Palladium-Catalyzed Multicomponent Synthesis of 2-Imidazolines from Imines and Acid Chlorides

Abstract
:1. Introduction
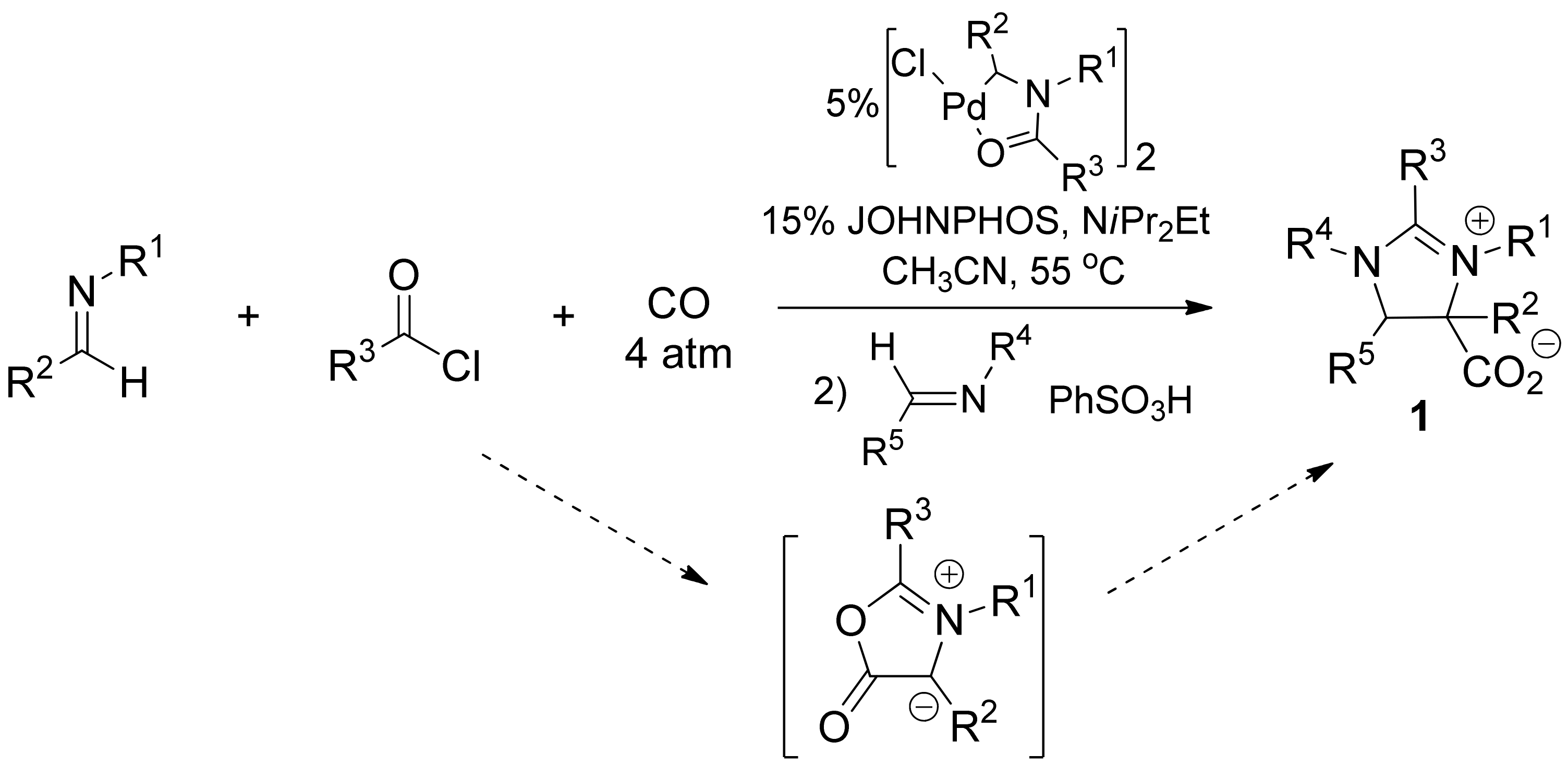
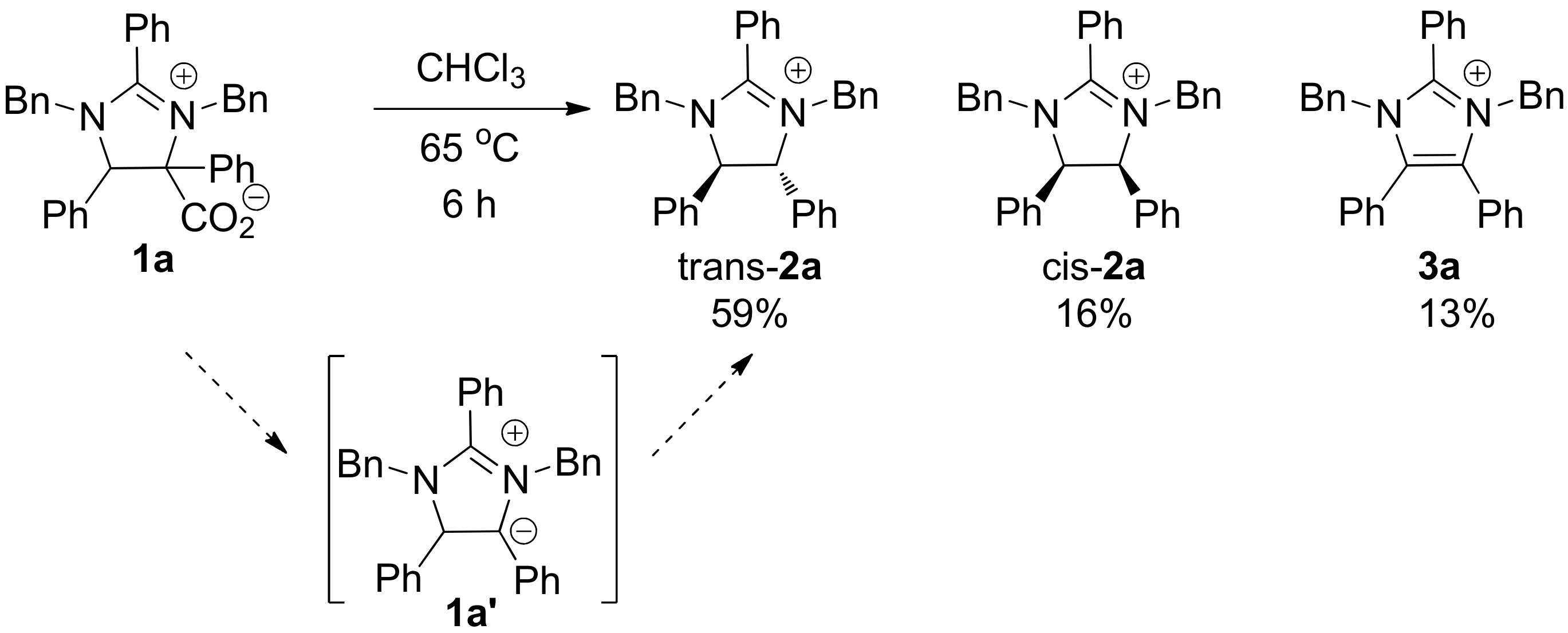
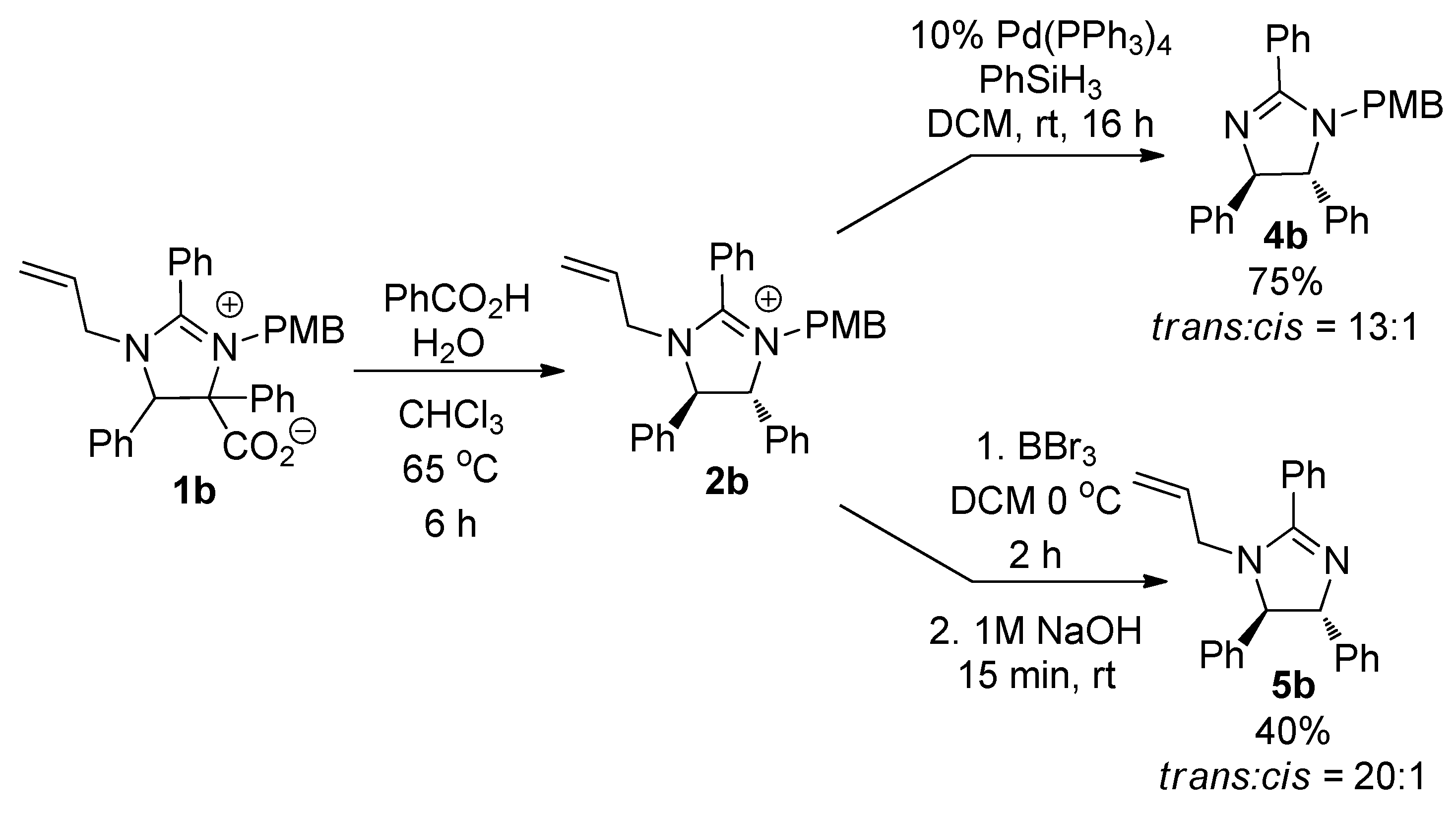
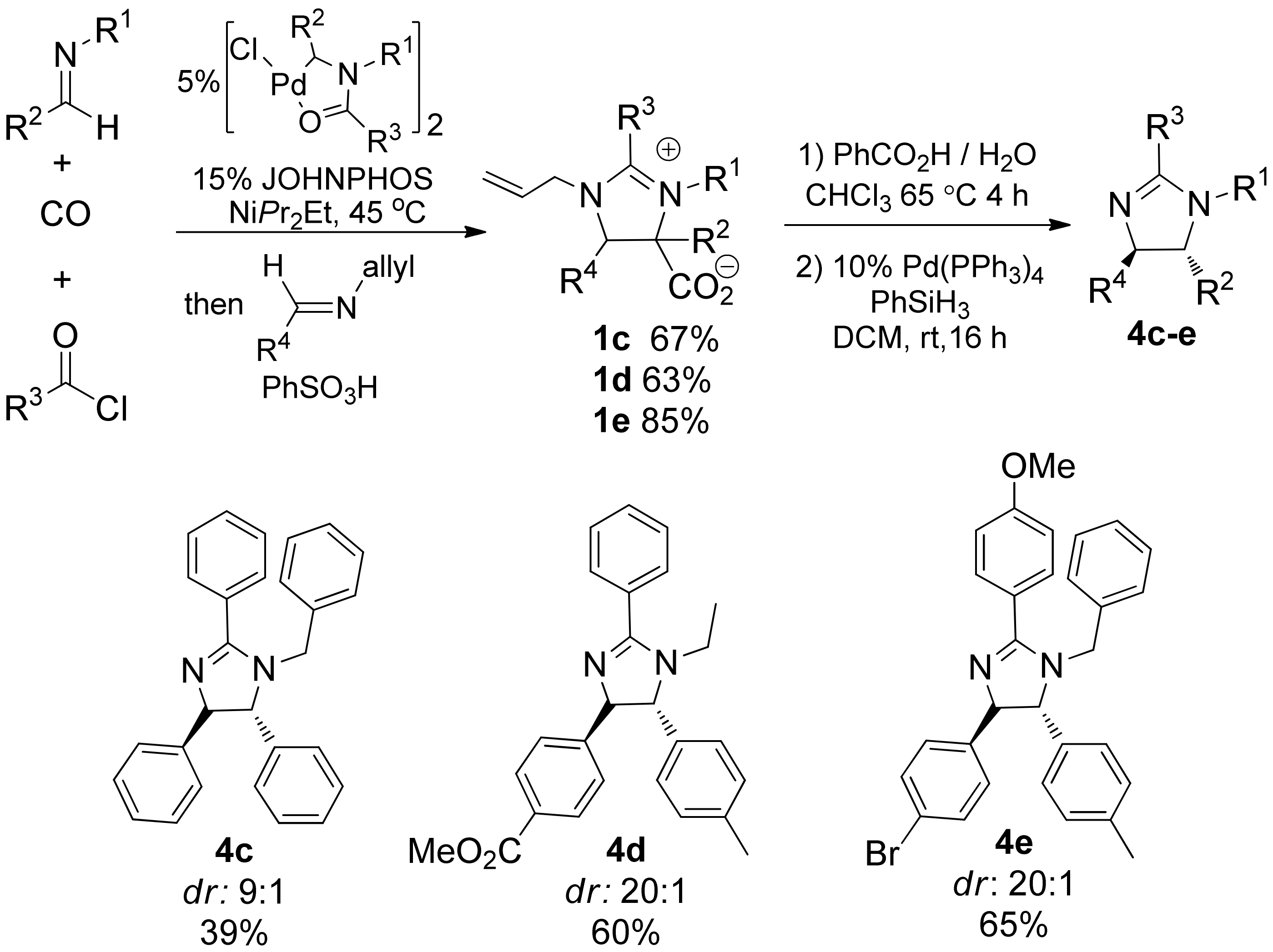
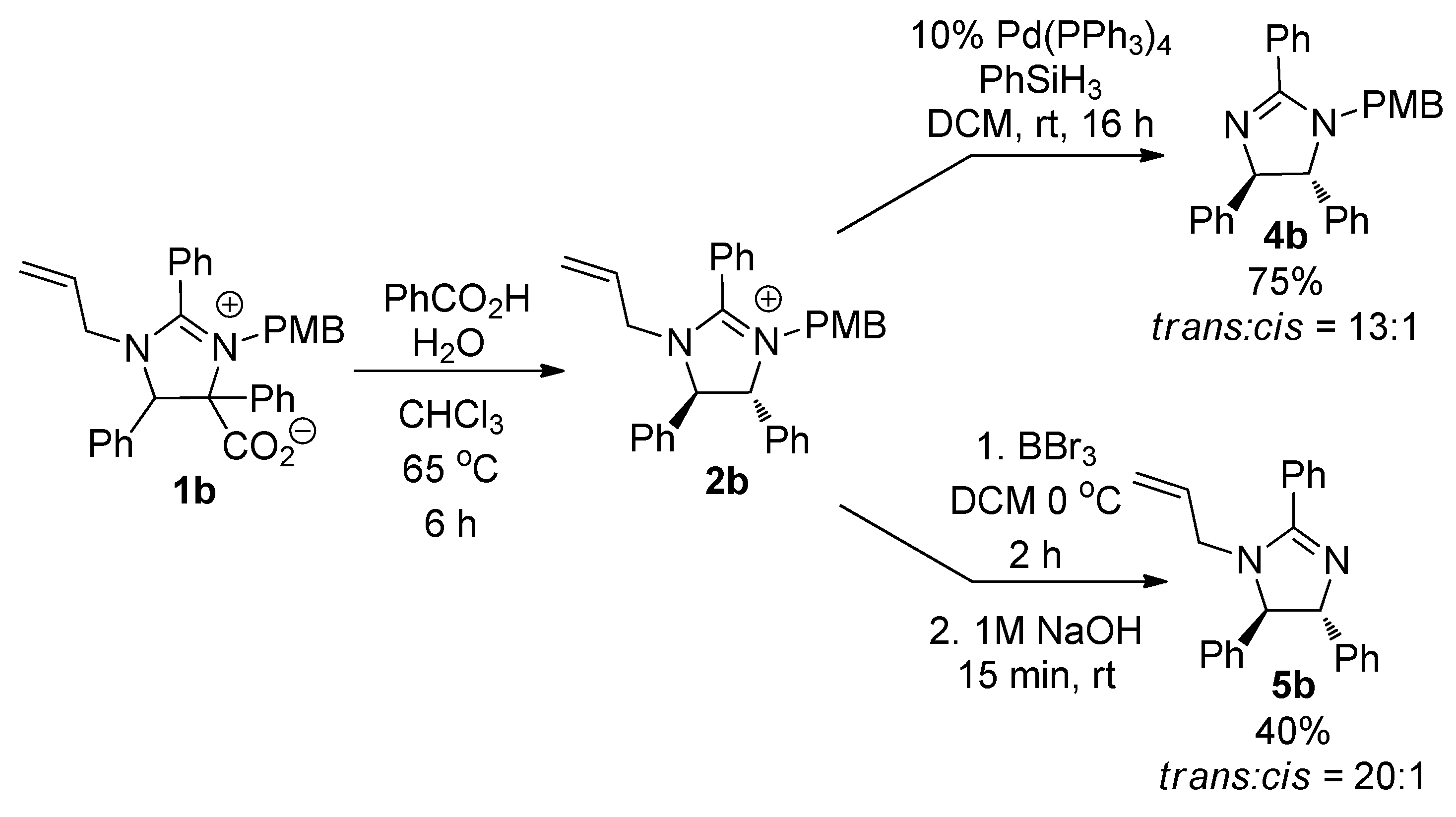
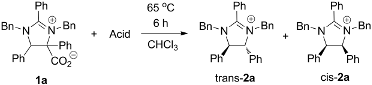
2. Results and Discussion
3. Experimental
3.1. General Considerations
3.2. Synthesis of trans-1,3-Dibenzyl-2,4,5-triphenyl-4,5-dihydro-3H-imidazol-1-ium (trans-2a) and cis-1,3-Dibenzyl-2,4,5-triphenyl-4,5-dihydro-3H-imidazol-1-ium (cis-2a)
3.3. Synthesis of 1,3-Dibenzyl-2,4,5-triphenyl-3H-imidazol-1-ium (3a)
3.4. Synthesis of 1-(4-Methoxy-benzyl)-2,4,5-triphenyl-4,5-dihydro-1H-imidazole (4b)
3.5. Synthesis of 1-Allyl-2,4,5-triphenyl-4,5-dihydro-1H-imidazole (5b)
4. Conclusions
Supplementary Materials
References and Notes
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Lansdell, T.A.; Peddibhotla, S.; Tepe, J.J. Sensitization of Tumor Cells toward Chemotherapy: Enhancing the Efficacy of Camptothecin with Imidazolines. Chem. Biol. 2004, 11, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Gentili, F.; Pizzinat, N.; Ordener, C.; Marchal-Victorion, S.; Maurel, A.; Hofmann, R.; Renard, P.; Delagrange, P.; Pigini, M.; Parini, A.; et al. 3-[5-(4,5-Dihydro-1H-imidazol-2-yl)-furan-2-yl]phenylamine (Amifuraline), a Promising Reversible and Selective Peripheral MAO-A Inhibitor. J. Med. Chem. 2006, 49, 5578–5586. [Google Scholar] [CrossRef] [PubMed]
- Hlasta, D.J.; Luttinger, D.; Perrone, M.H.; Silbernagel, M.J.; Ward, S.J.; Haubrich, D.R. α2-Adrenergic agonists/antagonists: the synthesis and structure-activity relationships of a series of indolin-2-yl and tetrahydroquinolin-2-yl imidazolines. J. Med. Chem. 1987, 30, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-X.; Zhang, Y. Imidazoline I2 receptors: Target for new analgesics? Eur. J. Pharmacol. 2011, 658, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-R.; Wang, C.; Zhang, Z.; Peng, S. A new class of analgesic agents toward prostacyclin receptor inhibition: Synthesis, Biological studies and QSAR analysis of 1-hydroxyl-2-substituted phenyl-4,4,5,5-tetramethylimidazolines. Eur. J. Med. Chem. 2008, 43, 1048–1058. [Google Scholar]
- Kahlon, D.K.; Lansdell, T.A.; Fisk, J.S.; Hupp, C.D.; Friebe, T.L.; Hovde, S.; Jones, A.D.; Dyer, R.D.; Henry, R.W.; Tepe, J.J. Nuclear Factor-кβ Mediated Inhibition of Cytokine Production by Imidazoline Scaffolds. J. Med. Chem. 2009, 52, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Merriman, G.H.; Ma, L.; Shum, P.; McGarry, D.; Volz, F.; Sabol, J.S.; Gross, A.; Zhao, Z.; Rampe, D.; Wang, L.; et al. Synthesis and SAR of novel 4,5-diarylimidazolines as potent P2X7 receptor antagonists. Bioorg. Chem. Med. Lett. 2005, 15, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.S.; Frederich, J.H.; Overman, L.E. Palladacyclic Imidazoline−Naphthalene Complexes: Synthesis and Catalytic Performance in Pd(II)-Catalyzed Enantioselective Reactions of Allylic Trichloroacetimidates. J. Org. Chem. 2012, 77, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Mei, L.; Wei, Y.; Shi, M.; Kattamuri, P.V.; McDowell, P.; Li, G. Asymmetric catalytic Mannich-type reaction of hydrazones with difluoroenoxysilanes using imidazoline-anchored phosphine ligand–zinc(II) complexes. Org. Biomol. Chem. 2012, 10, 2509–2513. [Google Scholar] [CrossRef] [PubMed]
- Parra-Hake, M.; Larter, M.L.; Gantzel, P.; Aguirre, G.; Ortega, F.; Somanathan, R.; Walsh, P.J. Synthesis and Structure of Dimeric [(cis-Py3Im)M]2 Complexes [M = Ni, Cu, Zn; (cis-Py3Im) = cis-2,4,5-Tri(2-pyridyl)imidazoline]. Inorg. Chem. 2000, 39, 5400–5403. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.P. (Ed.) N-Heterocyclic Carbenes in Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Benhamou, L.; Chardon, E.; Lavigne, G.; Bellemin-Laponnaz, S.; César, V. Synthetic Routes to N-Heterocyclic Carbene Precursors. Chem. Rev. 2011, 111, 2705–2733. [Google Scholar] [CrossRef] [PubMed]
- Neef, G.; Eder, U.; Sauer, G. One-step conversions of esters to 2-imidazolines, benzimidazoles and benzothiazoles by aluminum organic reagents. J. Org. Chem. 1981, 46, 2824–2826. [Google Scholar] [CrossRef]
- Chitwood, H.C.; Reid, E.E. Some Alkyl-glyoxalidines. J. Am. Chem. Soc. 1935, 57, 2424–2426. [Google Scholar] [CrossRef]
- Dauwe, C.; Buddrus, J. Synthesis of Enantiopure C2-Chiral Amidines. Synthesis 1995, 171–172. [Google Scholar] [CrossRef]
- Paliakov, E.; Elleboe, T.; Boykin, W.D. New Synthons for the Preparation of Arylimidazolines and Tetrahydropyrimidine Analogues. Synthesis 2007, 1475–1480. [Google Scholar] [CrossRef]
- Boland, N.A.; Casey, M.; Hynes, S.J.; Matthews, J.W.; Smyth, M.P. A Novel General Route for the Preparation of Enantiopure Imidazolines. J. Org. Chem. 2002, 67, 3919–3922. [Google Scholar] [CrossRef] [PubMed]
- Bender, H.S.; Heine, H.W. The Isomerization of Some Aziridine Derivatives. III. A New Synthesis of 2-Imidazolines. J. Org. Chem. 1960, 25, 461–463. [Google Scholar]
- Kuszpit, M.R.; Wulff, W.D.; Tepe, J.J. One-Pot Synthesis of 2-Imidazolines via the Ring Expansion of Imidoyl Chlorides with Aziridines. J. Org. Chem. 2011, 76, 2913–2919. [Google Scholar] [CrossRef] [PubMed]
- Elders, N.; Ruijter, E.; de Kanter, F.J.J.; Groen, M.B.; Orru, R.V.A. Selective Formation of 2-Imidazolines and 2-Substituted Oxazoles by Using a Three-Component Reaction. Chem. Eur. J. 2008, 14, 4961–4973. [Google Scholar] [CrossRef] [PubMed]
- Strassberger, Z.; Mooijman, M.; Ruijter, E.; Alberts, A.H.; de Graaff, C.; Orru, R.V.A.; Rothenberg, G. A Facile Route to Ruthenium-Carbene Complexes and their Application in Furfural Hydrogenation. Appl. Organomet. Chem. 2010, 24, 142–146. [Google Scholar]
- Aydin, J.; Kumar, K.S.; Eriksson, L.; Szabó, K.J. Palladium Pincer Complex-Catalyzed Condensation of Sulfonimines and Isocyanoacetate to Imidazoline Derivatives. Dependence of the Stereoselectivity on the Ligand Effects. Adv. Synth. Catal. 2007, 349, 2585–2594. [Google Scholar] [CrossRef]
- Bowman, R.K.; Johnson, J.S. Lewis Acid Catalyzed Dipolar Cycloadditions of an Activated Imidate. J. Org. Chem. 2004, 69, 8537–8540. [Google Scholar] [CrossRef] [PubMed]
- Worrall, K.; Xu, B.; Bontemps, S.; Arndtsen, B.A. A Palladium-Catalyzed Multicomponent Synthesis of Imidazolinium Salts and Imidazolines from Imines, Acid Chlorides, and Carbon Monoxide. J. Org. Chem. 2011, 76, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Bontemps, S.; Quesnel, J.S.; Worrall, K.; Arndtsen, B.A. Palladium-Catalyzed Aryl Iodide Carbonylation as a Route to Imidazoline Synthesis: Design of a Five-Component Coupling Reaction. Angew. Chem. Int. Ed. 2011, 50, 8949–8951. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, R.; Dghaym, R.D.; St. Cyr, D.J.; Arndtsen, B.A. Direct, Palladium-Catalyzed, Multicomponent Synthesis of β-Lactams from Imines, Acid Chloride, and Carbon Monoxide. Org. Lett. 2006, 8, 3927–3930. [Google Scholar] [CrossRef] [PubMed]
- Balme, G.; Bouyssi, D.; Monteiro, N. Metal-Catalyzed Multicomponent Reactions. In Multicomponent Reactions; Zhu, J., Bienayme, H., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp. 224–276. [Google Scholar]
- D’Souza, D.M.; Müller, T.J.J. Multi-component syntheses of heterocycles by transition-metal catalysis. Chem. Soc. Rev. 2007, 37, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Arndtsen, B.A. Metal-Catalyzed One-Step Synthesis: Towards Direct Alternatives to Multistep Heterocycle and Amino Acid Derivative Formation. Chem. Eur. J. 2009, 15, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Siamaki, A.R.; Black, D.A.; Arndtsen, B.A. Palladium-Catalyzed Carbonylative Cross-Coupling with Imines: A Multicomponent Synthesis of Imidazolones. J. Org. Chem. 2008, 73, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Black, D.A.; Arndtsen, B.A. Copper-Catalyzed Cross-Coupling of Imines, Acid Chlorides, and Organostannanes: A Multicomponent Synthesis of α-Substituted Amides. J. Org. Chem. 2005, 70, 5133–5138. [Google Scholar] [CrossRef] [PubMed]
- Black, D.A.; Arndtsen, B.A. General Approach to the Coupling of Organoindium Reagents with Imines via Copper Catalysis. Org. Lett. 2006, 8, 1991–1993. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, H.; Hüisgen, R.; Schaefer, F.C. Δ2-pyrroline aus mesoionischen oxazolen und olefinen. Tetrahedron Lett. 1964, 5, 487–491. [Google Scholar] [CrossRef]
- Gotthardt, H.; Hüisgen, R. 1.3-Dipolare Cycloaddition, LVII. Δ2-Pyrroline aus N-substituierten Oxazolium-5-oxiden und olefinischen Dipolarophilen. Chem. Ber. 1970, 103, 2625–2638. [Google Scholar] [CrossRef]
- Qu, K.; Fisk, J.S.; Tepe, J.J. Azomethine ylide mediated inversion of configuration of quaternary imidazoline carbon: Converting trans- to its cis-imidazolines. Tetrahedron Lett. 2011, 52, 4840–4842. [Google Scholar] [CrossRef] [PubMed]
- The relative stereochemistry of trans-2a and cis-2a was verified debenzylation of these compounds and comparing the resulting imidazolines to known literature compounds. The identity of imidazolium 3a was verified by synthesizing an authentic sample via the benzylation of imidazole. See supporting information for details.
- Layer, R.W. The Chemistry of Imines. Chem. Rev. 1963, 63, 489–510. [Google Scholar] [CrossRef]
- Dhawan, R.; Dghaym, R.D.; Arndtsen, B.A. The Development of a Catalytic Synthesis of Münchnones: A Simple Four-Component Coupling Approach to α-Amino Acid Derivatives. J. Am. Chem. Soc. 2003, 125, 1474–1475. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liao, Q.; Xi, C. Convenient One-Step Synthesis of cis-2,4,5-Triarylimidazolines from Aromatic Aldehydes with Urea. Synth. Commun. 2012, 42, 905–913. [Google Scholar] [CrossRef]
- The parent imidazole was synthesized according to: Siamaki, A.R.; Arndtsen, B.A. A Direct, One Step Synthesis of Imidazoles from Imines and Acid Chlorides: A Palladium Catalyzed Multicomponent Coupling Approach. J. Am. Chem. Soc. 2006, 128, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Acid | Yield 2a | trans : cis 2a |
|---|---|---|---|
| 1 | 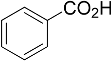 | 78% | 3.2:1 |
| 2 | 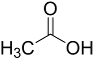 | 78% | 2.7:1 |
| 3 |  | 87% | 2.7:1 |
| 4 | HCl | - a | - |
| 5 | 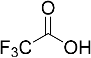 | - a | - |
| 6 | 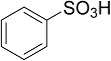 | - a | - |
| 7 |  | 24% | 1.5:1 |
| 8 | 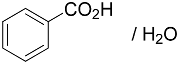 | 86% | 20:1 |
| 9 | H2O (20 equiv.) | - b | - |
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, B.; Worrall, K.; Arndtsen, B.A. Palladium-Catalyzed Multicomponent Synthesis of 2-Imidazolines from Imines and Acid Chlorides. Molecules 2012, 17, 13759-13768. https://doi.org/10.3390/molecules171213759
Xu B, Worrall K, Arndtsen BA. Palladium-Catalyzed Multicomponent Synthesis of 2-Imidazolines from Imines and Acid Chlorides. Molecules. 2012; 17(12):13759-13768. https://doi.org/10.3390/molecules171213759
Chicago/Turabian StyleXu, Boran, Kraig Worrall, and Bruce A. Arndtsen. 2012. "Palladium-Catalyzed Multicomponent Synthesis of 2-Imidazolines from Imines and Acid Chlorides" Molecules 17, no. 12: 13759-13768. https://doi.org/10.3390/molecules171213759
APA StyleXu, B., Worrall, K., & Arndtsen, B. A. (2012). Palladium-Catalyzed Multicomponent Synthesis of 2-Imidazolines from Imines and Acid Chlorides. Molecules, 17(12), 13759-13768. https://doi.org/10.3390/molecules171213759