
2. Results and Discussion
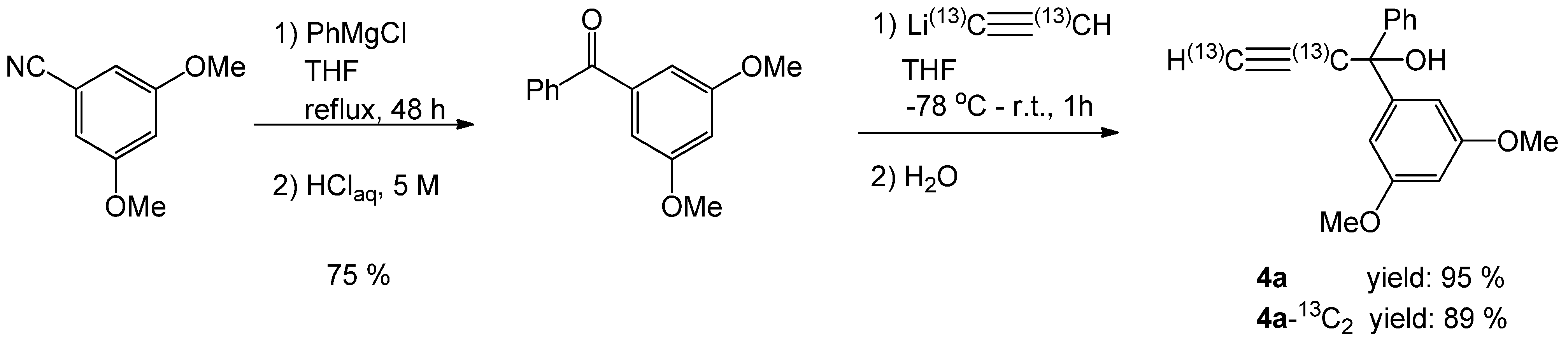
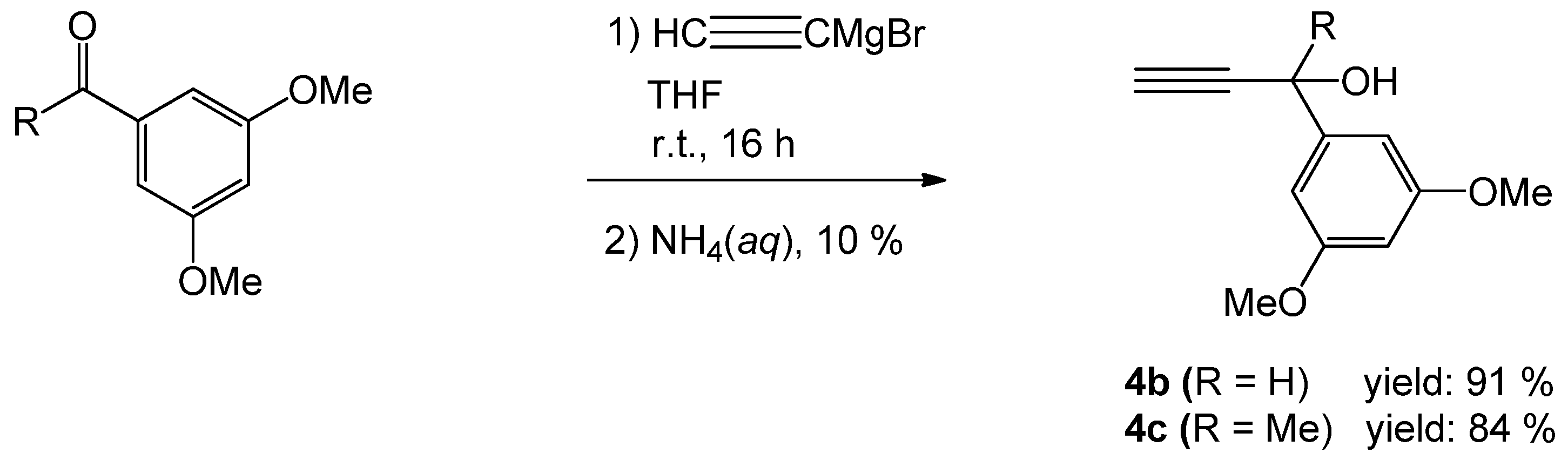
2.1. Synthesis of the 1-Phenylprop-2-yn-1-ol Derivatives
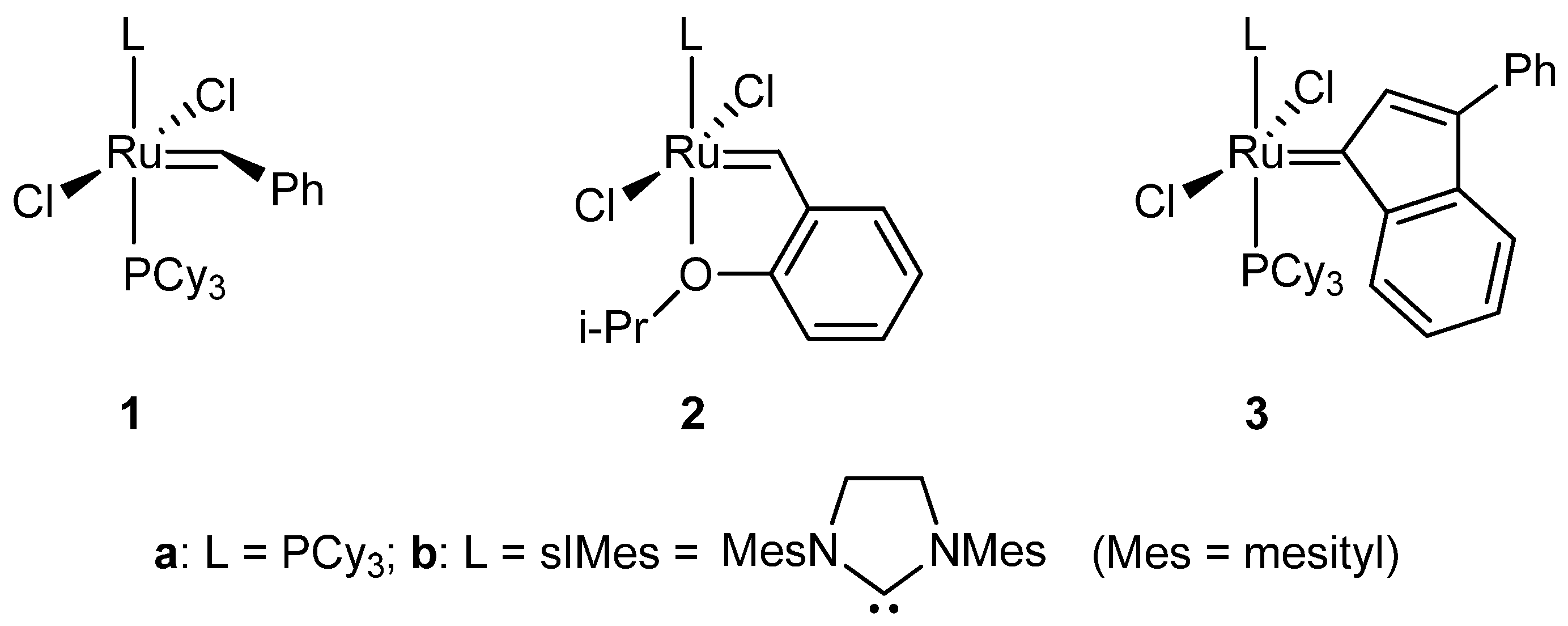
2.2. Reaction of 1-Phenylprop-2-yn-1-ol Derivatives with Different Ruthenium Starting Materials
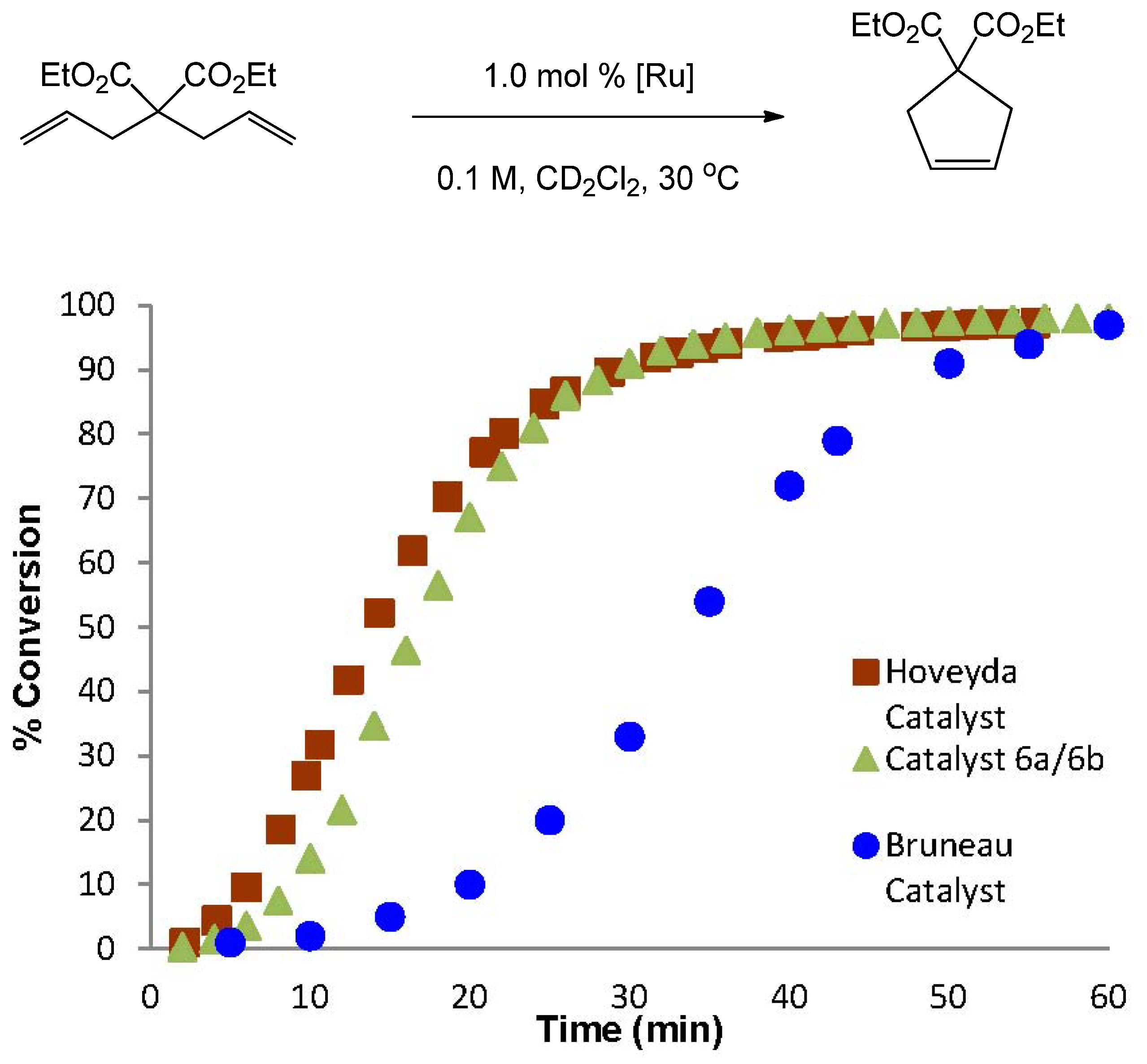
2.3. RCM Activity of the Ruthenium Indenylidene-Ether Complexes
3. Experimental
3.1. General
3.2. Preparation of the 1-Phenylprop-2-yn-1-ol Derivatives 4b and 4c
3.2.1. Preparation of 1-(3,5-Dimethoxyphenyl)-prop-2-yn-1-ol (4b)
3.2.2. Preparation of 1-(3,5-Dimethoxyphenyl)-1-methylprop-2-yn-1-ol (4c)
3.3. Reaction between 1-Phenylprop-2-yn-1-ol Derivatives and Different Ruthenium Starting Materials
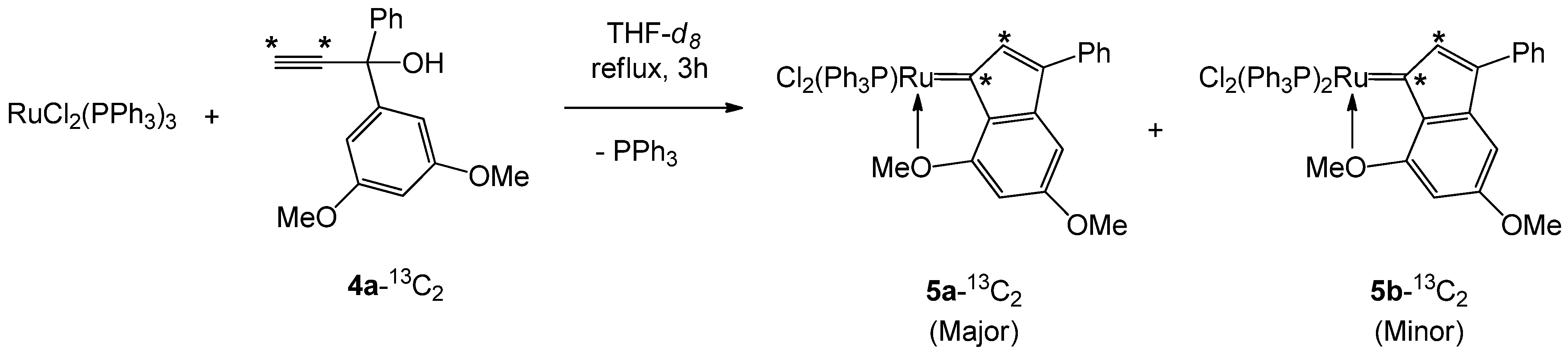
3.3.1. NMR Study of the Reaction between RuCl2(PPh3)3 and 1-(3,5-dimethoxyphenyl)-1-phenylprop-2-yn-1-ol (4a)
3.3.2. NMR study of the Reaction between RuCl2(PPh3)3 and 1-(3,5-Dimethoxyphenyl)-1-Phenylprop-2-yn-1-ol (4a-13C2)
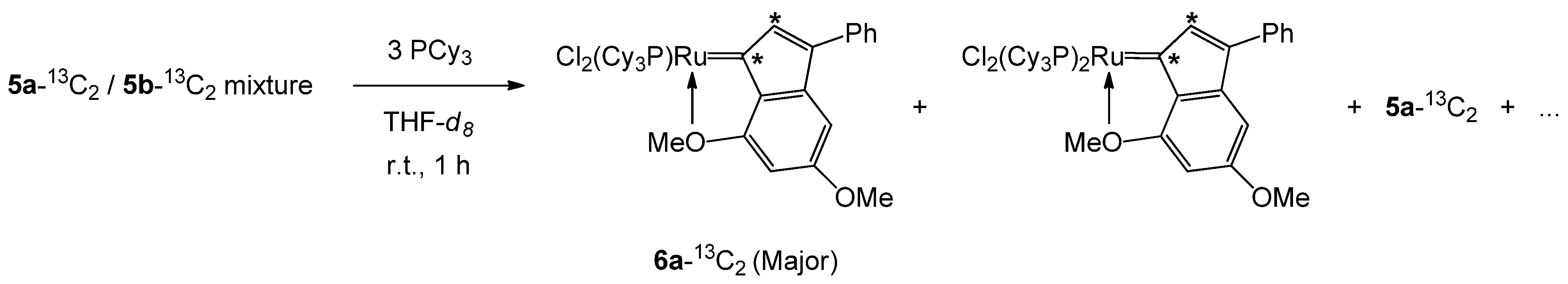
3.3.3. Gram-Scale Preparation of the 6a/6b Mixture
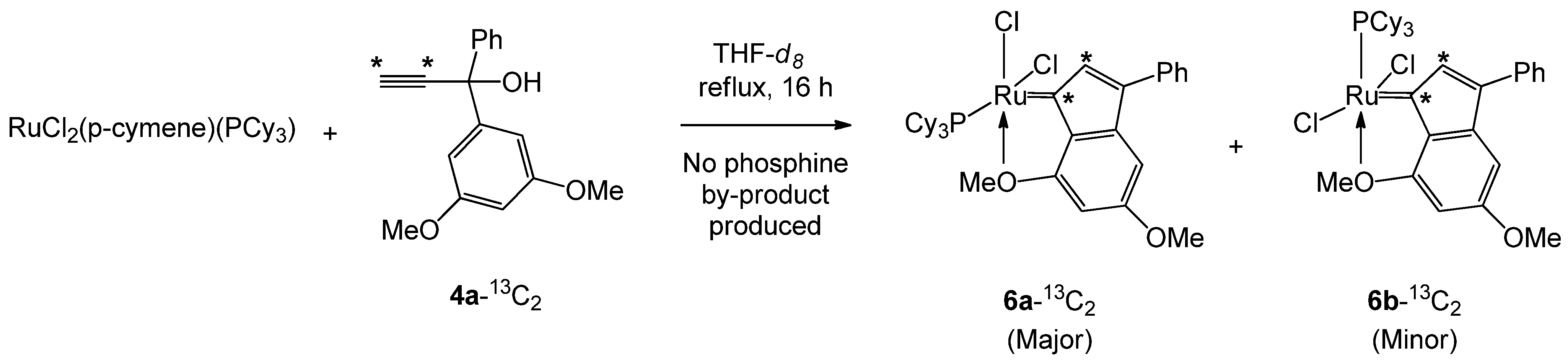
3.3.4. General Procedure for the Reaction between RuCl2(p-cymene)(L) and Organic Precursors 4a
3.3.5. General Procedure for the Reaction between RuCl2(p-cymene)(PCy3) and Organic Precursors 4b and 4c
3.3.6. General Procedure for the Reaction between RuCl2(PPh3)3 and Organic Precursors 4b and 4c
3.4. General Procedure for the RCM of Diethyl Diallylmalonate (DEDAM)
4. Conclusions
Acknowledgments
References and Notes
- Herndon, J.W. The chemistry of the carbon-transition metal double and triple bond. Annual survey covering the year 2009. Coord. Chem. Rev. 2011, 255, 3–100. [Google Scholar] [CrossRef]
- Schrock, R.R. Recent advances in high oxidation state Mo and W imido alkylidene chemistry. Chem. Rev. 2009, 109, 3211–3226. [Google Scholar] [CrossRef] [PubMed]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, R.H. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Hoveyda, A.H.; Malcolmson, S.J.; Meek, S.J.; Zhugralin, A.R. Catalytic enantioselective olefin metathesis in natural product synthesis. chiral metal-based complexes that deliver high enantioselectivity and more. Angew. Chem. Int. Ed. Engl. 2010, 49, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Peryshkov, D.V.; Schrock, R.R.; Takase, M.K.; Muller, P.; Hoveyda, A.H. Z-Selective olefin metathesis reactions promoted by tungsten oxo alkylidene complexes. J. Am. Chem. Soc. 2011, 133, 20754–20757. [Google Scholar] [CrossRef] [PubMed]
- Seiders, T.J.; Ward, D.W.; Grubbs, R.H. Enantioselective ruthenium-catalyzed ring-closing metathesis. Org. Lett. 2001, 3, 3225–3228. [Google Scholar] [CrossRef] [PubMed]
- van Veldhuizen, J.J.; Garber, S.B.; Kingsbury, J.S.; Hoveyda, A.H. A recyclable chiral Ru catalyst for enantioselective olefin metathesis. Efficient catalytic asymmetric ring-opening/cross metathesis in air. J. Am. Chem. Soc. 2002, 124, 4954–4955. [Google Scholar] [CrossRef] [PubMed]
- Funk, T.W.; Berlin, J.M.; Grubbs, R.H. Highly active chiral ruthenium catalysts for asymmetric ring-closing olefin metathesis. J. Am. Chem. Soc. 2006, 128, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Grubbs, R.H. Chelated ruthenium catalysts for z-selective olefin metathesis. J. Am. Chem. Soc. 2011, 133, 8525–8527. [Google Scholar] [CrossRef] [PubMed]
- Schwab, P.; Grubbs, R.H.; Ziller, J.W. Synthesis and applications of RuCl2(=CHR')(PR3)2: The influence of the alkylidene moiety on metathesis activity. J. Am. Chem. Soc. 1996, 118, 100–110. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, P.J., Jr.; Hoveyda, A.H. A recyclable ru-based metathesis catalyst. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Fürstner, A.; Thiel, O.R.; Ackermann, L.; Schanz, H.-J.; Nolan, S.P. Ruthenium carbene complexes with N,N′-bis(mesityl)imidazol-2-ylidene ligands: RCM catalysts of extended scope. J. Org. Chem. 2000, 65, 2204–2207. [Google Scholar] [CrossRef] [PubMed]
- Monsaert, S.; Drozdzak, R.; Dragutan, V.; Dragutan, I.; Verpoort, F. Indenylidene-ruthenium complexes bearing saturated N-heterocyclic carbenes: Synthesis and catalytic investigation in olefin metathesis reactions. Eur. J. Inorg. Chem. 2008, 2008, 432–440. [Google Scholar] [CrossRef]
- Harlow, K.J.; Hill, A.F.; Wilton-Ely, J.D.E.T. The first co-ordinatively unsaturated Group 8 allenylidene complexes: Insights into Grubbs’ vs. Dixneuf-Furstner olefin metathesis catalysts. J. Chem. Soc. Dalton Trans. 1999, 285–292. [Google Scholar] [CrossRef]
- Schanz, H.-J.; Jafarpour, L.; Stevens, E.D.; Nolan, S.P. Coordinatively unsaturated 16-electron ruthenium allenylidene complexes: Synthetic, structural, and catalytic studies. Organometallics 1999, 18, 5187–5190. [Google Scholar] [CrossRef]
- Fuerstner, A.; Guth, O.; Duffels, A.; Seidel, G.; Liebl, M.; Gabor, B.; Mynott, R. Indenylidene complexes of ruthenium: Optimized synthesis, structure elucidation, and performance as catalysts for olefin metathesis-application to the synthesis of the ADE-ring system of nakadomarin A. Chem.-Eur. J. 2001, 7, 4811–4820. [Google Scholar] [CrossRef]
- Shaffer, E.A.; Chen, C.-L.; Beatty, A.M.; Valente, E.J.; Schanz, H.-J. Synthesis of ruthenium phenylindenylidene, carbyne, allenylidene and vinylmethylidene complexes from (PPh3)3-4RuCl2: A mechanistic and structural investigation. J. Organomet. Chem. 2007, 692, 5221–5233. [Google Scholar] [CrossRef]
- Castarlenas, R.; Vovard, C.; Fischmeister, C.; Dixneuf, P.H. Allenylidene-to-indenylidene rearrangement in arene-ruthenium complexes: A key step to highly active catalysts for olefin metathesis reactions. J. Am. Chem. Soc. 2006, 128, 4079–4089. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L.R.; Gallon, B.J.; Schrodi, Y. A most convenient and atom-economic preparation of a highly active ring-closing metathesis catalyst. Organometallics 2010, 29, 3471–3473. [Google Scholar] [CrossRef]
- Kabro, A.; Roisnel, T.; Fischmeister, C.; Bruneau, C. Ruthenium-Indenylidene olefin metathesis catalyst with enhanced thermal stability. Chem.-Eur. J. 2010, 16, 12255–12261. [Google Scholar] [CrossRef] [PubMed]
- Ung, T.; Hejl, A.; Grubbs, R.H.; Schrodi, Y. Latent ruthenium olefin metathesis catalysts that contain an n-heterocyclic carbene ligand. Organometallics 2004, 23, 5399–5401. [Google Scholar] [CrossRef]
- Ritter, T.; Hejl, A.; Wenzel, A.G.; Funk, T.W.; Grubbs, R.H. A standard system of characterization for olefin metathesis catalysts. Organometallics 2006, 25, 5740–5745. [Google Scholar] [CrossRef]
- Amoroso, D.; Yap, G.P.A.; Fogg, D.E. Deactivation of ruthenium metathesis catalysts via facile formation of face-bridged dimers. Organometallics 2002, 21, 3335–3343. [Google Scholar] [CrossRef]
- Bustelo, E.; Tenorio, M.J.; Puerta, M.C.; Valerga, P. Activation of alkynols by [Cp*RuCl(PEt3)2]: new intermediates and alternative dehydration products. X-ray crystal structures of [Cp*Ru{=C=CHC(=CH2)Ph}(PEt3)2][BPh4], [Cp*Ru(=C=C=CPh2)(PEt3)2][BPh4], and [Cp*Ru{C≡CC(PEt3)Me2}(PEt3)2][BPh4]. Organometallics 1999, 18, 4563–4573. [Google Scholar] [CrossRef]
- Vorfalt, T.; Wannowius, K.-J.; Plenio, H. Probing the mechanism of olefin metathesis in grubbs-hoveyda and grela type complexes. Angew. Chem. Int. Ed. Engl. 2010, 49, 5533–5536. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, I.W.; Hillier, I.H.; Nelson, D.J.; Percy, J.M.; Vincent, M.A. What is the initiation step of the Grubbs-Hoveyda olefin metathesis catalyst? Chem. Commun. 2011, 47, 5428–5430. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Hendann, M.; Wannowius, K.-J.; Plenio, H. On the mechanism of the initiation reaction in Grubbs-Hoveyda complexes. J. Am. Chem. Soc. 2012, 134, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Dias, E.L.; Nguyen, S.T.; Grubbs, R.H. Well-defined ruthenium olefin metathesis catalysts: Mechanism and activity. J. Am. Chem. Soc. 1997, 119, 3887–3897. [Google Scholar] [CrossRef]
- Sanford, M.S.; Ulman, M.; Grubbs, R.H. New insights into the mechanism of ruthenium-catalyzed olefin metathesis reactions. J. Am. Chem. Soc. 2001, 123, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Mathew, J.; Suresh, C.H. Assessment of stereoelectronic effects in grubbs first-generation olefin metathesis catalysis using molecular electrostatic potential. Organometallics 2011, 30, 1438–1444. [Google Scholar] [CrossRef]
- Occhipinti, G.; Bjorsvik, H.-R.; Jensen, V.R. Quantitative structure-activity relationships of ruthenium catalysts for olefin metathesis. J. Am. Chem. Soc. 2006, 128, 6952–6964. [Google Scholar] [CrossRef] [PubMed]
- Sanford, M.S.; Love, J.A.; Grubbs, R.H. Mechanism and activity of ruthenium olefin metathesis catalysts. J. Am. Chem. Soc. 2001, 123, 6543–6554. [Google Scholar] [CrossRef] [PubMed]
- Demonceau, A.; Stumpf, A.W.; Saive, E.; Noels, A.F. Novel ruthenium-based catalyst systems for the ring-opening metathesis polymerization of low-strain cyclic olefins. Macromolecules 1997, 30, 3127–3136. [Google Scholar] [CrossRef]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand (L) | RuCl2(p-cymene)(L) δ (ppm) | Major Product δ (ppm) |
| P(tBu)2Me | 43.8 | 61.1 |
| P(Cy)2Ph | 19.5 | 50.0 |
| P(iBu)3 | 22.9 | 44.2 |
| P(iPr)3 | 35.7 | 58.1 |
| P(nPr)3 | 16.1 | 40.8 |

| Entry | Ligand (L) | Ru loading (mol %) | Time (min) | Conversion (%) b |
| 1 | PCy3 | 1.0 | 30 | > 97 |
| 2 | P(tBu)2Me | 1.0 | 60 | 63 |
| 3 | P(tBu)2Me | 2.0 | 60 | 92 |
| 4 | P(Cy)2Ph | 2.0 | 60 | > 3 |
| 5 | P(iBu)3 | 2.0 | 60 | 4 |
| 6 | P(iPr)3 | 1.0 | 60 | 68 |
| 7 | P(iPr)3 | 2.0 | 60 | 95 |
| 8 | P(nPr)3 | 2.0 | 60 | > 3 |
© 2012 by the authors; Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jimenez, L.R.; Tolentino, D.R.; Gallon, B.J.; Schrodi, Y. Development of a Method for the Preparation of Ruthenium Indenylidene-Ether Olefin Metathesis Catalysts. Molecules 2012, 17, 5675-5689. https://doi.org/10.3390/molecules17055675
Jimenez LR, Tolentino DR, Gallon BJ, Schrodi Y. Development of a Method for the Preparation of Ruthenium Indenylidene-Ether Olefin Metathesis Catalysts. Molecules. 2012; 17(5):5675-5689. https://doi.org/10.3390/molecules17055675
Chicago/Turabian StyleJimenez, Leonel R., Daniel R. Tolentino, Benjamin J. Gallon, and Yann Schrodi. 2012. "Development of a Method for the Preparation of Ruthenium Indenylidene-Ether Olefin Metathesis Catalysts" Molecules 17, no. 5: 5675-5689. https://doi.org/10.3390/molecules17055675