Discovery and Validation of SIRT2 Inhibitors Based on Tenovin-6: Use of a 1H-NMR Method to Assess Deacetylase Activity

Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
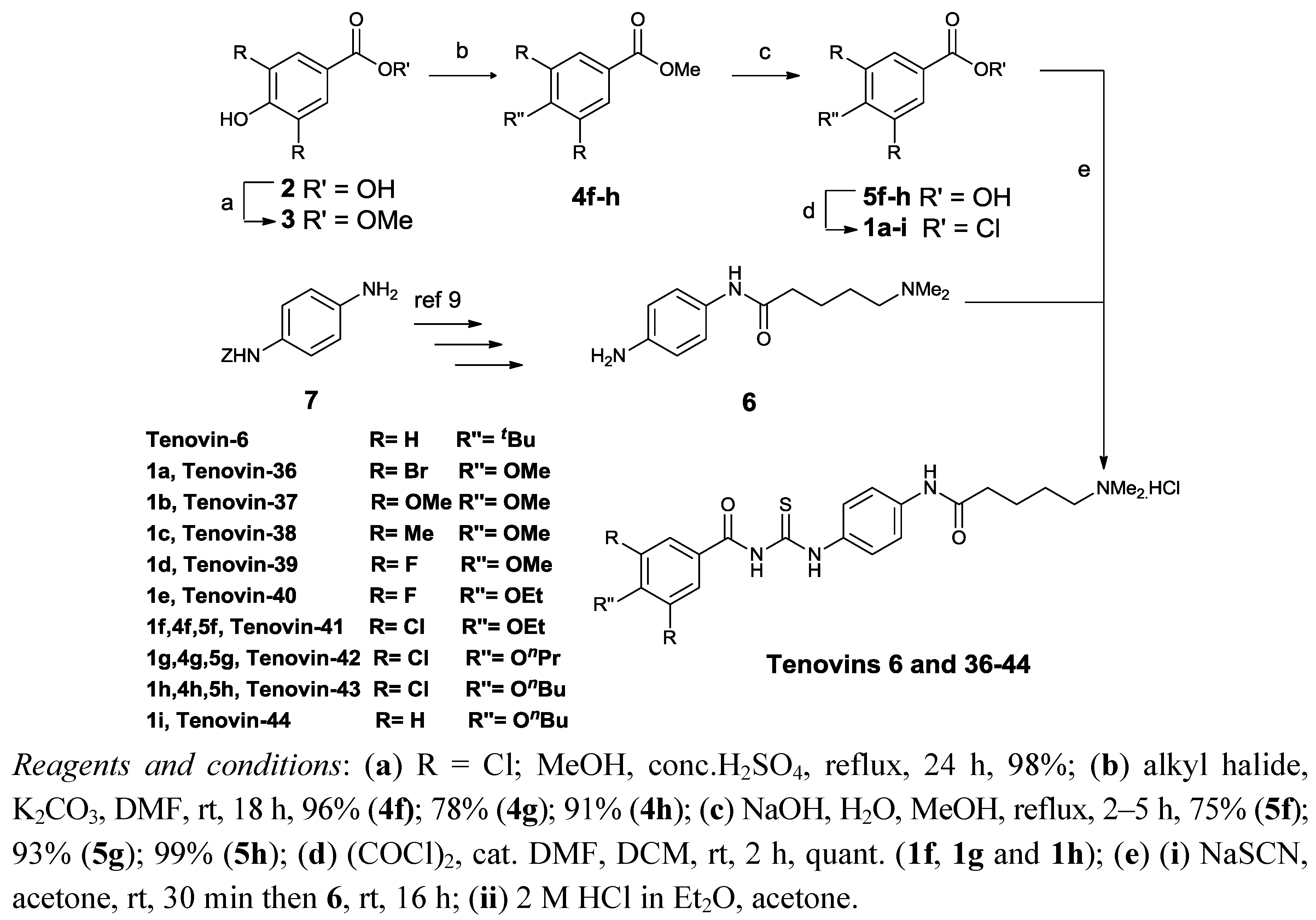

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
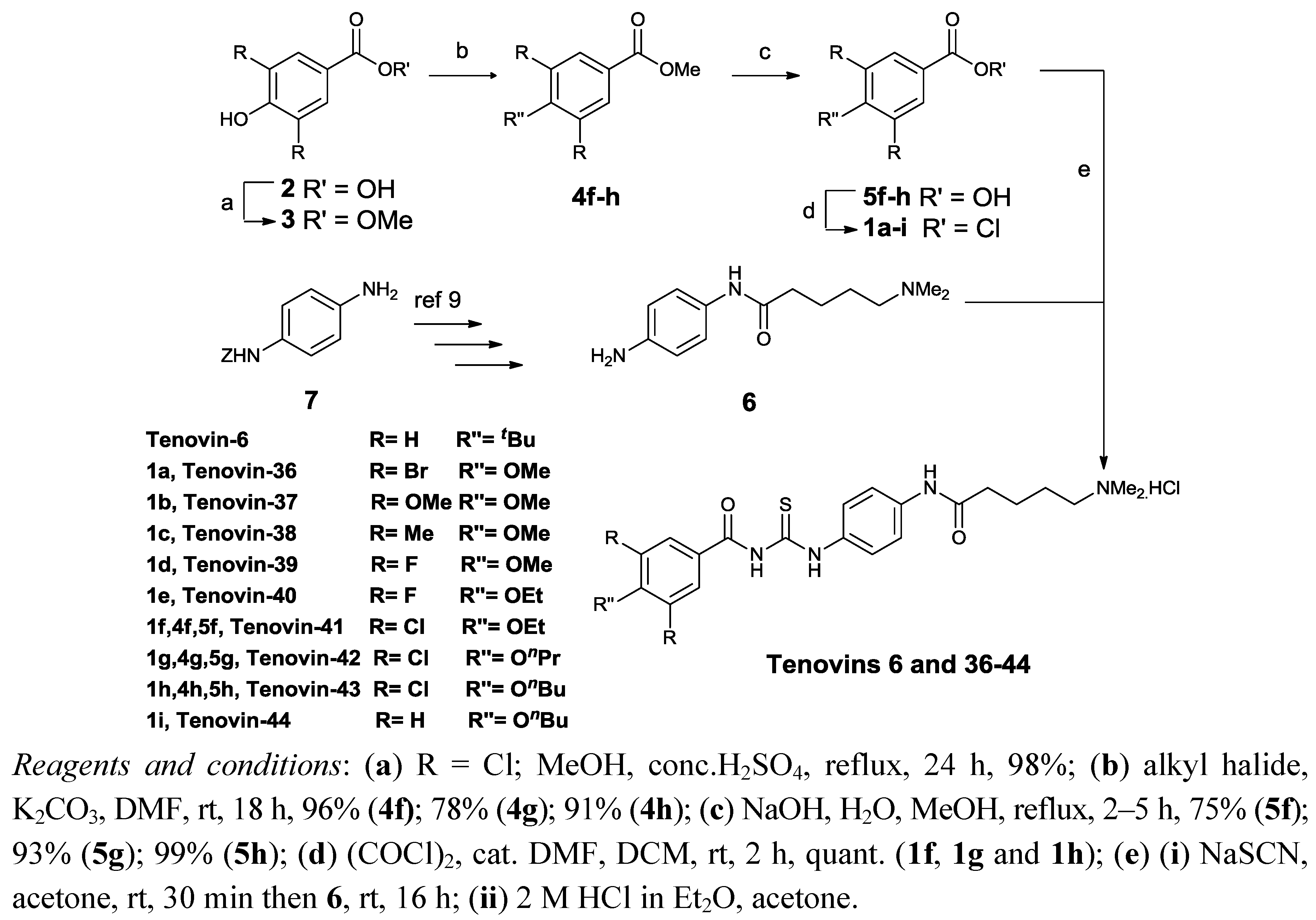{kind=link}
| Tenovin | R | R′′ | SIRT1 | IC50 | SIRT2 | IC50 | Selectivity Factor |
|---|---|---|---|---|---|---|---|
| % at 60 µm a | % at 60 μM a | ||||||
| 6 | H | tBu | 73.9 ± 1 | 21b | 94.9 ± 1 | 10 b | 2.1 |
| 36 | Br | OMe | 66.2 ± 2 | 51.6 ± 2 | 82.7 ± 1 | 16.0 ± 3 | 3.2 |
| 37 | OMe | OMe | 12.5 ± 1 | n.d. | 18.1 ± 1 | n.d. | n.d. |
| 38 | Me | OMe | 34.2 ± 3 | 65.4 ± 1 | 54.7 ± 1 | 44.9 ± 1 | 1.5 |
| 39 | F | OMe | 79.5 ± 4 | 47.2 ± 1 | 81.2 ± 5 | 18.0 ± 3 | 2.6 |
| 40 | F | OEt | 72.0 ± 4 | 35.3 ± 3 | 89.1 ± 1 | 17.5 ± 2 | 2.0 |
| 41 | Cl | OEt | 79.4 ± 1 | 28.5 ± 1 | 81.5 ± 2 | 12.9 ± 1 | 2.2 |
| 42 | Cl | OnPr | 81.0 ± 3 | 23.5 ± 2 | 96.1 ± 1 | 4.7 ± 3 | 5.0 |
| 43 | Cl | OnBu | 89.2 ± 2 | 21.5 ± 1 | 99.6 ± 3 | 0.8 ± 0.4 | 26.9 |
| 44 | H | OnBu | 87.7 ± 1 | 36.5 ± 5 | 88.2 ± 2 | 7.2 ± 1 | 5.1 |
2.2. In Vitro Inhibition of SIRT1 and SIRT2
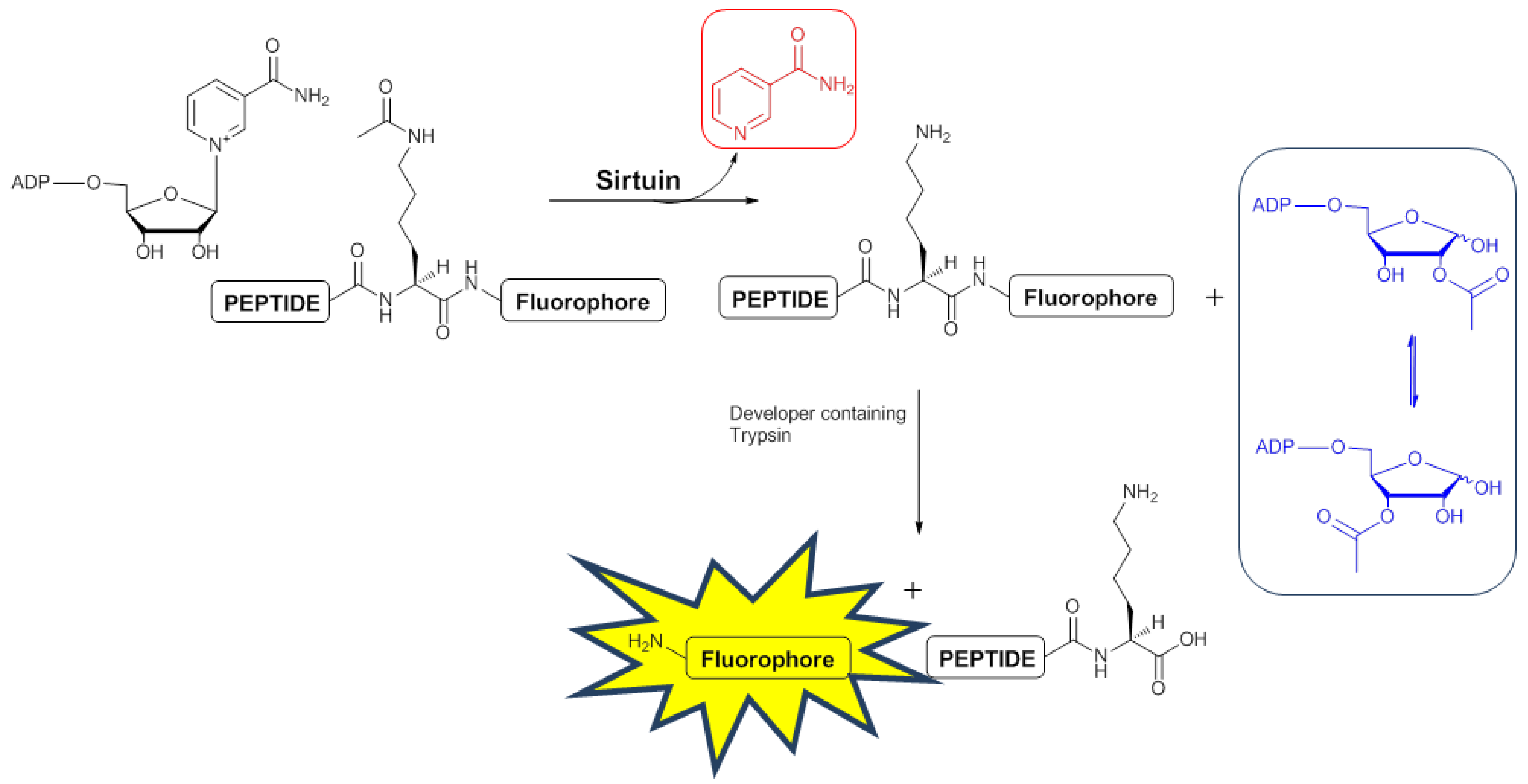
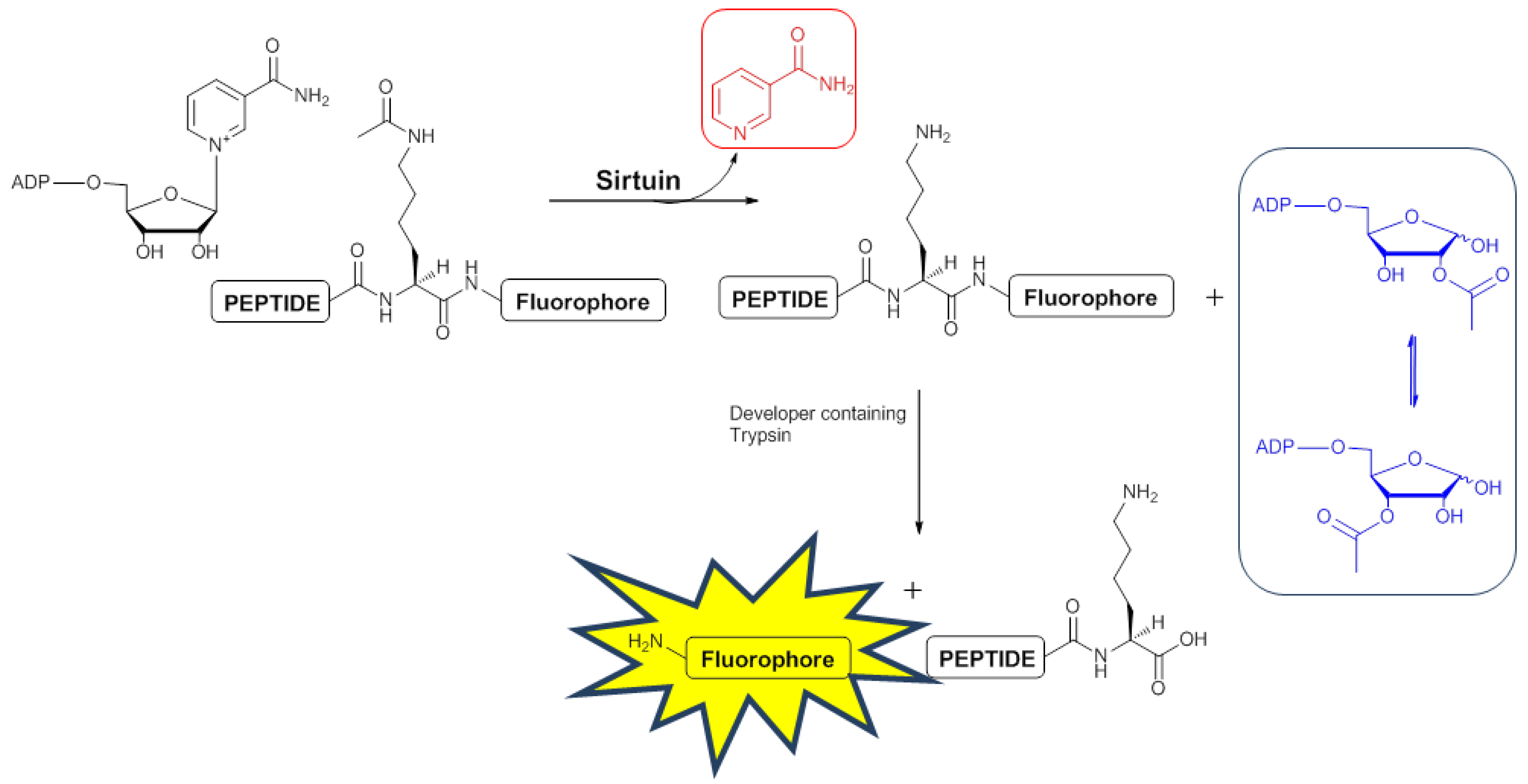
2.3. Development of a 1H-NMR Assay for Deacetylase Activity
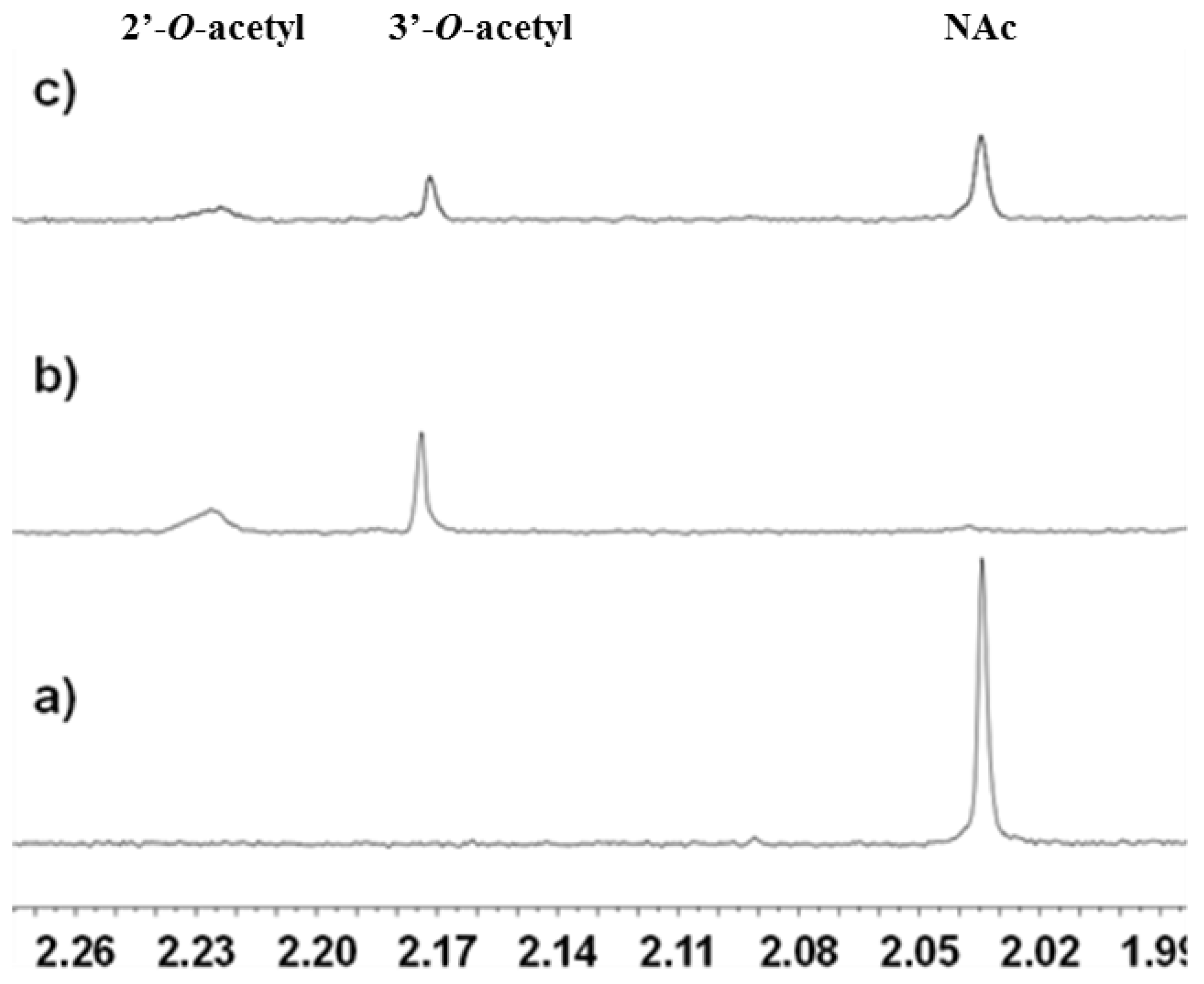
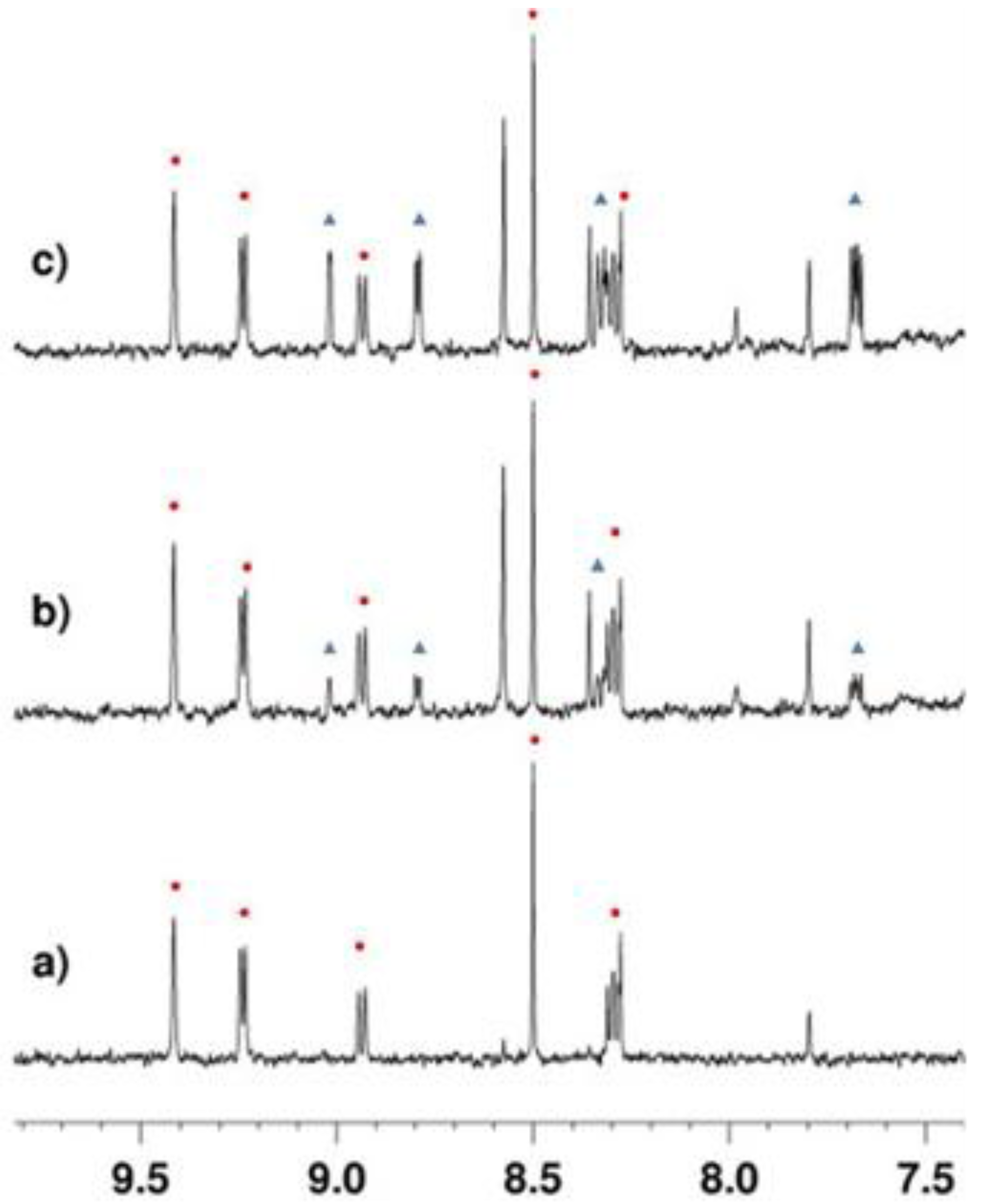
2.3.1. Deacetylation of a Histone H4 Peptide by SIRT2
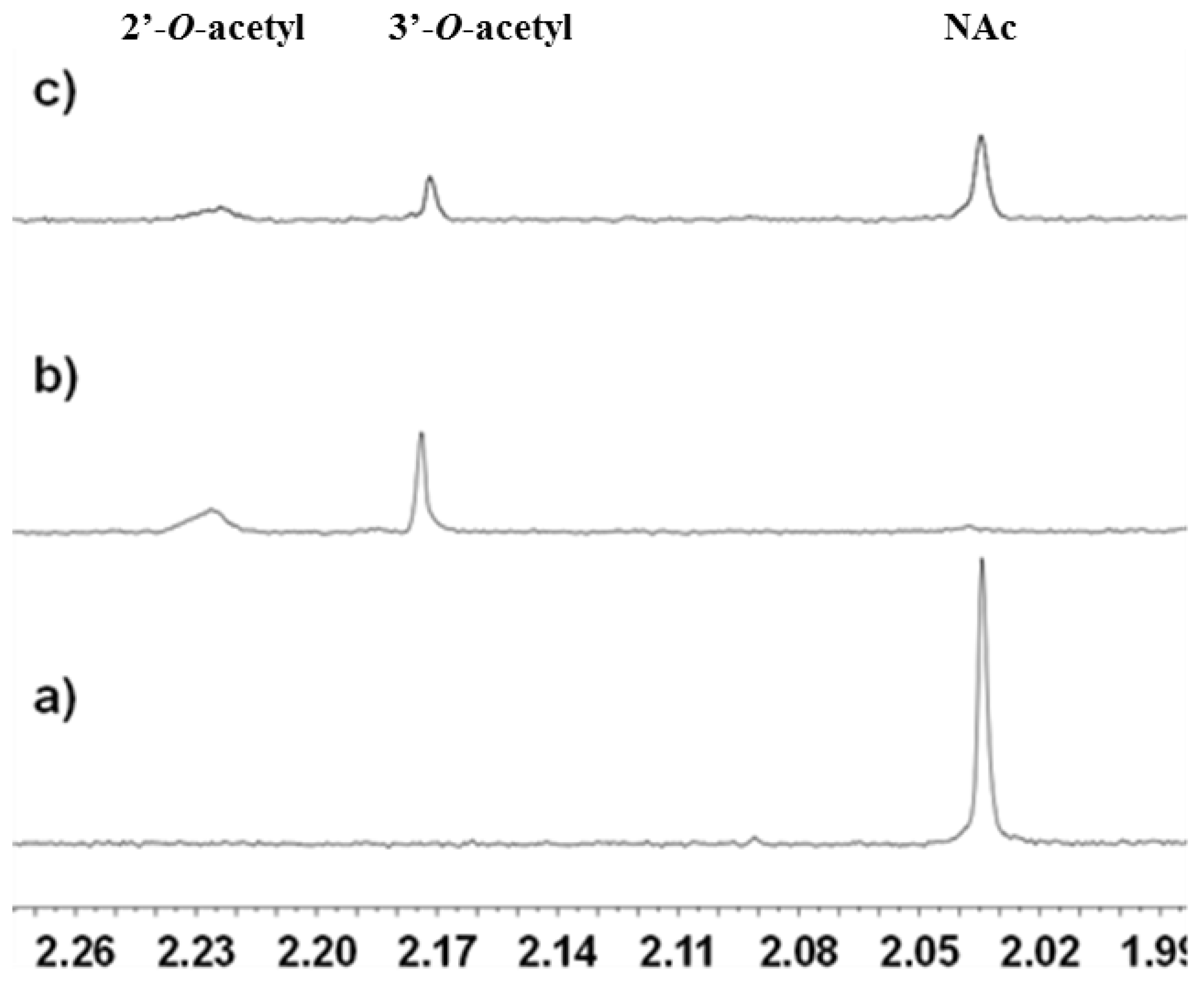
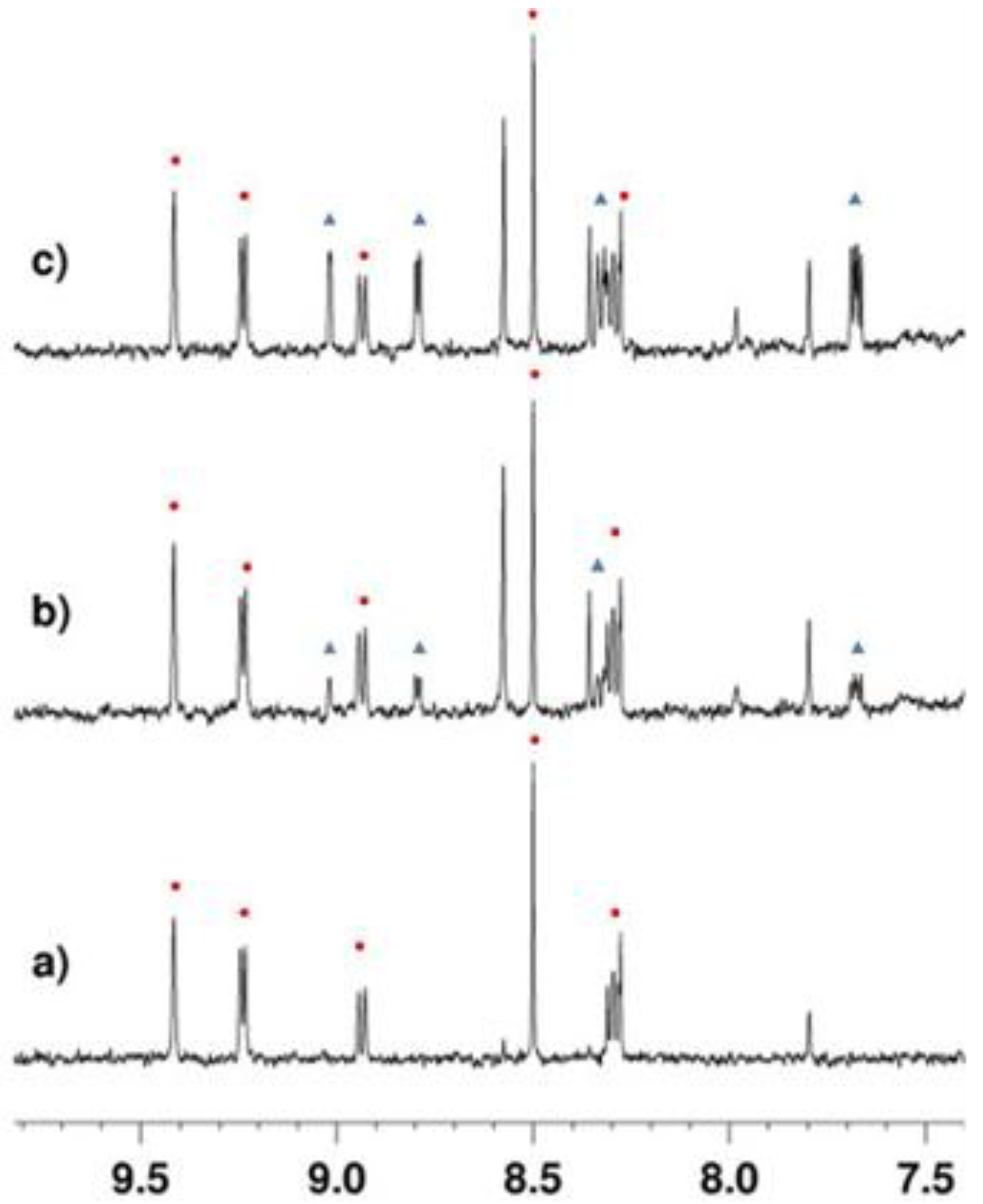
2.3.2. Inhibition of the Deacetylase Reaction of SIRT2 by the Tenovins
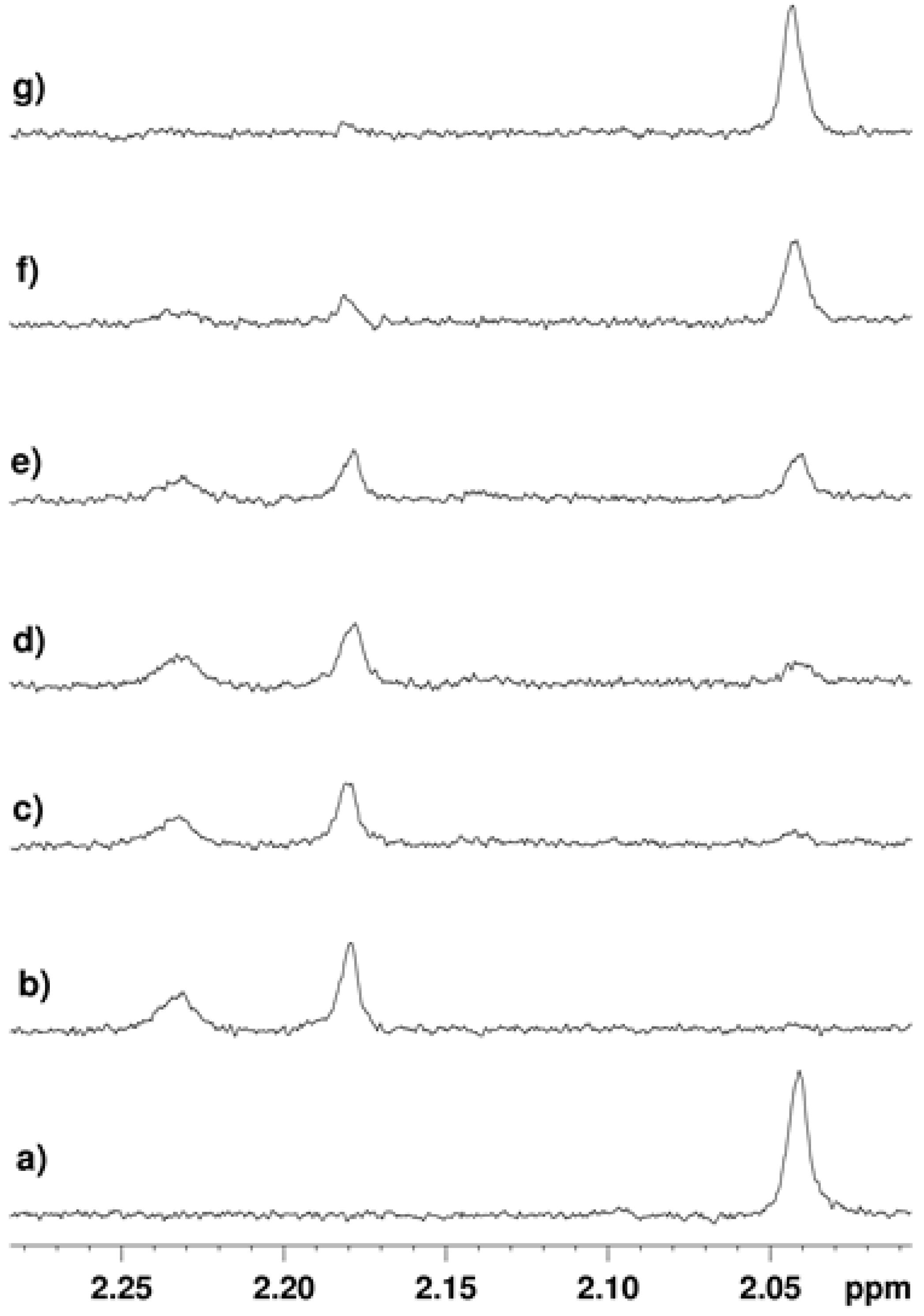

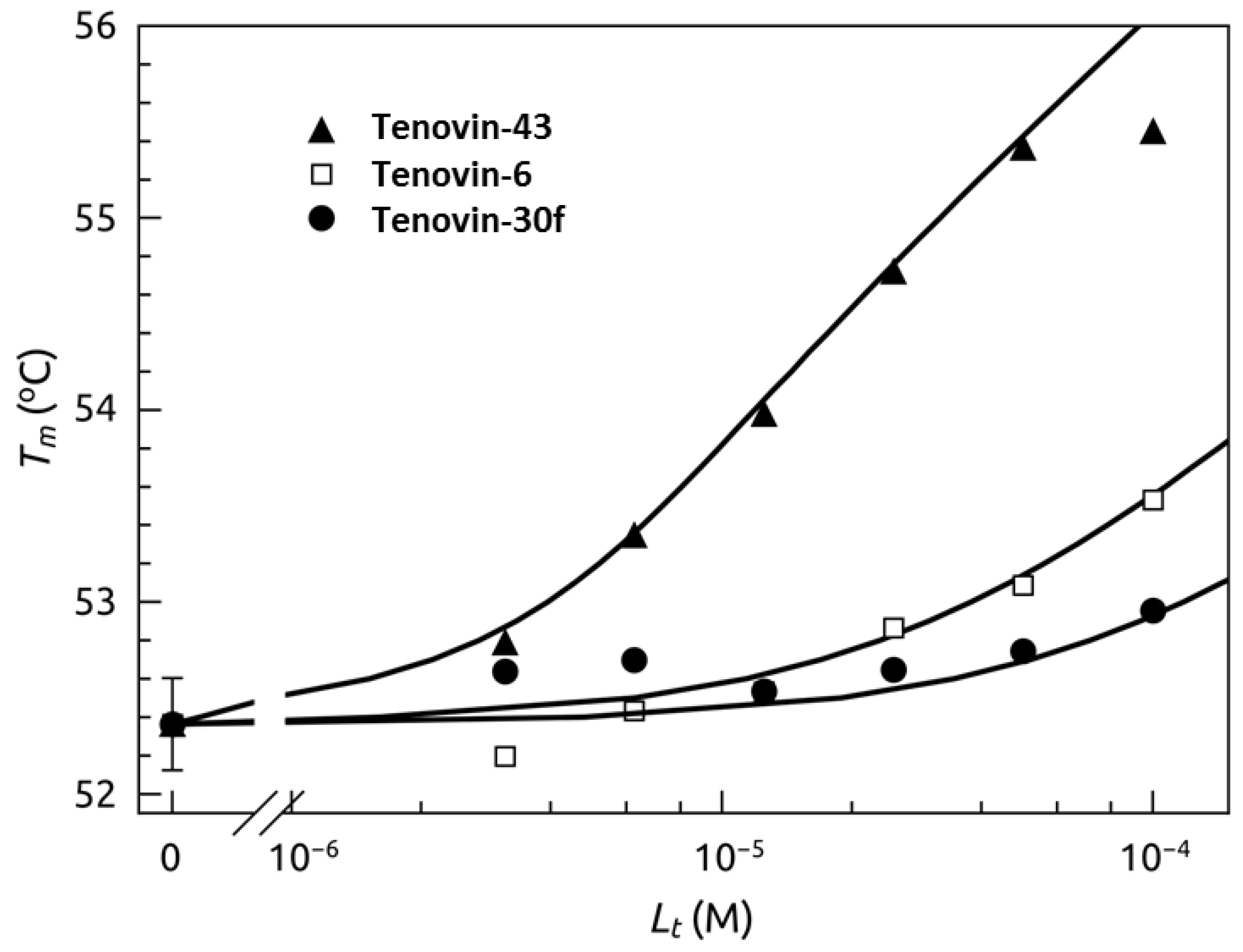
2.4. Thermal Shift Analysis of SIRT2 in the Presence of the Tenovins and AGK2
3. Experimental
3.1. SIRT2 Expression and Purification
3.2. SIRT1 and SIRT2 Inhibition Assay
3.3. 1H-NMR Experiments
3.4. Thermal Shift Experiments
3.5. General
3.6. Synthesis
3.6.1. General Procedure for the alkylation of 3
3.6.2. General Procedure for Ester Hydrolysis
3.6.3. General Procedure for Synthesis of Acid Chlorides 1a–1
3.6.4. General Procedure for Sodium Thiocyanate Coupling Reaction
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Alcain, F.J.; Villalba, J.M. Sirtuin inhibitors. Expert Opin. Ther. Pat. 2009, 19, 283–294. [Google Scholar] [CrossRef]
- Cen, Y. Sirtuins inhibitors: The approach to affinity and selectivity. Biochim. Biophys. Acta 2010, 1804, 1635–1644. [Google Scholar] [CrossRef]
- Grubisha, O.; Smith, B.C.; Denu, J.M. Small molecule regulation of Sir2 protein deacetylases. FEBS J. 2005, 272, 4607–4616. [Google Scholar] [CrossRef]
- Itoh, Y.; Suzuki, T.; Miyata, N. Isoform-selective histone deacetylase inhibitors. Curr. Pharm. Des. 2008, 14, 529–544. [Google Scholar] [CrossRef]
- Neugebauer, R.C.; Sippl, W.; Jung, M. Inhibitors of NAD(+) dependent histone deacetylases (sirtuins). Curr. Pharm. Des. 2008, 14, 562–573. [Google Scholar] [CrossRef]
- Medda, F.; Joseph, T.L.; Pirrie, L.; Higgins, M.; Slawin, A.M.Z.; Lain, S.; Verma, C.; Westwood, N.J. N1-Benzyl substituted cambinol analogues as isozyme selective inhibitors of the sirtuin family of protein deacetylases. Med. Chem. Comm. 2011, 2, 611–615. [Google Scholar]
- Medda, F.; Russell, R.J.M.; Higgins, M.; McCarthy, A.R.; Campbell, J.; Slawin, A.M.Z.; Lane, D.P.; Lain, S.; Westwood, N.J. Novel Cambinol Analogs as Sirtuin Inhibitors: Synthesis, Biological Evaluation, and Rationalization of Activity. J. Med. Chem. 2009, 52, 2673–2682. [Google Scholar] [CrossRef]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, A.R.; Pirrie, L.; Hollick, J.J.; Ronseaux, S.; Campbell, J.; Higgins, M.; Staples, O.D.; Tran, F.; Slawin, A.M.Z.; Lain, S.; et al. Synthesis and biological characterisation of sirtuin inhibitors based on the tenovins. Bioorg. Med. Chem. 2012, 20, 1779–1793. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The Critical Role of the Class III Histone Deacetylase SIRT1 in Cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, Z.; Li, L.; Zhang, H.; Modi, H.; Horne, D.; Bhatia, R.; Chen, W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 2012, 119, 1904–1914. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, H.; Roth, M.; Stark, J.M.; Bhatia, R.; Chen, W.Y. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene 2012, in press. [Google Scholar]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef]
- Harting, K.; Knoll, B. SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur. J. Cell Biol. 2010, 89, 262–269. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar]
- Fluor de Lys SIRT2 fluorometric drug discovery kit, Enzo Life Sciences, BML-AK556-0001.
- Suzuki, T.; Khan, M.N.A.; Sawada, H.; Imai, E.; Itoh, Y.; Yamatsuta, K.; Tokuda, N.; Takeuchi, J.; Seko, T.; Nakagawa, H.; et al. Design, Synthesis, and Biological Activity of a Novel Series of Human Sirtuin-2-Selective Inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [Google Scholar]
- Tenovin-43 was tested in cell-based assays that read out on sirtuin inhibitory activity. It was found to be inactive in a p53-dependant transcription assay. Studies in an acetylated tubulin assay were hampered by tenovin-43’s apparent toxicity at concentrations of greater than 20 µM although an increase in the levels of K40-acetylated tubulin was observed when tenovin-43 was used at a final concentration of 10 µM (see Figure S1).
- Auld, D.S.; Southall, N.T.; Jadhav, A.; Johnson, R.L.; Diller, D.J.; Simeonov, A.; Austin, C.P.; Inglese, J. Characterization of Chemical Libraries for Luciferase Inhibitory Activity. J. Med. Chem. 2008, 51, 2372–2386. [Google Scholar] [CrossRef]
- Kongkamnerd, J.; Milani, A.; Cattoli, G.; Terregino, C.; Capua, I.; Beneduce, L.; Gallotta, A.; Pengo, P.; Fassina, G.; Monthakantirat, O.; et al. The Quenching Effect of Flavonoids on 4-Methylumbelliferone, a Potential Pitfall in Fluorimetric Neuraminidase Inhibition Assays. J. Biomol. Screen. 2011, 16, 755–764. [Google Scholar] [CrossRef]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and Resveratrol Are Not Direct Activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef]
- Cimmperman, P.; Baranauskien, L.; Jachimovičiūtė, S.; Jachno, J.; Torresan, J.; Michailovien, V.; Matulien, J.; Sereikait, J.; Bumelis, V.; Matulis, D. A Quantitative Model of Thermal Stabilization and Destabilization of Proteins by Ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar]
- Baranauskienė, L.; Hilvo, M.; Matulienė, J.; Golovenko, D.; Manakova, E.; Dudutienė, V.; Michailovienė, V.; Torresan, J.; Jachno, J.; Parkkila, S.; et al. Inhibition and binding studies of carbonic anhydrase isozymes I, II and IX with benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides. J. Enzyme Inhib. Med. Chem. 2010, 25, 863–870. [Google Scholar] [CrossRef]
- Matulis, D.; Kranz, J.K.; Salemme, F.R.; Todd, M.J. Thermodynamic Stability of Carbonic Anhydrase: Measurements of Binding Affinity and Stoichiometry Using ThermoFluor. Biochemistry 2005, 44, 5258–5266. [Google Scholar]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protocols 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Petrikaite, V.M. Thermodynamics of Natural and Synthetic Inhibitor Binding to Human Hsp90. In Application of Thermodynamics to Biological and Materials Science; Tadashi, M., Ed.; In Tech: New York, NY, USA, 2011. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pirrie, L.; McCarthy, A.R.; Major, L.L.; Morkūnaitė, V.; Zubrienė, A.; Matulis, D.; Lain, S.; Lebl, T.; Westwood, N.J. Discovery and Validation of SIRT2 Inhibitors Based on Tenovin-6: Use of a 1H-NMR Method to Assess Deacetylase Activity. Molecules 2012, 17, 12206-12224. https://doi.org/10.3390/molecules171012206
Pirrie L, McCarthy AR, Major LL, Morkūnaitė V, Zubrienė A, Matulis D, Lain S, Lebl T, Westwood NJ. Discovery and Validation of SIRT2 Inhibitors Based on Tenovin-6: Use of a 1H-NMR Method to Assess Deacetylase Activity. Molecules. 2012; 17(10):12206-12224. https://doi.org/10.3390/molecules171012206
Chicago/Turabian StylePirrie, Lisa, Anna R. McCarthy, Louise L. Major, Vaida Morkūnaitė, Asta Zubrienė, Daumantas Matulis, Sonia Lain, Tomas Lebl, and Nicholas J. Westwood. 2012. "Discovery and Validation of SIRT2 Inhibitors Based on Tenovin-6: Use of a 1H-NMR Method to Assess Deacetylase Activity" Molecules 17, no. 10: 12206-12224. https://doi.org/10.3390/molecules171012206