Synthesis and NMR Spectral Studies of the 7-C60-Adduct of N,N-(Tetrachlorophthaloyl) Dehydroabietylamine

Abstract
:1. Introduction
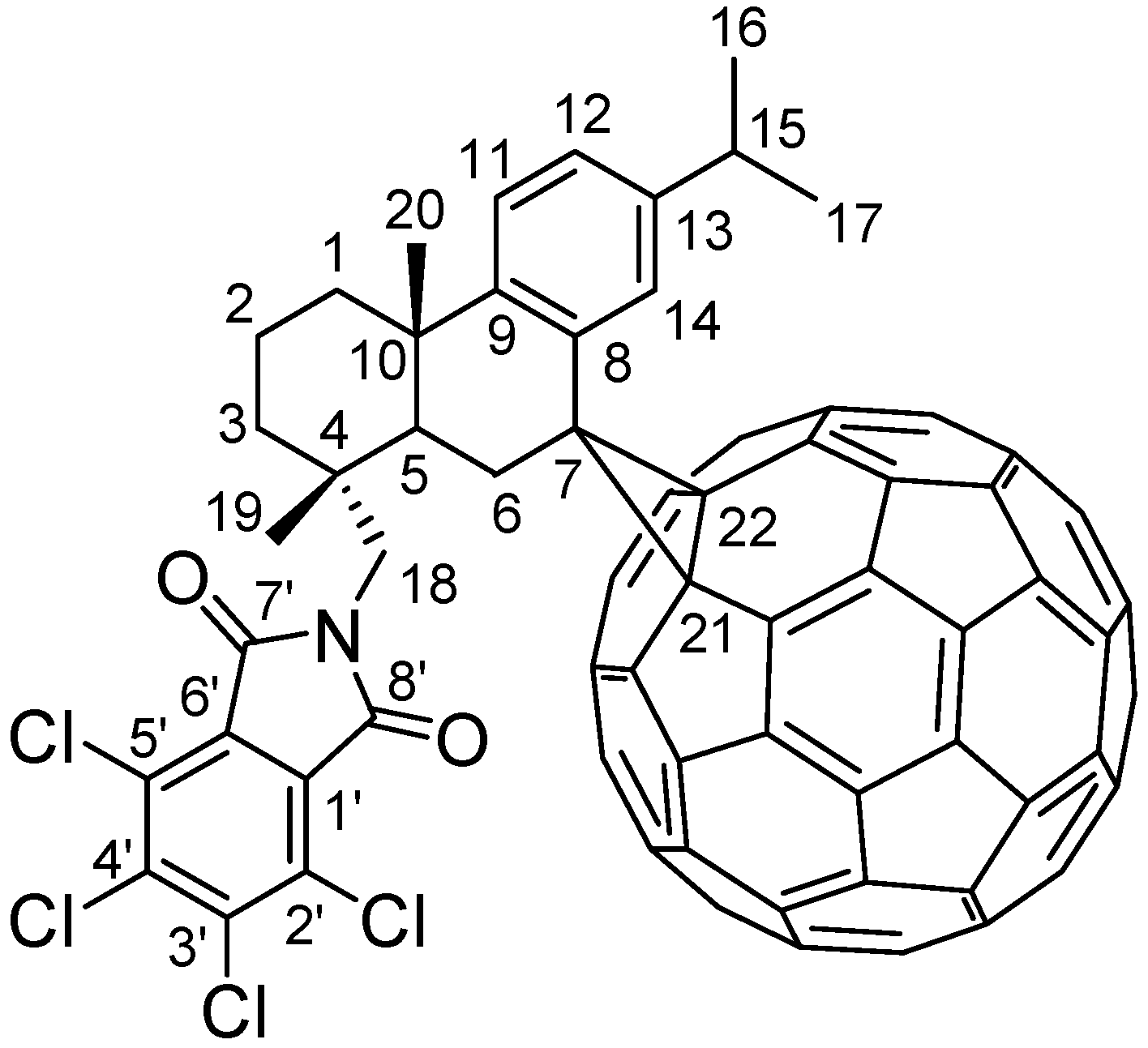
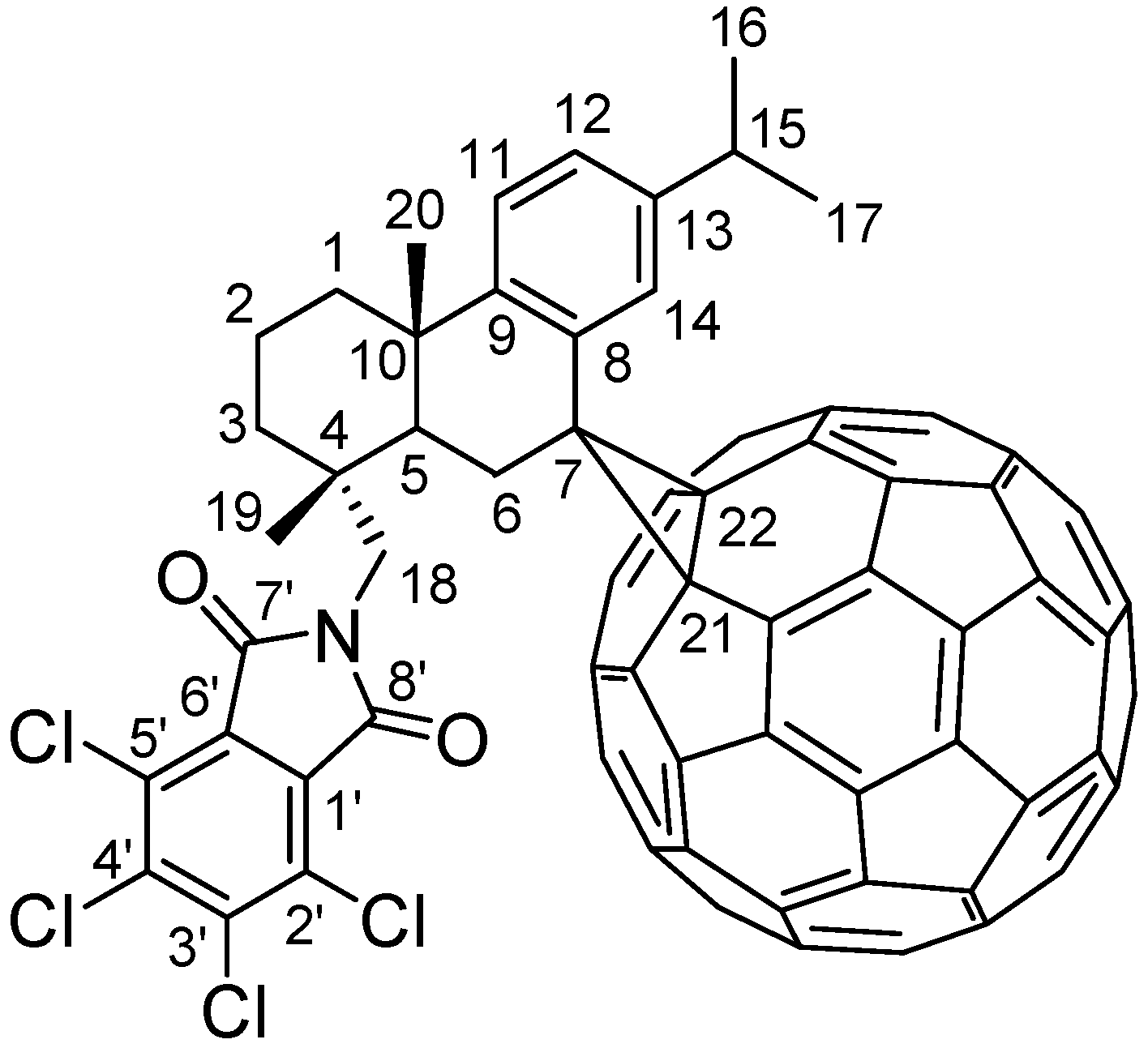
2. Results and Discussion
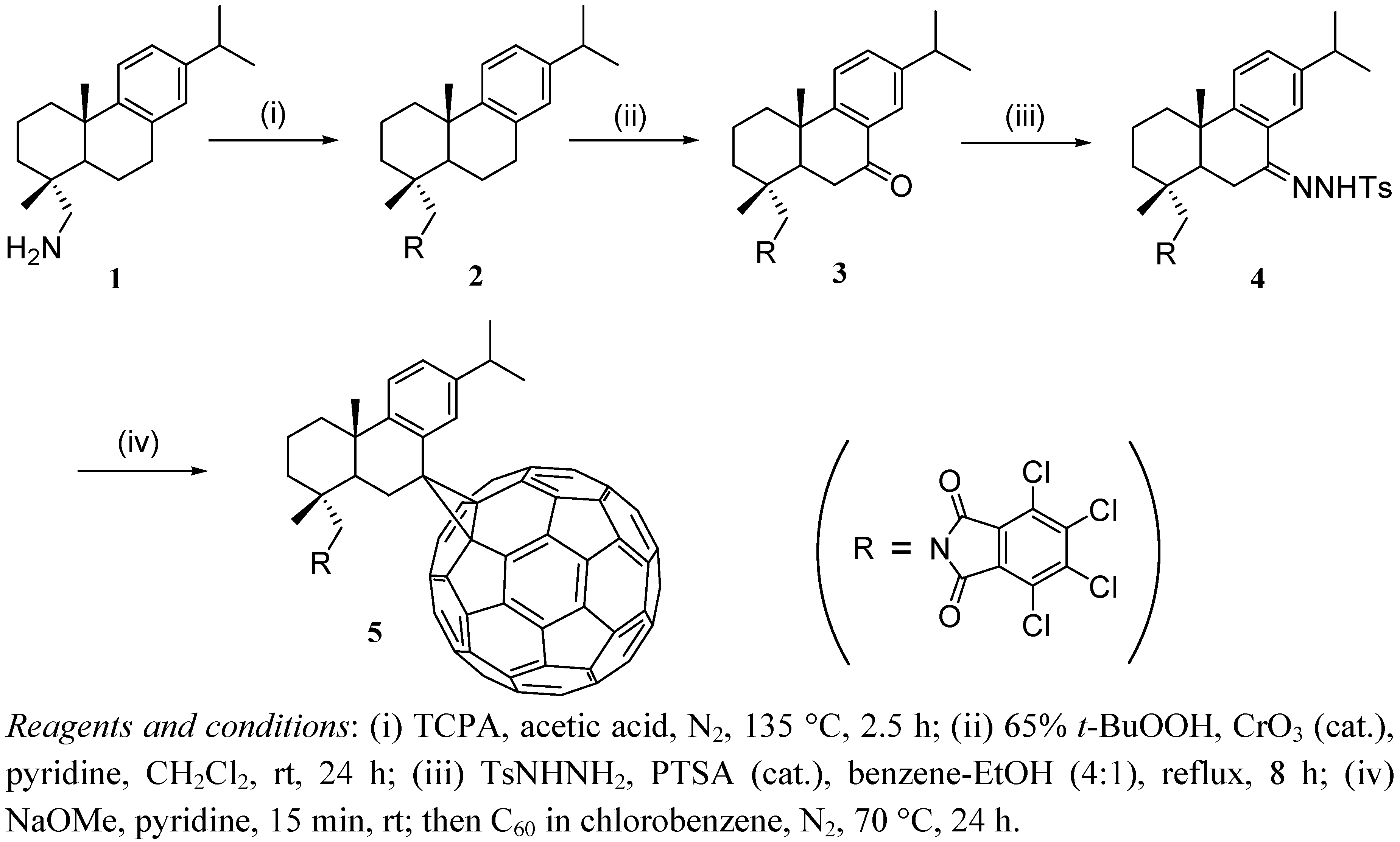
2.1. Synthesis Procedures
2.2. Analysis of Compound 5
2.2.1. IR, UV-Vis and Mass Spectrum Analysis of Compound 5

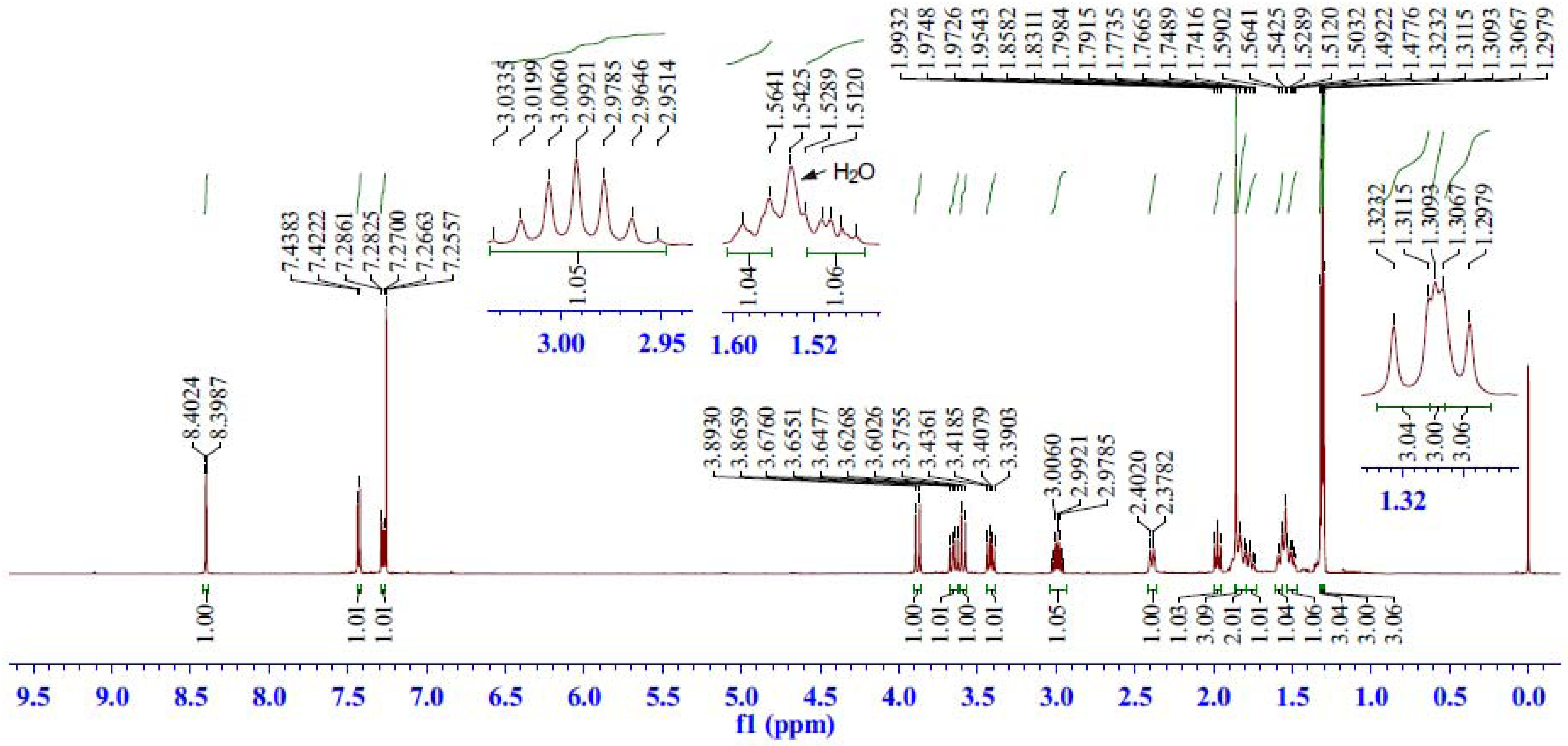
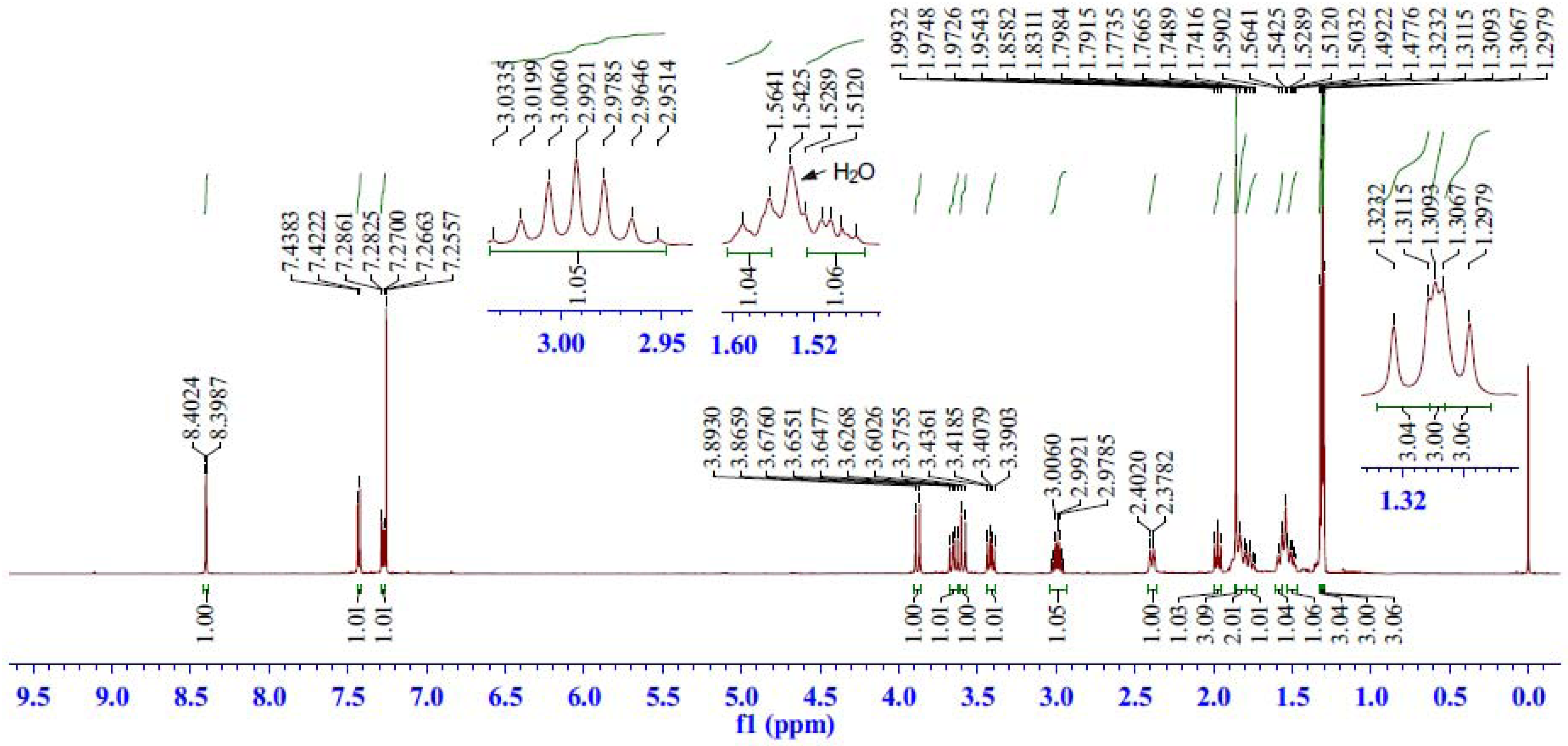
2.2.2. 1H-NMR Spectrum Analysis of Compound 5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | δC (ppm) | H | δH (ppm) | HSQC | HMBC | COSY | ROESY |
|---|---|---|---|---|---|---|---|
| 1 | 39.48 | 1β | 2.39 (br d, J = 11.9 Hz) | H-1β, 1α | H-20, 2, 3α | H-1α, 2 | H-20, 11, 2, 1α |
| 1α | 1.79–1.74 (m) | H-1β, 2 | H-5, 11, 1β | ||||
| 2 | 18.18 | 2 | 1.85–1.79 (m, 2H) | H-2 | H-3β, 3α, 1β, 1α | H-19, 3β, 3α, 1β | |
| 3 | 37.52 | 3β | 1.58 (br d, J = 13.0 Hz) | H-3β, 3α | H-19, 18α, 18β | H-3α, 2 | H-19, 3α, 2 |
| 3α | 1.53–1.47 (m) | H-3β, 2 | H-5, 18α, 3β, 2 | ||||
| 4 | 40.89 | ― | ― | ― | H-19, 5, 18α, 18β, 6β | ― | ― |
| 5 | 48.67 | 5 | 1.97 (dd, J = 10.3, 9.2 Hz) | H-5 | H-19, 20, 18α, 18β, 6α, 6β | H-6β, 6α | H-3α, 1α, 6α, 18α, 2 |
| 6 | 28.35 | 6β | 3.65 (dd, J = 14.1, 10.5 Hz) | H-6β, 6α | C-6/H-5 | H-5, 6α | H-19, 20, 6α |
| 6α | 3.41 (dd, J = 14.1, 8.8 Hz) | H-5, 6β | H-5, 6β, 18α | ||||
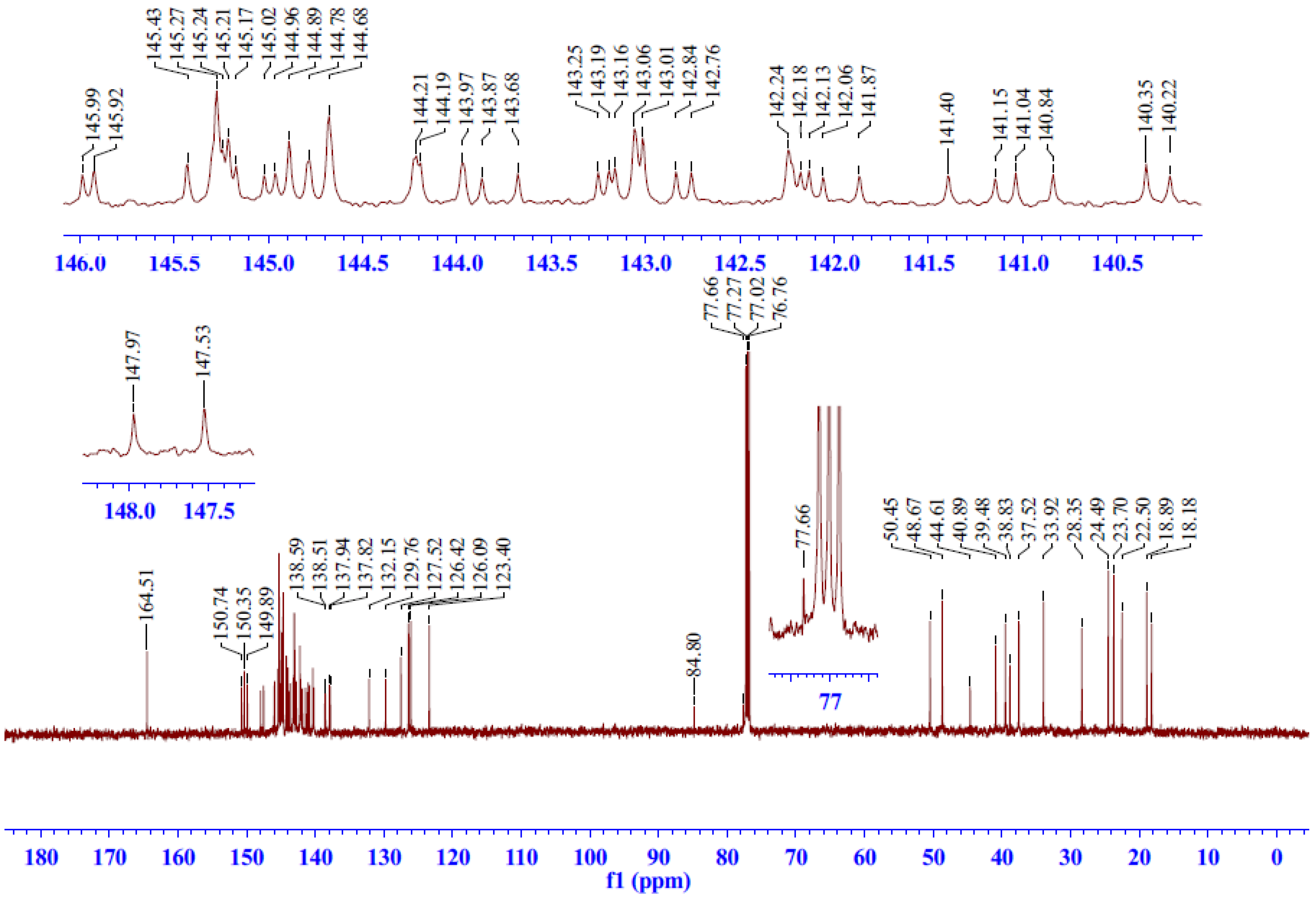
| 7 | 44.61 | ― | ― | ― | H-6α, 6β, 14, 11 | ― | ― |
| 8 | 132.15 | ― | ― | ― | H-6β, 11, | ― | ― |
| 9 | 150.35 | ― | ― | ― | H-14, 12, 20, 5 | ― | ― |
| 10 | 38.83 | ― | ― | ― | H-20, 5, 6α, 2, 11 | ― | ― |
| 11 | 123.40 | 11 | 7.43 (d, J = 8.1 Hz) | H-11 | H-12 | H-12 | H-1β, 1α, 12 |
| 12 | 126.09 | 12 | 7.28 (dd, J = 8.1, 1.8 Hz) | H-12 | H-15, 11, 14 | H-11 | H-15, 11, 16 |
| 13 | 145.43 | ― | ― | ― | H-16, 17, 15, 11 | ― | ― |
| 14 | 126.42 | 14 | 8.40 (d, J = 1.8 Hz) | H-14 | H-12, 15 | ― | ― |
| 15 | 33.92 | 15 | 2.99 (septet, J = 6.7 Hz) | H-15 | H-16, 17, 12, 14 | H-16, 17 | H-16, 17, 12 |
| 16 | 23.70 | 16 | 1.30 (d, J = 6.8 Hz, 3H) | H-16 | H-15, 17, | H-15 | H-15, 12 |
| 17 | 24.49 | 17 | 1.32 (d, J = 7.0 Hz, 3H) | H-17 | H-15, 16 | H-15 | H-15 |
| 18 | 50.45 | 18β | 3.88 (d, J = 13.6 Hz) | H-18β, 18α | H-19, 5 | H-18α | H-18α, 19 |
| 18α | 3.59 (d, J = 13.6 Hz) | H-18β | H-18β, 5, 3α, 6α | ||||
| 19 | 18.89 | 19 | 1.31 (s, 3H) | H-19 | H-5, 18α, 18β | ― | H-20, 6β, 18β, 3β, 2 |
| 20 | 22.50 | 20 | 1.86 (s, 3H) | H-20 | H-1α, 5 | ― | H-19, 6β, 1β |
| 21,22 | 84.80 | ― | ― | ― | H-6α, 6β | ― | ― |
| 77.66 | ― | ― | ― | H-6α | ― | ― | |
| 1',6' | 127.52 | ― | ― | ― | ― | ― | ― |
| 2',5' | 129.76 | ― | ― | ― | ― | ― | ― |
| 3',4' | 140.35 | ― | ― | ― | ― | ― | ― |
| 7',8' | 164.51 | ― | ― | ― | H-18α, 18β | ― | ― |
| C60 | 150.74–137.82 | ― | ― | ― | ― | ― | ― |
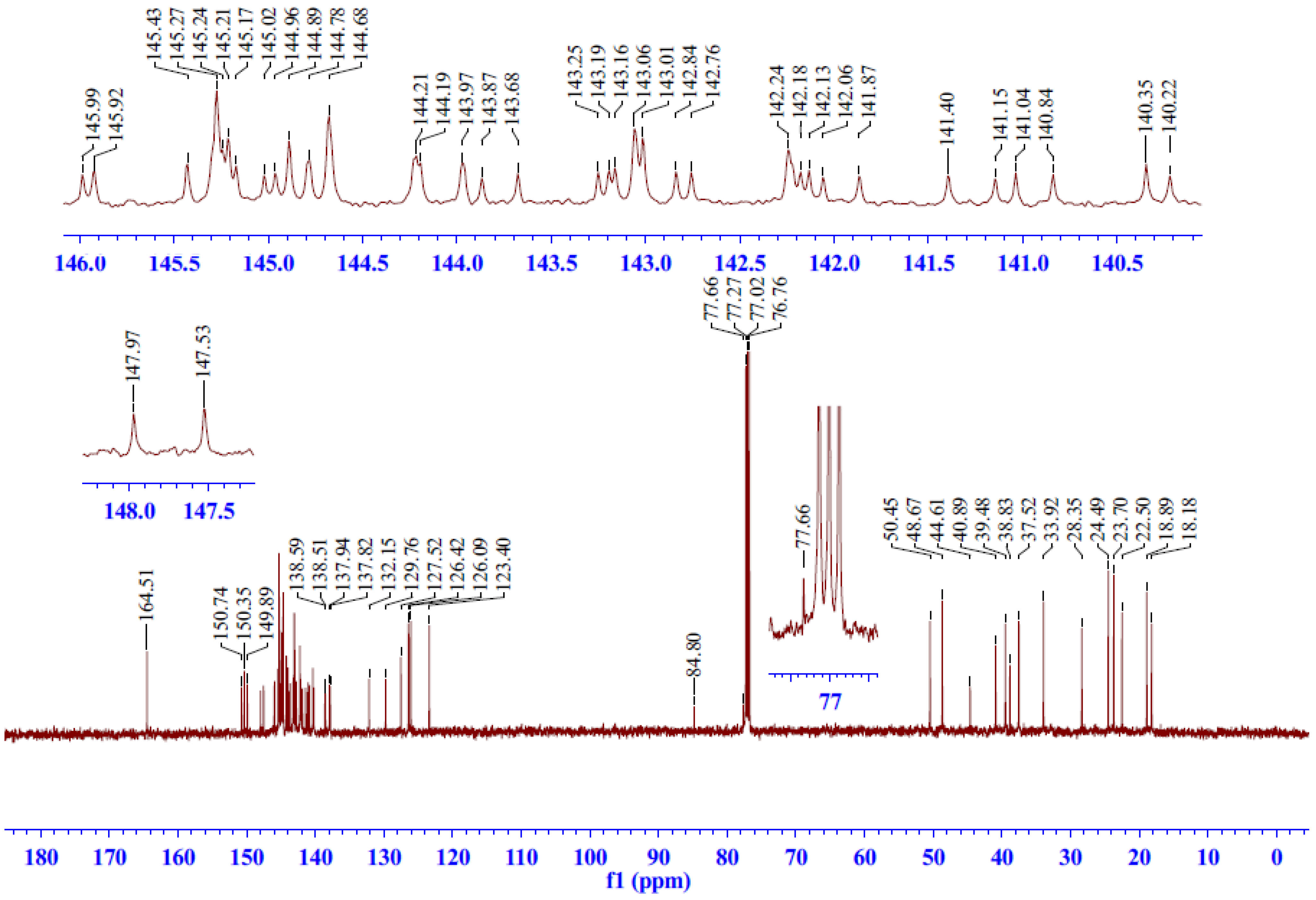
2.2.3. 13C-NMR Spectrum Analysis of Compound 5
3. Experimental
3.1. General
3.2. Preparation of N,N-(Tetrachlorophthaloyl)dehydroabietylamine (2)
3.3. Preparation of 7-oxo-N,N-(Tetrachlorophthaloyl)dehydroabietylamine (3)
3.4. Preparation of N,N-(Tetrachlorophthaloyl)dehydroabietylamine p-Tosylhydrazone (4)
3.5. Preparation of 7-C60-Adduct of N,N-(Tetrachlorophthaloyl)dehydroabietylamine (5)
4. Conclusions
Acknowledgments
References and Notes
- Bolchi, C.; Pallavicini, M.; Fumagalli, L.; Rusconi, C.; Binda, M.; Valoti, E. Resolution of 2-substituted 1,4-benzodioxanes by entrainment. Tetrahedron Asymmetry 2007, 18, 1038–1041. [Google Scholar]
- Bolchi, C.; Fumagalli, L.; Moroni, B.; Pallavicini, M.; Valoti, E. A short entry to enantiopure 2-substituted 1,4-benzodioxanes by efficient resolution methods. Tetrahedron Asymmetry 2003, 14, 3779–3785. [Google Scholar]
- Wilkerson, W.W.; Galbraith, W.; Delucca, I. Topical antiinflammatory dehydroabietymine derivatives. IV. Bioorg. Med. Chem. Lett. 1993, 3, 2087–2092. [Google Scholar] [CrossRef]
- Wilkerson, W.W.; Delucca, I.; Galbraith, W.; Gans, K.; Harris, R.; Jaffee, B.; Kerr, J. Antiinflammatory phospholipase-A2 inhibitors. I. Eur. J. Med. Chem. 1991, 26, 667–676. [Google Scholar] [CrossRef]
- Wang, Y.; Song, Z.Q. Synthesis of zwitterionic surface active agents from dehydroabietic amine and correlation between their structures and performance. Chem. Ind. For. Prod. 1996, 16, 1–6. [Google Scholar]
- Wang, Y.; Song, Z.Q.; Liang, M.L. Synthesis and characteristics of quaternary ammonium cationic surfactants from N-dehydroabietyl-N,N-bis(hydroxyethyl)amine. Chem. Ind. For. Prod. 1997, 17, 6–10. [Google Scholar]
- Friedman, S.H.; DeCamp, D.L.; Sijbesma, R.P.; Srdanov, G.; Wudl, F.; Kenyon, G.L. Inhibition of the HIV-1 protease by fullerene derivatives: Model building studies and experimental verification. J. Am. Chem. Soc. 1993, 115, 6506–6509. [Google Scholar]
- Zhao, G.J.; He, Y.J.; Xu, Z.; Hou, J.H.; Zhang, M.J.; Min, J.; Chen, H.Y.; Ye, M.F.; Hong, Z.R.; Yang, Y.; et al. Effect of carbon chain length in the substituent of PCBM-like molecules on their photovoltaic properties. Adv. Funct. Mater. 2010, 20, 1480–1487. [Google Scholar]
- Sánchez, L.; Rispens, M.T.; Hummelen, J.C. A supramolecular array of fullerenes byquadruple hydrogen bonding. Angew. Chem. Weinheim Bergstr. Ger. 2002, 41, 838–840. [Google Scholar]
- Li, W.S.; Yamamoto, Y.; Fukushima, T.; Saeki, A.; Seki, S.; Tagawa, S.; Masunaga, H.; Sasaki, S.; Takata, M.; Aida, T. Amphiphilic molecular design as a rational strategy for tailoring bicontinuous electron donor and acceptor arrays: Photoconductive liquid crystalline oligothiophene-C60 dyads. J. Am. Chem. Soc. 2008, 130, 8886–8887. [Google Scholar]
- Gómez, R.; Segura, J.L. Synthesis of a π-conjugated oligomer-fullerene dyad through a versatile [6,6] diphenylmethanofullerene carboxylic acid. Tetrahedron 2009, 65, 540–546. [Google Scholar]
- Wang, D.L.; Niu, Z.M.; Liu, H.D. Purification and characterization of optically active resolving reagent dehydroabietylamine. Beijing Ligong Daxue Xuebao 2004, 24, 357–359. [Google Scholar]
- Hummelen, J.C.; Knight, B.W.; LePeq, F.; Wudl, F.; Yao, J.; Wilkins, C.L. Preparation and characterization of fulleroid and methanofullerene derivatives. J. Org. Chem. 1995, 60, 532–538. [Google Scholar]
- Liu, S.Z.; Tang, G.S. Symmetry, Cage Structure of [60]Fullerene Derivatives and 13C-NMR Spectroscopy (in Chinese). Prog. Chem. 2004, 16, 561–573. [Google Scholar]
- Khalilov, L.M.; Tulyabaev, A.R.; Tuktarov, A.R. Homo- and methano[60]fullerenes with chiral attached moieties—1H and 13C-NMR chemical shift assignments and diastereotopicity effects. Magn. Reson. Chem. 2011, 49, 768–774. [Google Scholar]
- Li, Z.J.; Zhang, S.N.; Cui, J.R.; Li, S.C.; Li, Q.; Huang, H.Q.; Cai, M.S. Glycosyl phthalic imidine compounds with anti-inflammatory activity (in Chinese). C.N. Patent 1,594,342A, 2005. [Google Scholar]
- Sample Availability: Samples of the compounds 2–5 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, Z.; Lin, Z. Synthesis and NMR Spectral Studies of the 7-C60-Adduct of N,N-(Tetrachlorophthaloyl) Dehydroabietylamine. Molecules 2012, 17, 4209-4218. https://doi.org/10.3390/molecules17044209
Zhou Z, Lin Z. Synthesis and NMR Spectral Studies of the 7-C60-Adduct of N,N-(Tetrachlorophthaloyl) Dehydroabietylamine. Molecules. 2012; 17(4):4209-4218. https://doi.org/10.3390/molecules17044209
Chicago/Turabian StyleZhou, Zhi, and Zhongxiang Lin. 2012. "Synthesis and NMR Spectral Studies of the 7-C60-Adduct of N,N-(Tetrachlorophthaloyl) Dehydroabietylamine" Molecules 17, no. 4: 4209-4218. https://doi.org/10.3390/molecules17044209
APA StyleZhou, Z., & Lin, Z. (2012). Synthesis and NMR Spectral Studies of the 7-C60-Adduct of N,N-(Tetrachlorophthaloyl) Dehydroabietylamine. Molecules, 17(4), 4209-4218. https://doi.org/10.3390/molecules17044209