1,3-Substituted Imidazolidine-2,4,5-triones: Synthesis and Inhibition of Cholinergic Enzymes

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
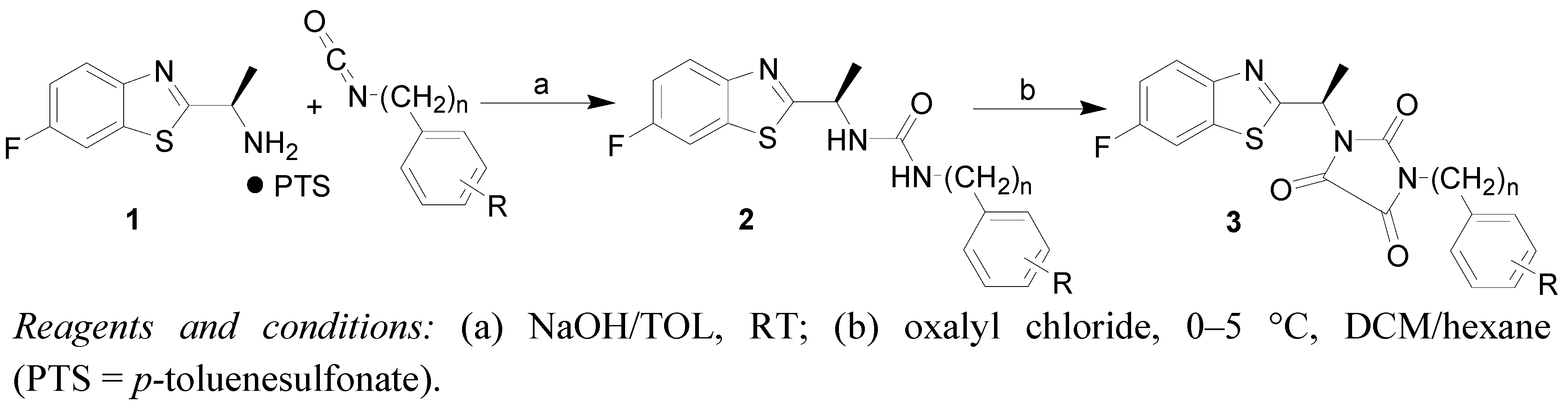
2.1. Chemistry

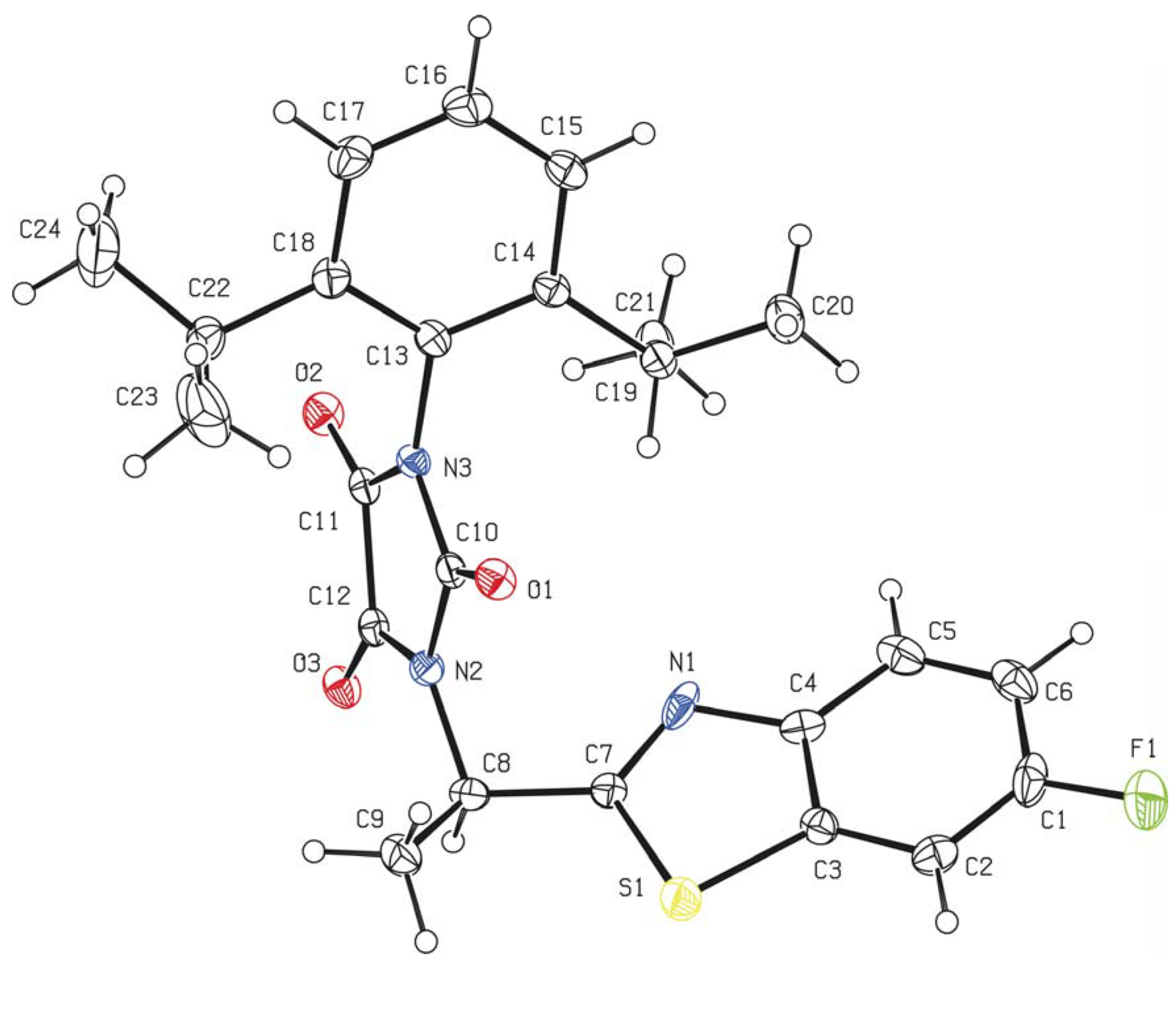
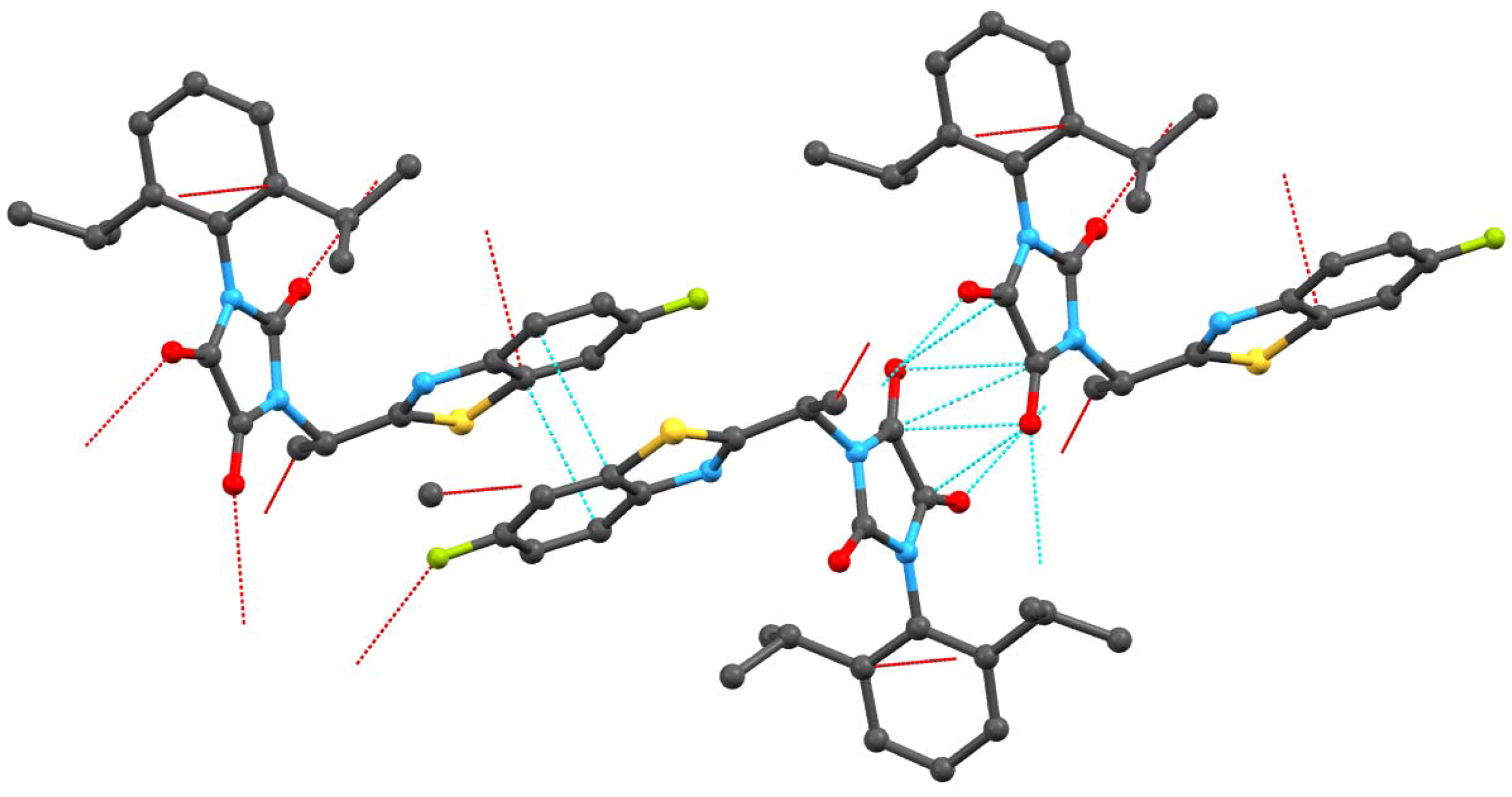
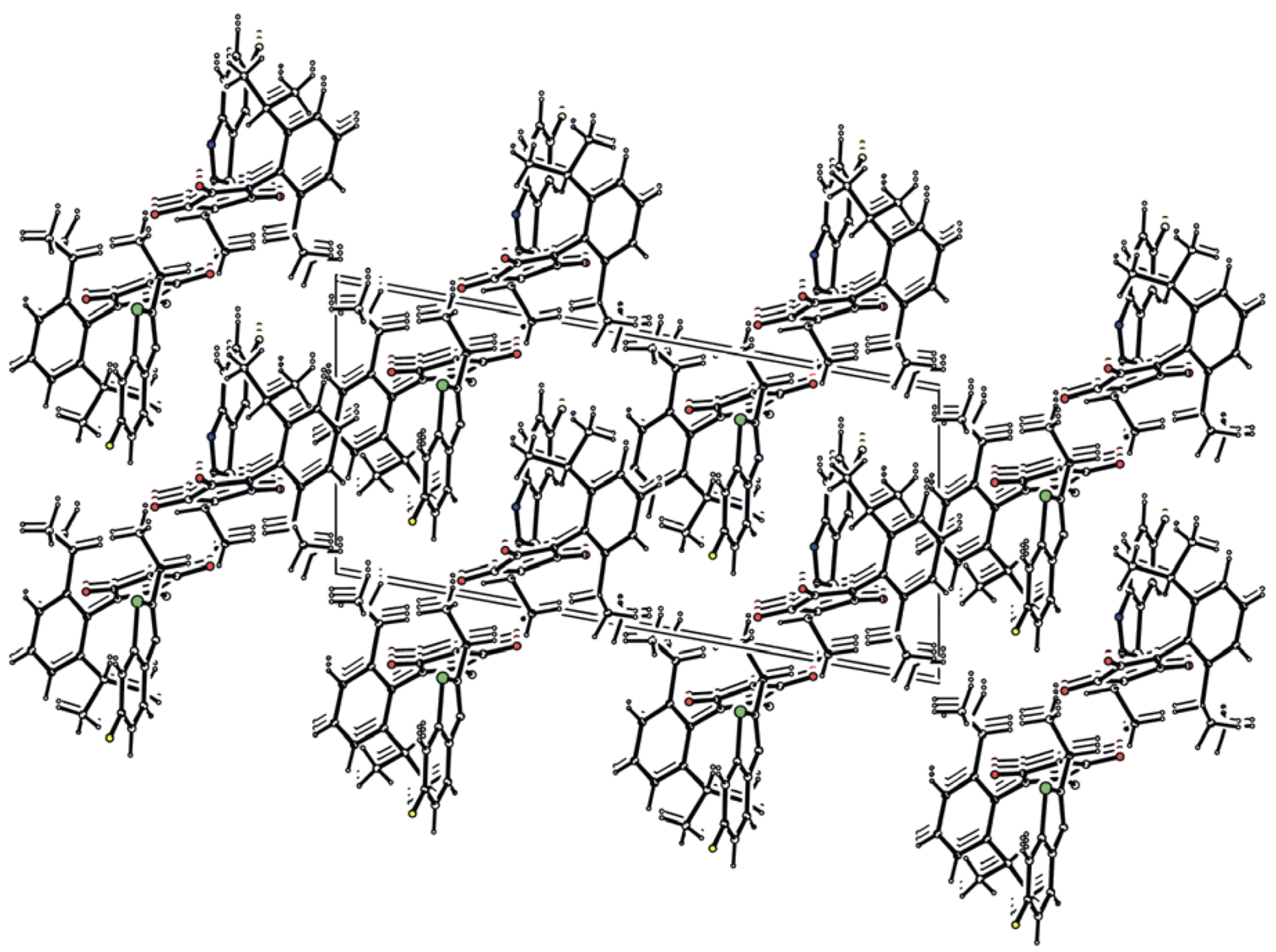
2.2. Crystallography



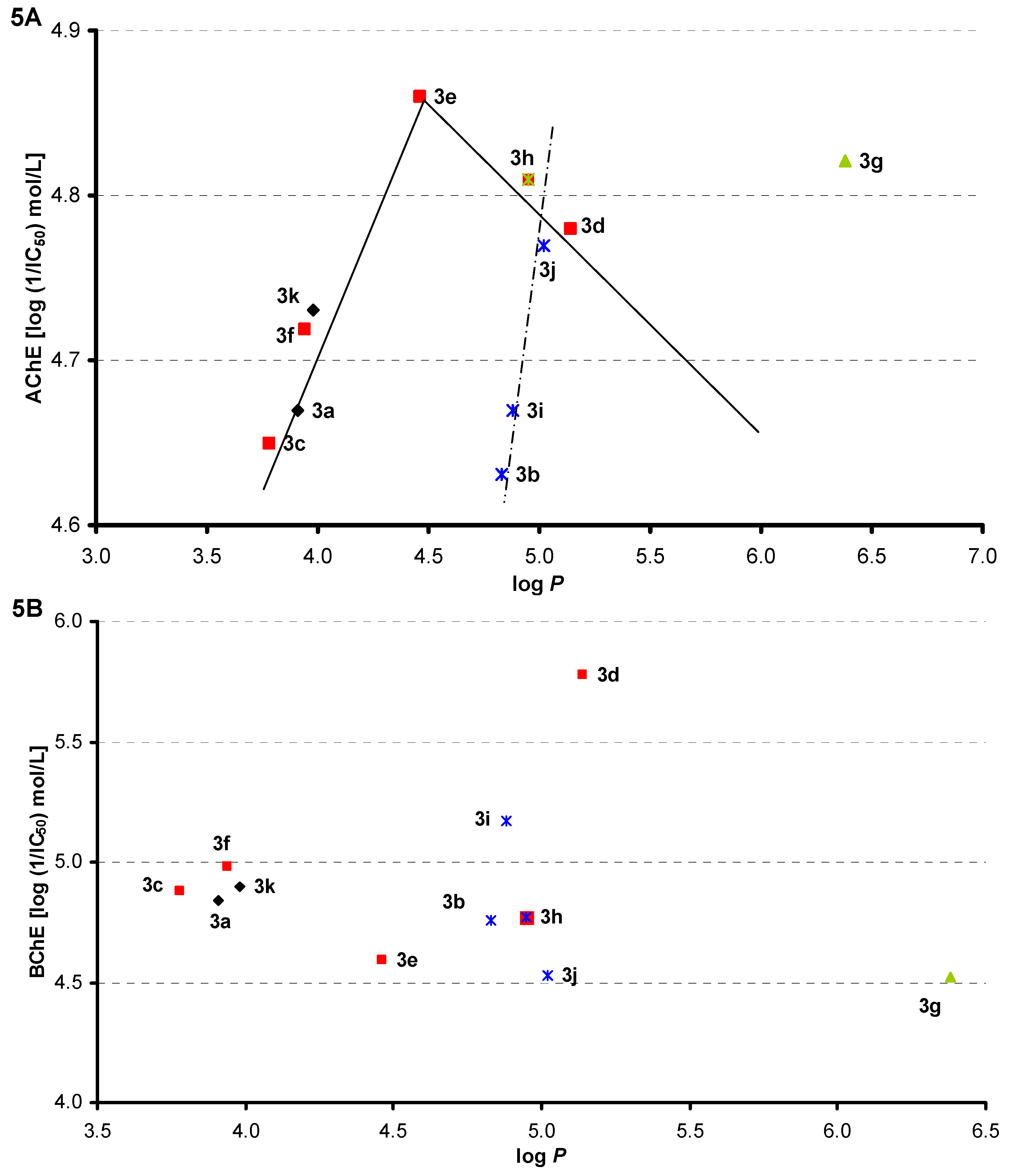
2.3. Lipophilicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
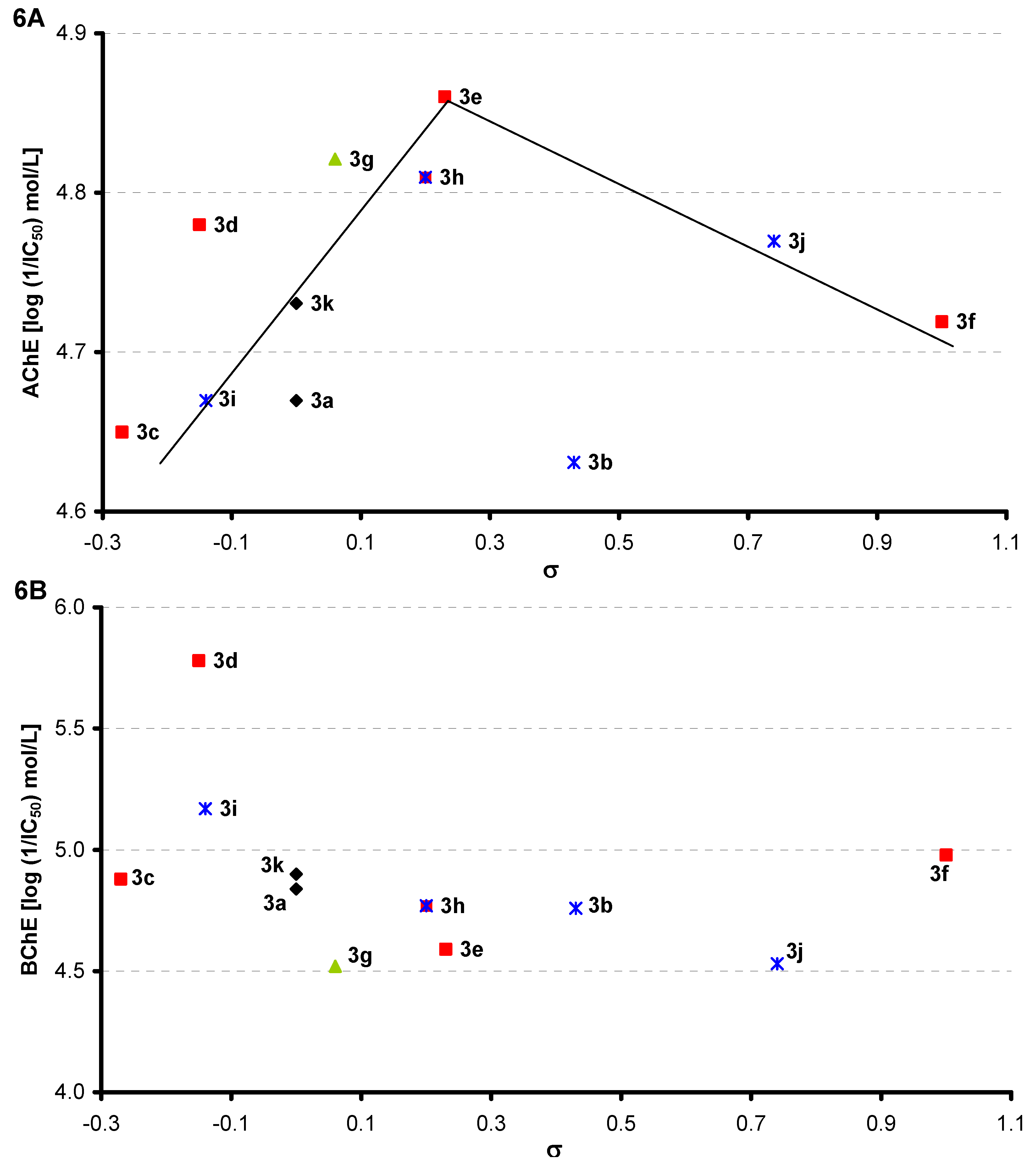
| Comp. | R | n | AChE | BChE | log Kow | log P/Clog P | σ [39] |
|---|---|---|---|---|---|---|---|
| IC50 [μmol/L] | |||||||
| 3a | H | 0 | 21.4 ± 0.19 | 14.5 ± 0.21 | 1.51 ± 0.03 | 3.91 / 2.769 | 0.00 |
| 3b | 3-CF3 | 0 | 23.4 ± 0.28 | 17.4 ± 0.37 | 1.12 ± 0.15 | 4.83 / 3.652 | 0.43 |
| 3c | 4-OCH3 | 0 | 22.4 ± 0.21 | 13.2 ± 0.22 | 0.69 ± 0.02 | 3.78 / 2.688 | −0.27 |
| 3d | 4-CH(CH3)2 | 0 | 16.6 ± 0.29 | 1.66 ± 0.14 | 1.64 ± 0.03 | 5.14 / 4.196 | −0.15 |
| 3e | 4-Cl | 0 | 13.8 ± 0.13 | 25.7 ± 0.29 | 0.41 ± 0.02 | 4.46 / 3.482 | 0.23 |
| 3f | 4-CN | 0 | 19.1 ± 0.27 | 10.5 ± 0.18 | 0.41 ± 0.02 | 3.94 / 2.202 | 1.00 |
| 3g | 2,6-CH(CH3)2 | 0 | 15.1 ± 0.29 | 30.2 ± 0.45 | 0.98 ± 0.08 | 6.38 / 5.623 | 0.06 |
| 3h | 3-Cl-4-CH3 | 0 | 15.5 ± 0.2 | 17.0 ± 0.1 | 0.50 ± 0.02 | 4.95 / 3.981 | 0.20 |
| 3i | 3,5-CH3 | 0 | 21.4 ± 0.19 | 6.76 ± 0.17 | 0.58 ± 0.03 | 4.88 / 3.767 | −0.14 |
| 3j | 3,5-Cl | 0 | 17.0 ± 0.38 | 29.5 ± 0.39 | 0.77 ± 0.03 | 5.02 / 4.195 | 0.74 |
| 3k | H | 1 | 18.6 ± 0.16 | 12.6 ± 0.11 | 1.06 ± 0.09 | 3.98 / 3.102 | 0.00 |
| RIV | – | – | 501 ± 3.08 | 19.95 ± 0.31 | – | 2.36 / 2.099 | – |
| GLT | – | – | 4.0 ± 0.13 | 7.96 ± 0.13 | – | 1.41 / 1.025 | – |
2.4. Inhibition of Cholinergic Enzymes



3. Experimental
3.1. General
3.2. Synthesis
General Procedure for the Synthesis of Compounds 3a–k
3.3. Determination of Crystallography
 and χ scan mode. Data reductions were performed with DENZO-SMN [42]. The absorption was corrected by integration methods [43]. Structures were solved by direct methods (Sir92) [44] and refined by full matrix least-square based on F2 (SHELXL97) [45]. Hydrogen atoms were mostly localized on a difference Fourier map, however to ensure uniformity of the treatment of the crystal, all hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2 Ueq(pivot atom) or of 1.5 Ueq for the methyl moiety with C–H = 0.96, 0.98 and 0.93 Å for methyl, methine and hydrogen atoms in the aromatic rings, respectively.
and χ scan mode. Data reductions were performed with DENZO-SMN [42]. The absorption was corrected by integration methods [43]. Structures were solved by direct methods (Sir92) [44] and refined by full matrix least-square based on F2 (SHELXL97) [45]. Hydrogen atoms were mostly localized on a difference Fourier map, however to ensure uniformity of the treatment of the crystal, all hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2 Ueq(pivot atom) or of 1.5 Ueq for the methyl moiety with C–H = 0.96, 0.98 and 0.93 Å for methyl, methine and hydrogen atoms in the aromatic rings, respectively.3.4. Determination of Partition Coefficient Kow
3.5. Lipophilicity Calculations
3.6. In Vitro Evaluation of AChE- and BChE-Inhibiting Activity
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgements
References
- Abbs Fen Rejia, T.F.; Rajasekharan, K.N. Synthesis of 2-[2,4-diaminothiazol-5-oyl]benzothiazoles. J. Het. Chem. 2010, 47, 994–997. [Google Scholar] [CrossRef]
- Huang, W.; Yang, G. Microwave-assisted, one-pot syntheses and fungicidal activity of polyfluorinated 2-benzylthiobenzothiazoles. Bioorg. Med. Chem. 2006, 14, 8280–8285. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Mosula, L.; Zimenkovsky, B.; Vasylenko, O.; Gzella, A.; Lesyk, R. Synthesis and anticancer activity evaluation of 4-thiazolidinones containing benzothiazole moiety. Eur. J. Med. Chem. 2010, 45, 5012–5021. [Google Scholar] [CrossRef]
- Bradshaw, T.D.; Westwell, A.D. The development of the antitumour benzothiazole prodrug, phortress, as a clinical candidate. Curr. Med. Chem. 2004, 11, 1241–1253. [Google Scholar]
- Song, H.; Oh, S.R.; Lee, H.K.; Han, G.; Kim, J.H.; Chang, H.W.; Don, K.E.; Rhee, H.K.; Choo, H.Y.P. Synthesis and evaluation of benzoxazole derivatives as 5-lipoxygenase inhibitors. Bioorg. Med. Chem. 2010, 18, 7580–7585. [Google Scholar]
- Paramashivappa, R.; Kumar, P.P.; Rao, P.V.S.; Rao, A.S. Design, synthesis and biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 657–660. [Google Scholar] [CrossRef]
- Kotani, T.; Ishii, A.; Nagaki, Y.; Toyomaki, Y.; Yago, H.; Suehiro, S.; Okukado, N.; Okamoto, K. Highly selective aldose reductase inhibitors. 2. Optimization of the aryl part of 3-(arylmethyl)-2,4,5-trioxoimidazolidine-1-acetic acids. Chem. Pharm. Bull. 1997, 45, 297–304. [Google Scholar] [CrossRef]
- Robin, M.; Mialhe, S.; Pique, V.; Faure, R.; Galy, J.P. Synthesis of two novel classes of tetracycles bearing tetrahydro ring system from benzothiazole 7,8,9,10-tetrahydrothiazolo[5,4-a]acridine and 1,2,3,4-tetrahydro-12H-benzothiazolo[2,3-b]quinazolin-12-one. J. Het. Chem. 2002, 39, 295–298. [Google Scholar] [CrossRef]
- Pejchal, V.; Stepankova, S.; Drabina, P. Synthesis of 1-[(1R)-1-(6-fluoro-1,3-benzothiazol-2-yl)ethyl]-3-substituted phenyl ureas and their inhibition activity to acetylcholinesterase and butyrylcholinesterase. J. Heterocycl. Chem. 2011, 48, 57–62. [Google Scholar] [CrossRef]
- Costanzo, M.J.; Almond, H.R.; Hecker, L.R.; Schott, M.R.; Yabut, S.C.; Zhang, H.C.; Andrade-Gordon, P.; Corcoran, T.W.; Giardino, E.C.; Kauffman, J.A. In-depth study of tripeptide-based α-ketoheterocycles as inhibitors of thrombin. Effective utilization of the S1' subsite and its implications to structure-based drug design. J. Med. Chem. 48, 2005, 1984–2008. [Google Scholar]
- Cygler, M.; Schrag, J.D.; Sussman, J.L.; Harel, M.; Silman, I.; Gentry, M.K.; Doctor, B.P. Relationship between sequence conservation and 3-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993, 2, 366–382. [Google Scholar]
- Groner, E.; Ashani, Y.; Schorer-Apelbaum, D.; Sterling, J.; Herzig, Y.; Weinstock, M. The kinetics of inhibition of human acetylcholinesterase and butyrylcholinesterase by two series of novel carbamates. Mol. Pharmacol. 2007, 71, 1610–1617. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Dvir, H.; Silman, I.; Sussman, J.L. Acetylcholinesterase—A multifaceted target for structure-based drug design of anticholinesterase agents for the treatment of Alzheimer’s dinase. J. Mol. Neurosci. 2003, 20, 369–383. [Google Scholar] [CrossRef]
- Soukup, J.E. Alzheimer’s Disease: A Guide to Diagnosis, Treatment, and Management; Greenwood Publishing Group: Westport, CT, USA, 1996. [Google Scholar]
- Lu, L.C.; Bludau, J. Alzheimer's Disease; Greenwood Publishing Group: Santa Barbara, CA, USA, 2011. [Google Scholar]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatr. 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Patel, N.B.; Rathod, R.D. Studies on synthesis and microbial activity of novel benzothiazoles containing 2-hydroxy benzoic acid. Int. J. Chem. Sci. 2006, 4, 569–575. [Google Scholar]
- Menges, M.; Hamprecht, G.; Menke, O.; Reinhard, R.; Schafer, P.; Zagar, C.; Westphalen, K.O.; Otten, M.; Walter, H.; Basf, A.G. Substituted 2-(benzoaryl)pyridines. WO/1999/006394 A1 (PCT/EP1998/003833), 11 February 1999. [Google Scholar]
- Reuveni, M. Activity of the new fungicide benthiavalicarb against Plasmopara viticola and its efficacy in controlling downy mildew in grapevines. Eur. J. Plant. Pathol. 2003, 109, 243–251. [Google Scholar] [CrossRef]
- Ishii, A.; Kotani, T.; Nagaki, Y.; Shibayama, Y.; Toyomaki, Y.; Okukado, N.; Ienaga, K.; Okamoto, K. Highly selective aldose reductase inhibitors. 1. 3-(Arylalkyl)-2,4,5-trioxoimidazolidine-1-acetic acids. J. Med. Chem. 1996, 39, 1924–1927. [Google Scholar] [CrossRef]
- Hijikata, C. (Ihara Chemical Industry Co., Ltd.). Process for producing benzothiazolylalkylamine derivatives. WO/2001/074794 A1 (PCT/JP2001/002848), 11 October 2001. [Google Scholar]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lenghts determined by X-ray and neutron-difraction. 1. Bond lenghts in organic-compounds. J. Chem. Soc. Perkin Trans. 2 1987, 2, 1–19. [Google Scholar]
- Yoshihara, R.; Hosomi, H.; Aoyama, H.; Ohba, S. N-Propylimidazolidinetrione and N-methyl-N'-phenylethylimidazolidinetrione. Acta Crystallogr. C 1999, 55, 594–596. [Google Scholar] [CrossRef]
- Rodriguez, M.A.; Andrews, N.L.; Boyle, T.J.; Frazer, C.S. N-Methylimidazolidinetrione. Acta Crystallogr. E 2005, 61, o2288–o2290. [Google Scholar]
- Davies, D.R. The crystal structure of parabanic acid. Acta Crystallogr. 1955, 8, 129–136. [Google Scholar] [CrossRef]
- Craven, B.M.; McMullan, R.K. Charge density in parabanic acid from X-ray and neutron diffraction. Acta Crystallogr. B 1979, 35, 934–945. [Google Scholar] [CrossRef]
- He, X.M.; Swaminathan, S.; Craven, B.M.; McMullan, R.K. Thermal vibrations and electrostatic properties of parabanic acid at 123 and 298 K. Acta Crystallogr. B 1988, 44, 271–281. [Google Scholar] [CrossRef]
- Blackman, A.G.; Buckingham, D.A.; Simpson, J. Reactions of coordinated imidazole. Oxidation products and ring cleavage in the reactions of RImH3+ (R = pentaamminecobalt) with acetyl hypobromite and hypobromous acid. Inorg. Chem. 1991, 30, 1635–1642. [Google Scholar] [CrossRef]
- Weber, H.P.; Craven, B.M. Structure and charge density of the 1:1 complex of thiourea with parabanic acid at 298 K. Acta Crystallogr. B. 1987, 43, 202–209. [Google Scholar] [CrossRef]
- Colman, P.M.; Medlin, E.H. The crystal structure of thiourea parabanic acid. Acta Crystallogr. B 1970, 26, 1553–1559. [Google Scholar] [CrossRef]
- Weber, H.P.; Ruble, J.R.; Craven, B.M.; McMullan, R.K. The neutron structure at 116 K of the 1:1 complex of perdeuterated parabanic acid and urea. Acta Crystallogr. B 1980, 36, 1121–1126. [Google Scholar] [CrossRef]
- Colman, P.M.; Medlin, E.H. The crystal structure of urea parabanic acid. Acta Crystallogr. B 1970, 26, 1547–1553. [Google Scholar] [CrossRef]
- Sarker, S.R.; Stone, D.M.; Evain, E.J.; Cooley, J.H.; Willett, R.D. Reaction of oxalyl and malonyl chloride with 1,1-dimethyl-2-substituted hydrazides. J. Heterocycl. Chem. 1994, 31, 1535–1539. [Google Scholar] [CrossRef]
- Volkova, Y.A.; Averina, E.B.; Rybakov, V.B.; Kuznetsova, T.S. Private Communication. Available online: http://www.ccdc.cam.ac.uk/products/csd/deposit/communications.php/ (accessed on 20 August 2011).
- Zarzyka-Niemiec, I.; Lubczak, J.; Ciunik, Z.; Wolowiec, S.; Ruman, T. Bis(hydroxyalkylated) derivates of parabanic acid. Heterocycl. Commun. 2002, 8, 559–564. [Google Scholar]
- Forrester, A.R.; Howie, R.A.; Stephen, K. Structure of N,N'-diacetylparabanic acid. Acta Crystallogr. C 1988, 860–862. [Google Scholar]
- Kerns, E.H.; Li, D. Drug-like Properties: Concept, Structure Design and Methods; Elsevier: San Diego, CA, USA, 2008. [Google Scholar]
- Darvesh, S.; McDonald, R.S.; Darvesh, K.V.; Mataija, D.; Conrad, S.; Gomez, G.; Walsh, R.; Martin, E. Selective reversible inhibition of human butyrylcholinesterase by aryl amide derivatives of phenotiazine. Bioorg. Med. Chem. 2007, 15, 6367–6378. [Google Scholar] [CrossRef]
- Norrington, F.E.; Hyde, R.M.; Williams, S.G.; Wotton, R. Physicochemical-activity relations in practice. 1. Rational and self-consistent data bank. J. Med. Chem. 1975, 18, 604–607. [Google Scholar] [CrossRef]
- Chiou, S.Y.; Huang, C.F.; Hwang, M.T.; Lin, G. Comparison of active sites of butyrylcholinesterase and acetylcholinesterase based on inhibition by geometric isomers of benzene-di-N-substituted carbamates. J. Biochem. Mol. Tox. 2009, 5, 303–308. [Google Scholar]
- Berger, S.; Braun, S.; Kalinowski, H.O. NMR Spectroscopy of the Non-Metallic Elements; John Wiley: Chichester, UK, 1997. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzym. 1997, 276, 307–326. [Google Scholar]
- Ahmed, F.R.; Hall, S.R.; Huber, C.P. Crystallographic Computing; Munksgaard: Copenhagen, Denmark, 1970. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR92. J. Appl. Crystallogr. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- OECD guideline for the testing of chemicals 107—Partition coefficient (n-octanol/water): Shake Flask Method. Available online: http://www.oecd.org/ (accessed on 17 August 2011).
- Kwok, S.O.; Wang, K.C.; Kwok, H.B. An improved method to determine SH and –S–S– group content in soymilk protein. Food Chem. 2004, 88, 317–320. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Naturforsch. 2004, 59, 293–296. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pejchal, V.; Stepankova, S.; Padelkova, Z.; Imramovsky, A.; Jampilek, J. 1,3-Substituted Imidazolidine-2,4,5-triones: Synthesis and Inhibition of Cholinergic Enzymes. Molecules 2011, 16, 7565-7582. https://doi.org/10.3390/molecules16097565
Pejchal V, Stepankova S, Padelkova Z, Imramovsky A, Jampilek J. 1,3-Substituted Imidazolidine-2,4,5-triones: Synthesis and Inhibition of Cholinergic Enzymes. Molecules. 2011; 16(9):7565-7582. https://doi.org/10.3390/molecules16097565
Chicago/Turabian StylePejchal, Vladimir, Sarka Stepankova, Zdenka Padelkova, Ales Imramovsky, and Josef Jampilek. 2011. "1,3-Substituted Imidazolidine-2,4,5-triones: Synthesis and Inhibition of Cholinergic Enzymes" Molecules 16, no. 9: 7565-7582. https://doi.org/10.3390/molecules16097565