Phenylmethimazole Blocks dsRNA-Induced IRF3 Nuclear Translocation and Homodimerization

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
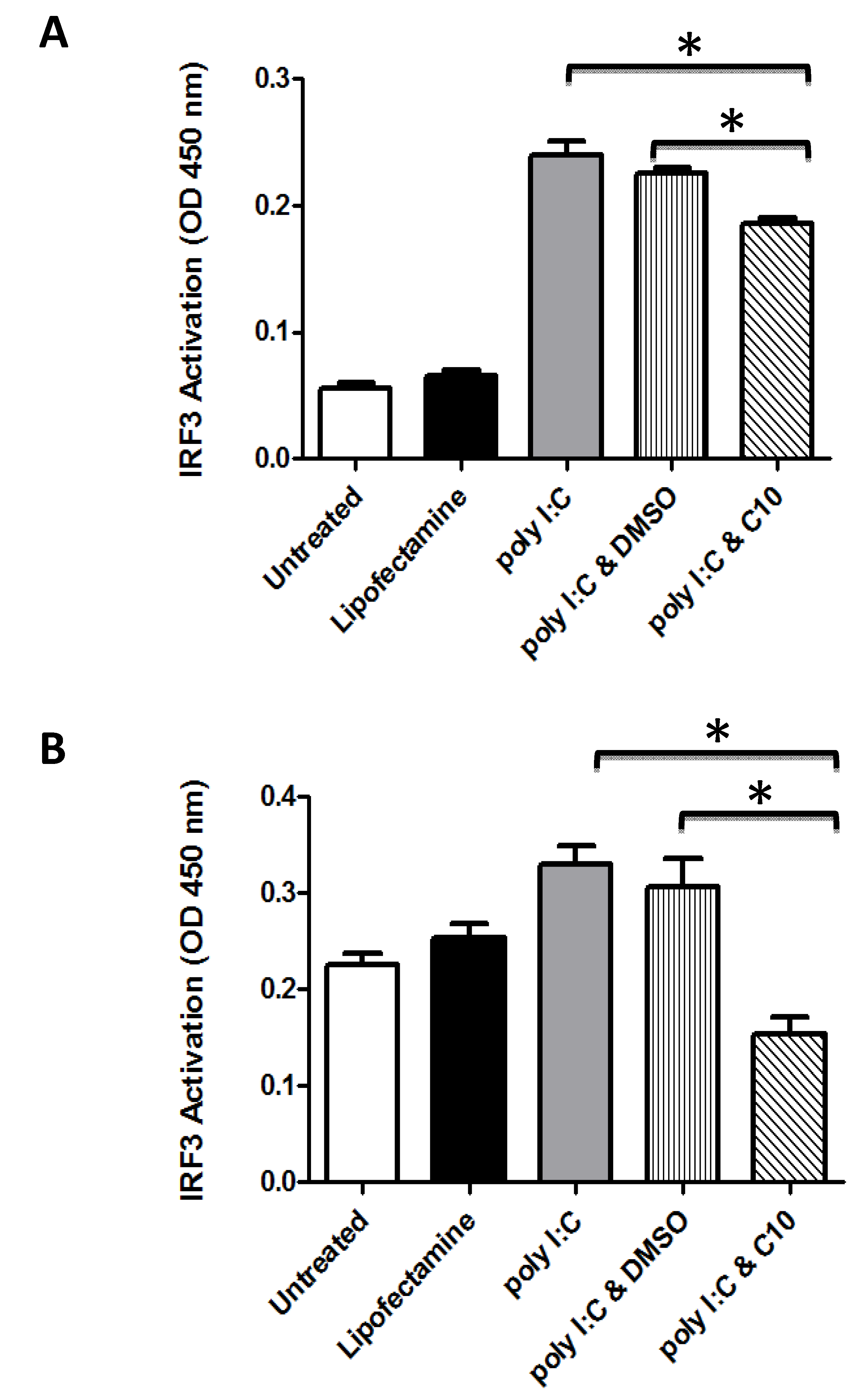
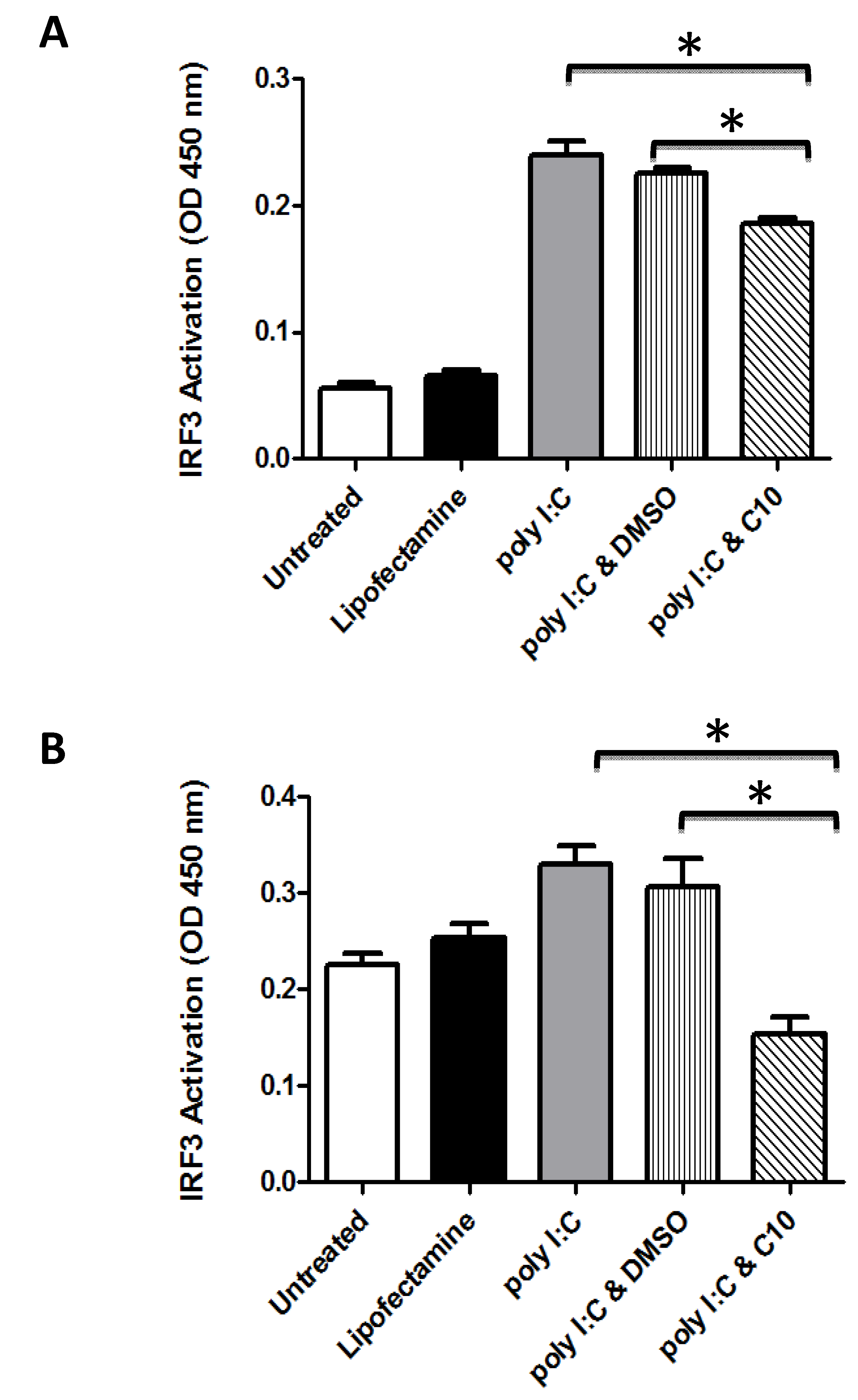
2.1. C10 Inhibits IRF3 DNA Binding in Human Embryonic Kidney and Pancreatic Cancer Cells
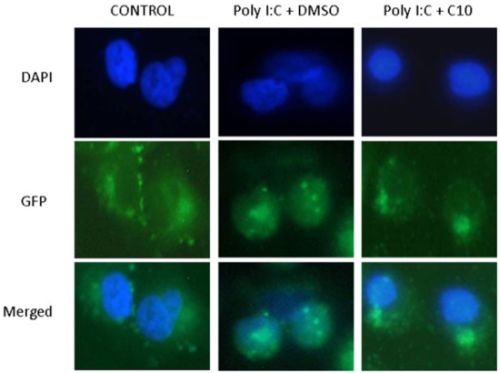
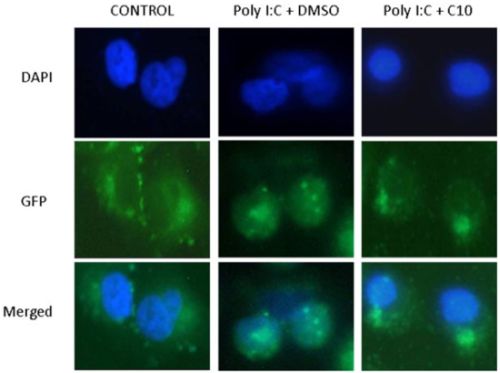
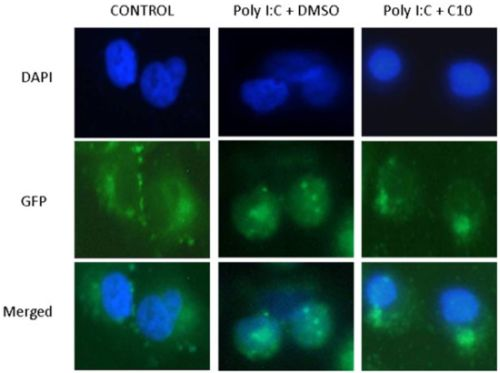
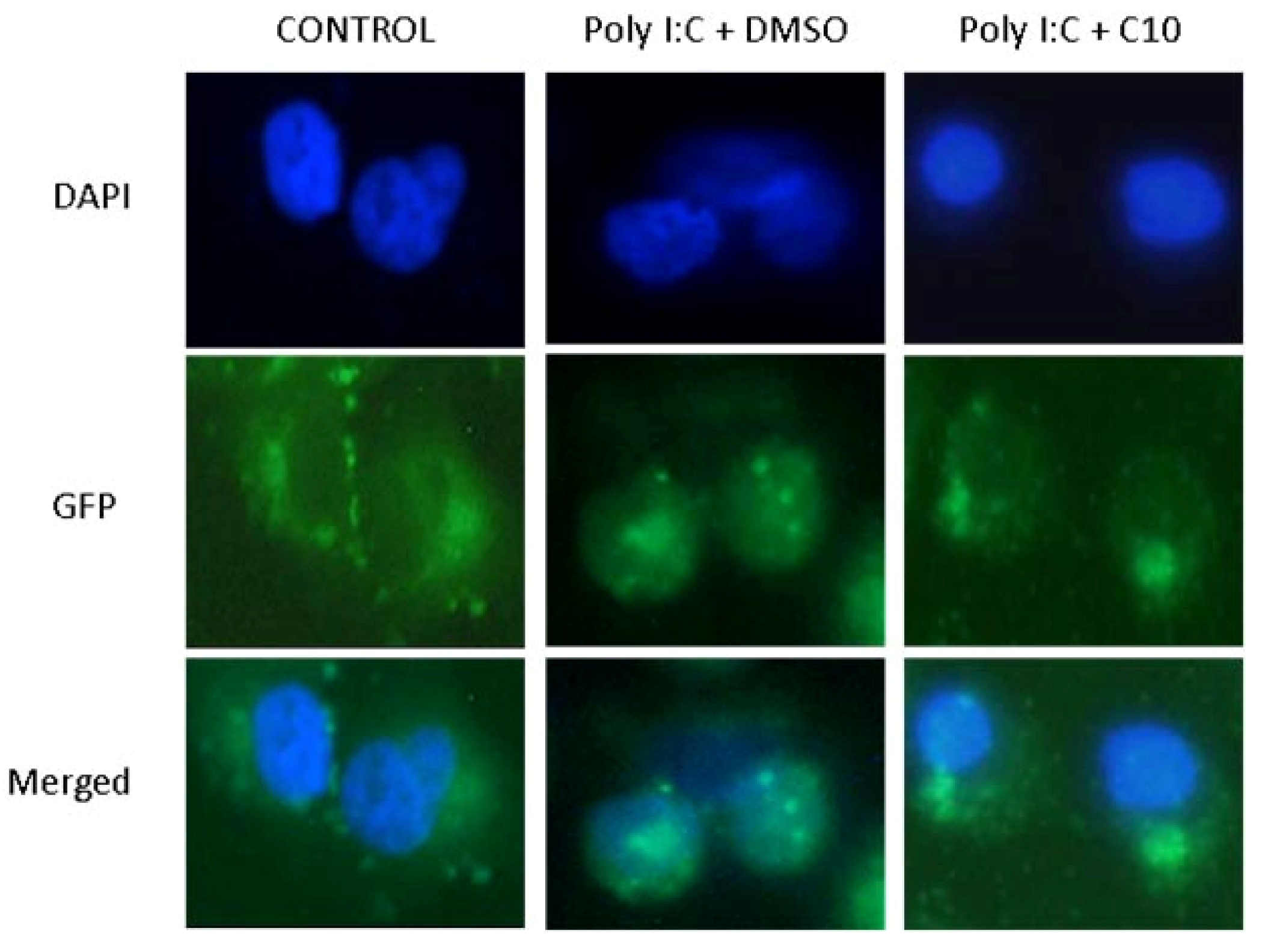
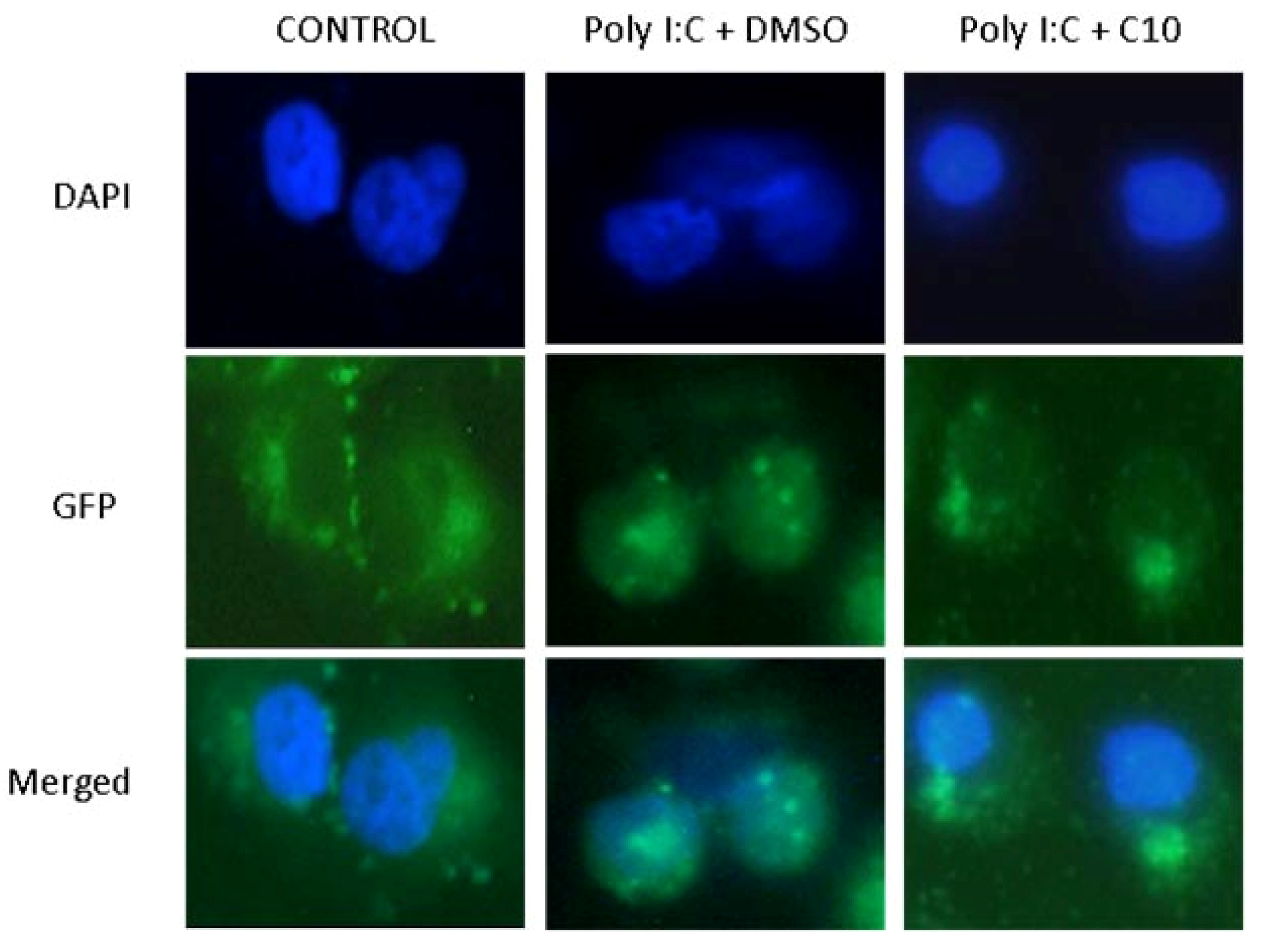
2.2. C10 Inhibits IRF3 Translocation to the Nucleus
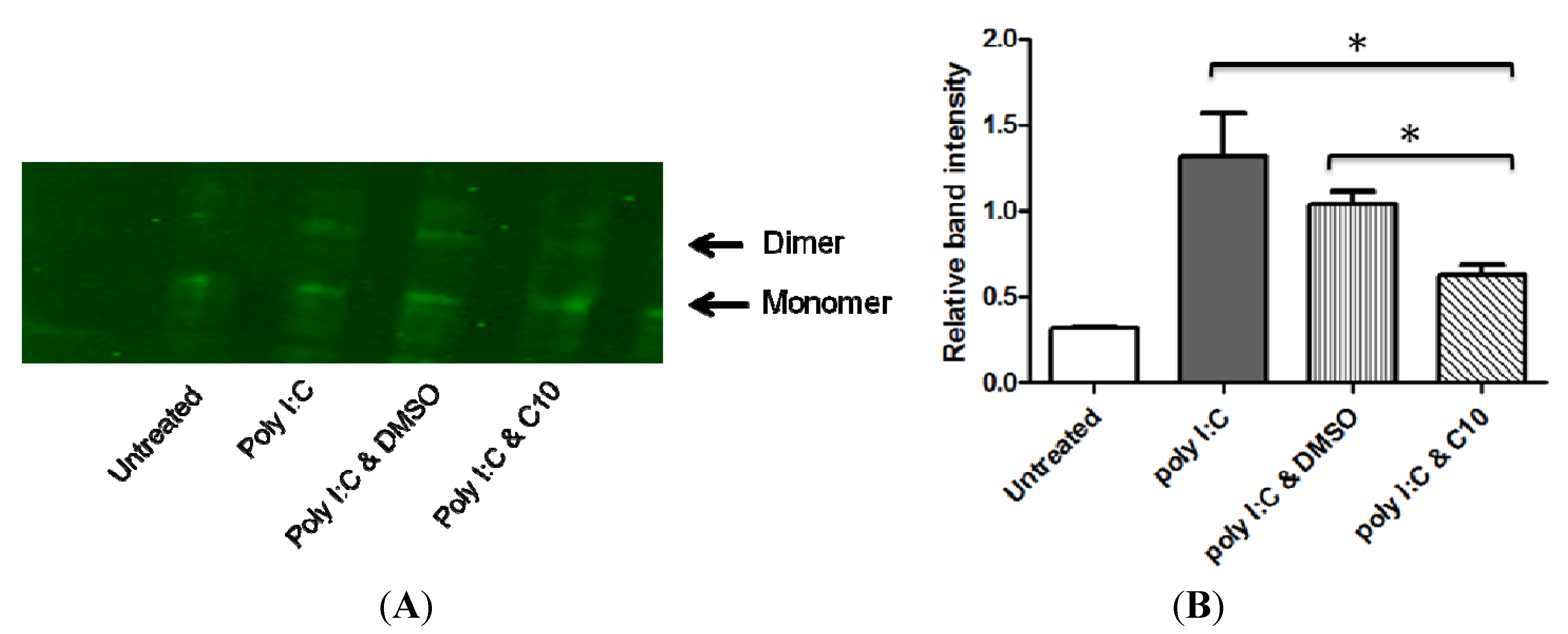
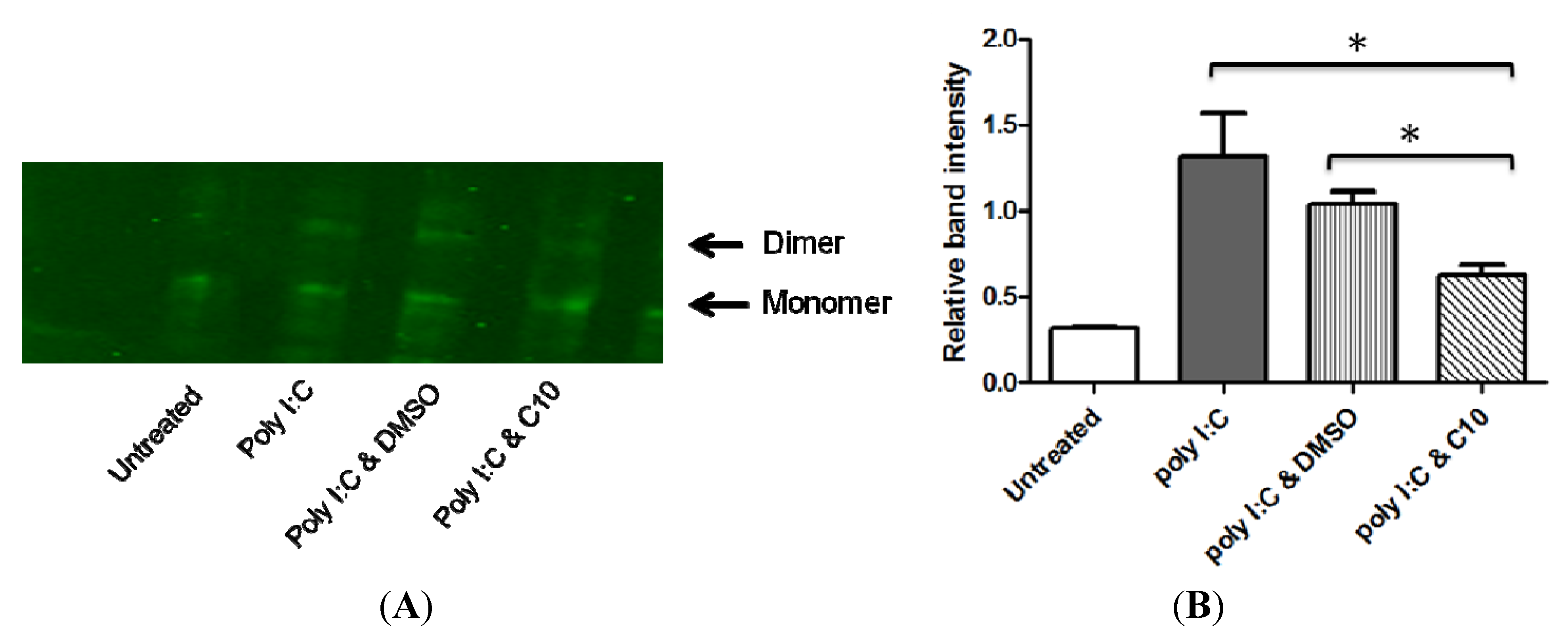
2.3. C10 Inhibits IRF3 Homodimer Formation
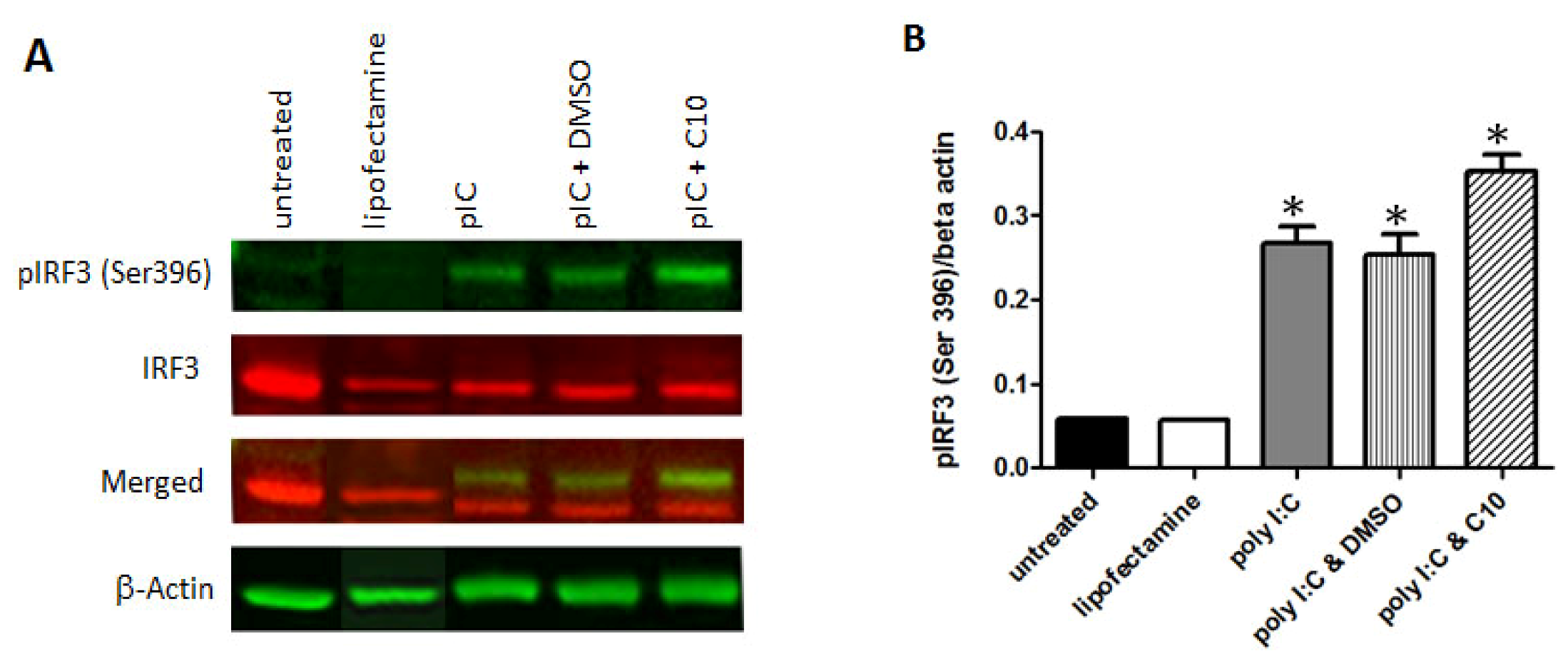
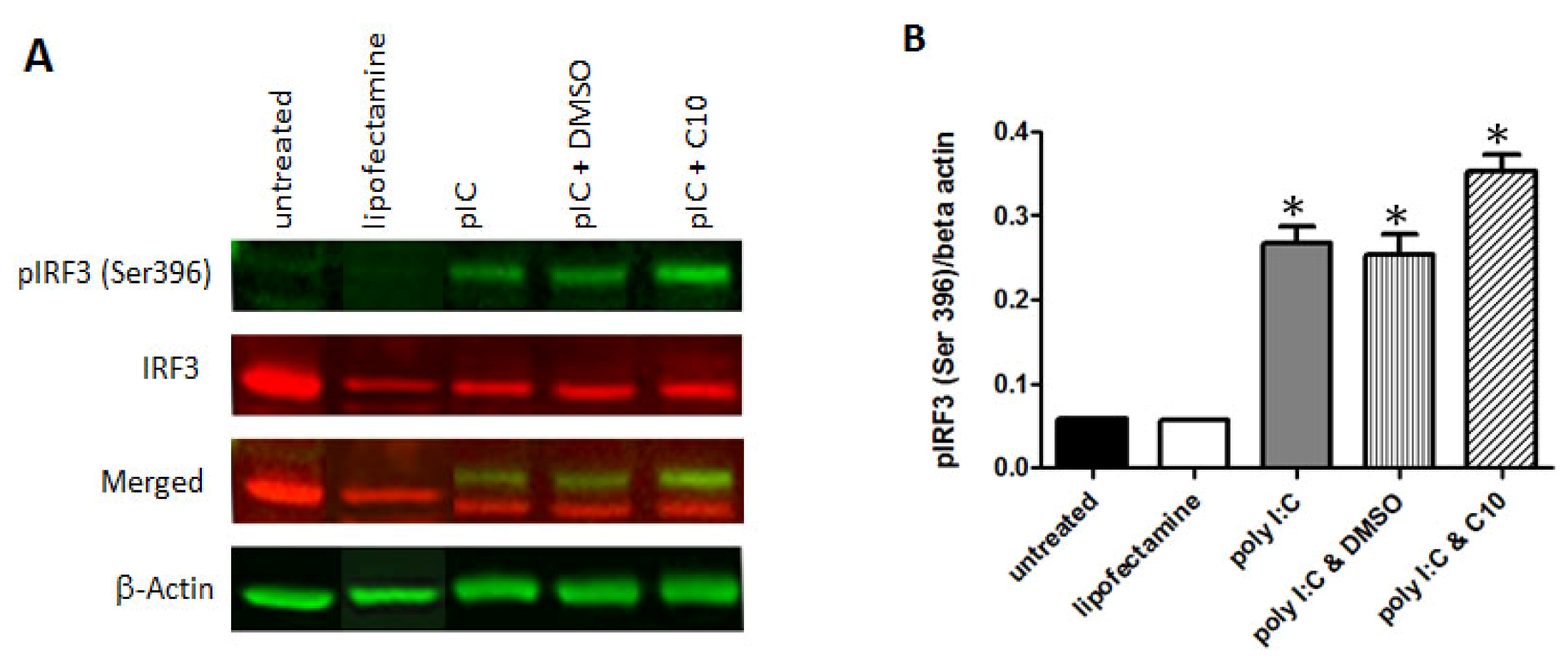
2.4. C10 Does Not Affect IRF-3 Phosphorylation
3. Experimental
3.1. Phenylmethimazole (C10) Solutions
3.2. Cell Culture
3.3. Plasmids and Transfections
3.4. Preparation of Nuclear Extracts for IRF-3 DNA-Binding Activity
3.5. Detection of IRF-3 DNA-Binding Activity
3.6. Translocation of IRF-3 to the Nucleus
3.7. SDS and Native PAGE Analyses
3.8. Statistical Analysis
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compound (C10) are available from the Interthyr Corporation.
References
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, Transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 1998, 18, 2986–2996. [Google Scholar]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef]
- Lin, R.; Mamane, Y.; Hiscott, J. Structural and functional analysis of interferon regulatory factor 3:Localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 1999, 19, 2465–2474. [Google Scholar]
- Grandvaux, N.; Servant, M.J.; tenOever, B.; Sen, G.C.; Balachandran, S.; Barber, G.N.; Lin, R.; Hiscott, J. Transcriptional profiling of interferon regulatory factor 3 target genes: Direct involvement in the regulation of interferon-stimulated genes. J. Virol. 2002, 76, 5532–5539. [Google Scholar]
- Kumar, K.P.; McBride, K.M.; Weaver, B.K.; Dingwall, C.; Reich, N.C. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, A subunit of double-stranded RNA-activated factor 1. Mol. Cell Biol. 2000, 20, 4159–4168. [Google Scholar] [CrossRef]
- Navarro, L.; Mowen, K.; Rodems, S.; Weaver, B.; Reich, N.; Spector, D.; David, M. Cytomegalovirus activates interferon immediate-early response gene expression and an interferon regulatory factor 3-containing interferon-stimulated response element-binding complex. Mol. Cell Biol. 1998, 18, 3796–3802. [Google Scholar]
- Wathelet, M.G.; Lin, C.H.; Parekh, B.S.; Ronco, L.V.; Howley, P.M.; Maniatis, T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1998, 1, 507–518. [Google Scholar] [CrossRef]
- Servant, M.J.; ten Oever, B.; LePage, C.; Conti, L.; Gessani, S.; Julkunen, I.; Lin, R.; Hiscott, J. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J. Biol. Chem. 2001, 276, 355–363. [Google Scholar]
- Servant, M.J.; Grandvaux, N.; Hiscott, J. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem. Pharmacol. 2002, 64, 985–992. [Google Scholar] [CrossRef]
- Chen, H.; Jiang, Z. The essential adaptors of innate immune signalin. Protein Cell 2012. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Hacker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef]
- Tseng, P.H.; Matsuzawa, A.; Zhang, W.; Mino, T.; Vignali, D.A.; Karin, M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 2010, 11, 70–75. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004, 199, 1641–1650. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, An adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, A mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Kato, H.; Sato, S.; Takahashi, K.; Coban, C.; Yamamoto, M.; Uematsu, S.; Ishii, K.J.; Takeuchi, O.; et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006, 203, 1795–1803. [Google Scholar]
- Sun, Q.; Sun, L.; Liu, H.H.; Chen, X.; Seth, R.B.; Forman, J.; Chen, Z.J. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006, 24, 633–642. [Google Scholar] [CrossRef]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998, 9, 143–150. [Google Scholar] [CrossRef]
- Schwartz, A.L.; Malgor, R.; Dickerson, E.; Weeraratna, A.T.; Slominski, A.; Wortsman, J.; Harii, N.; Kohn, A.D.; Moon, R.T.; Schwartz, F.L.; et al. Phenylmethimazole decreases Toll-like receptor 3 and noncanonical Wnt5a expression in pancreatic cancer and melanoma together with tumor cell growth and migration. Clin. Cancer Res. 2009, 15, 4114–4122. [Google Scholar] [CrossRef]
- McCall, K.D.; Harii, N.; Lewis, C.J.; Malgor, R.; Kim, W.B.; Saji, M.; Kohn, A.D.; Moon, R.T.; Kohn, L.D. High Basal Levels of Functional Toll-Like Receptor 3 (TLR3) and Non-Cannonical Wnt5a Are Expressed in Papillary Thyroid Cancer (PTC) and Are Coordinately Decreased by Phenylmethimazole Together with Cell Proliferation and Migration. Endocrinology 2007, 148, 4226–4237. [Google Scholar]
- Harii, N.; Lewis, C.; Vasko, V.; McCall, K.; Benavides-Peralta, U.; Sun, X.; Ringel, M.; Saji, M.; Kohn, L. Thyrocytes express a functional toll-like receptor 3(TLR3): Overexpression can be induced by viral infection, Reversed by Phenylmethimazole, and is associated with Hashimoto’s autoimmune thyroiditis. Mol. Endocrinol. 2005, 19, 1231–1250. [Google Scholar] [CrossRef]
- Jacobson, D.L.; Gange, S.J.; Rose, N.R.; Graham, N.M. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 1997, 84, 223–243. [Google Scholar] [CrossRef]
- McCall, K.D.; Holliday, D.; Dickerson, E.; Wallace, B.; Schwartz, A.L.; Schwartz, C.; Lewis, C.J.; Kohn, L.D.; Schwartz, F.L. Phenylmethimazole blocks palmitate-mediated induction of inflammatory cytokine pathways in 3T3L1 adipocytes and RAW 264.7 macrophages. J. Endocrinol. 2010, 207, 343–353. [Google Scholar] [CrossRef]
- Benavides, U.; Gonzalez-Murguiondo, M.; Harii, N.; Lewis, C.J.; Schwartz, A.L.; Giuliani, C.; Napolitano, G.; Dagia, N.M.; Malgor, R.; McCall, K.D.; et al. Phenylmethimazole inhibits production of proinflammatory mediators and is protective in an experimental model of endotoxic shock*. Crit. Care Med. 2012, 40, 886–894. [Google Scholar] [CrossRef]
- Michelsen, K.S.; Doherty, T.M.; Shah, P.K.; Arditi, M. TLR signaling: An emerging bridge from innate immunity to atherogenesis. J. Immunol. 2004, 173, 5901–5907. [Google Scholar]
- Beutler, B. Inferences, Questions and possibilities in Toll-like receptor signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef]
- Doyle, S.E.; O’Connell, R.; Vaidya, S.A.; Chow, E.K.; Yee, K.; Cheng, G. Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4. J. Immunol. 2003, 170, 3565–3571. [Google Scholar]
- Wen, L.; Peng, J.; Li, Z.; Wong, F.S. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J. Immunol. 2004, 172, 3173–3180. [Google Scholar]
- Devendra, D.; Eisenbarth, G.S. Interferon alpha—A potential link in the pathogenesis of viral-induced type 1 diabetes and autoimmunity. Clin. Immunol. 2004, 111, 225–233. [Google Scholar] [CrossRef]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar]
- Pasterkamp, G.; Van Keulen, J.K.; de Kleijn, D.P. Role of Toll-like receptor 4 in the initiation and progression of atherosclerotic disease. Eur. J. Clin. Invest. 2004, 34, 328–334. [Google Scholar] [CrossRef]
- Andonegui, G.; Bonder, C.S.; Green, F.; Mullaly, S.C.; Zbytnuik, L.; Raharjo, E.; Kubes, P. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J. Clin. Invest. 2003, 111, 1011–1020. [Google Scholar]
- Fiocchi, C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology 1998, 115, 182–205. [Google Scholar] [CrossRef]
- Kohn, L.D.; Goetz, D.J.; McCall, K.D.; Schwartz, A.L.; Schwartz, F.L.; Malgor, R. Methods and compositions for the treatment of melanoma and pancreatic cancer. U.S. Patent 11/130,922, 2008. [Google Scholar]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Basler, C.F.; Mikulasova, A.; Martinez-Sobrido, L.; Paragas, J.; Muhlberger, E.; Bray, M.; Klenk, H.D.; Palese, P.; Garcia-Sastre, A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003, 77, 7945–7956. [Google Scholar]
- Iwamura, T.; Yoneyama, M.; Yamaguchi, K.; Suhara, W.; Mori, W.; Shiota, K.; Okabe, Y.; Namiki, H.; Fujita, T. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells 2001, 6, 375–388. [Google Scholar] [CrossRef]
- Servant, M.J.; Grandvaux, N.; tenOever, B.R.; Duguay, D.; Lin, R.; Hiscott, J. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 2003, 278, 9441–9447. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Courreges, M.C.; Kantake, N.; Goetz, D.J.; Schwartz, F.L.; McCall, K.D. Phenylmethimazole Blocks dsRNA-Induced IRF3 Nuclear Translocation and Homodimerization. Molecules 2012, 17, 12365-12377. https://doi.org/10.3390/molecules171012365
Courreges MC, Kantake N, Goetz DJ, Schwartz FL, McCall KD. Phenylmethimazole Blocks dsRNA-Induced IRF3 Nuclear Translocation and Homodimerization. Molecules. 2012; 17(10):12365-12377. https://doi.org/10.3390/molecules171012365
Chicago/Turabian StyleCourreges, Maria C., Noriko Kantake, Douglas J. Goetz, Frank L. Schwartz, and Kelly D. McCall. 2012. "Phenylmethimazole Blocks dsRNA-Induced IRF3 Nuclear Translocation and Homodimerization" Molecules 17, no. 10: 12365-12377. https://doi.org/10.3390/molecules171012365