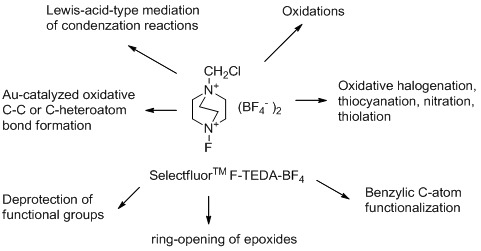
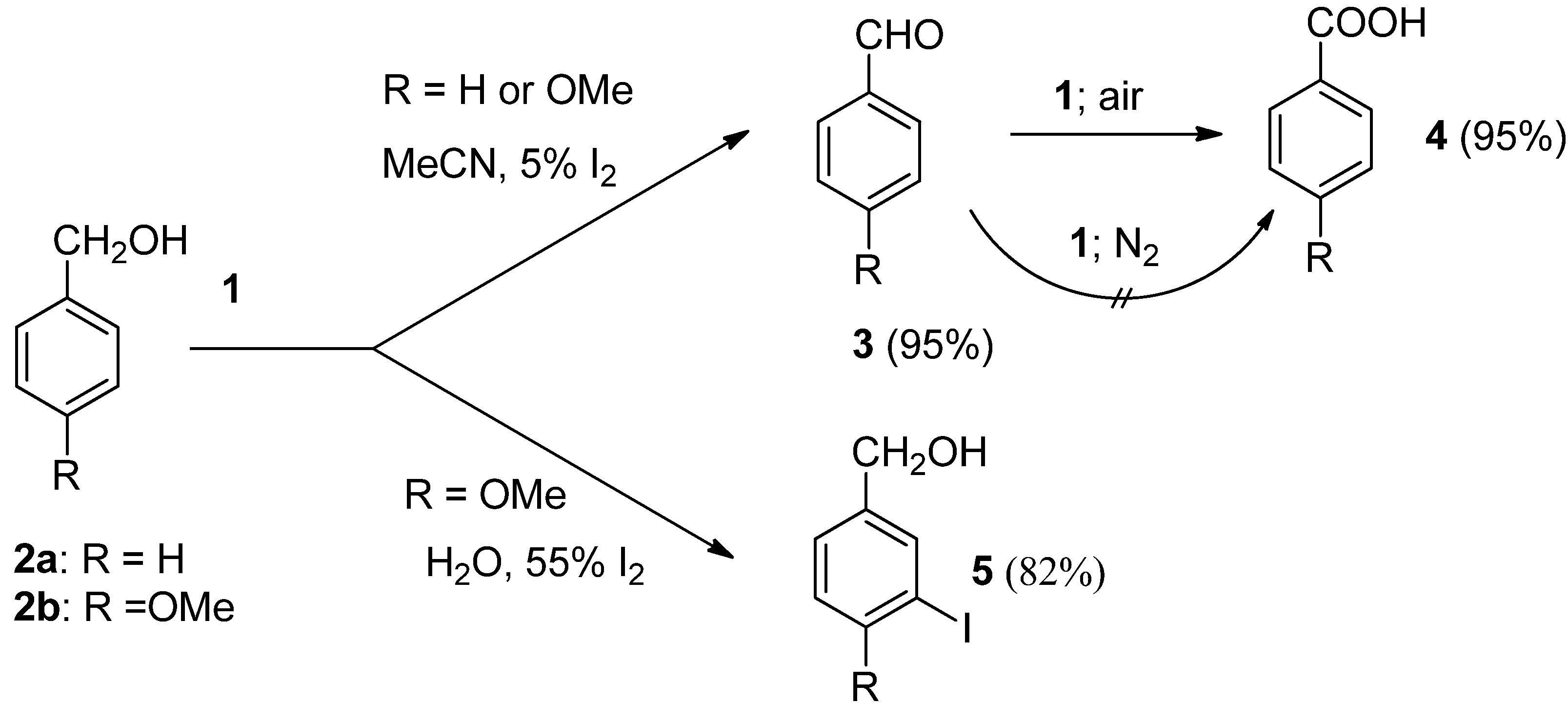
2.1. Transformations of Oxidizable Functional Groups
In the presence of chemicals having oxidizing power the hydroxyl functional group could often be transformed to various kinds of carbonyl functionalitiy. Primary benzylic alcohols were found to be relatively stable towards
1 since their transformations with
1 in acetonitrile media to moderate amounts of corresponding aldehydes, and further to benzoic acid derivatives, needs long reaction times (15-435 hours) and reflux temperature. It was also established that aromatic aldehydes could be transformed with
1 to benzamides or benzoates after reaction in the presence of amines or alcohols, but again the long reaction time (40-70 hours) required for these functionalisations makes them less attractive [
20]. On the other hand, catalytic amounts of molecular iodine enhance the reaction and its efficiency considerably. Benzyl alcohol (
2a) and 4-methoxybenzyl alcohol (
2b) were thus readily transformed to their aldehydes and further to benzoic acid derivatives (
3 and
4) after 2 hours treatment with
1 in MeCN solution in the presence of 5 mol% of I
2 under an air atmosphere (
Scheme 1), while in the case of the treatment of
2b in aqueous media and in the presence of 55 mol% of iodine, the benzylic hydroxyl group remained unattached and iodo-functionalization of the aromatic ring to
5 took place [
21]. Alkyl alcohols could also be readily transformed by
1 to their carbonyl derivatives [
22].
Scheme 1.
Reactions of benzylic alcohols with Selectfluor F-TEDA-BF2 1.
Scheme 1.
Reactions of benzylic alcohols with Selectfluor F-TEDA-BF2 1.
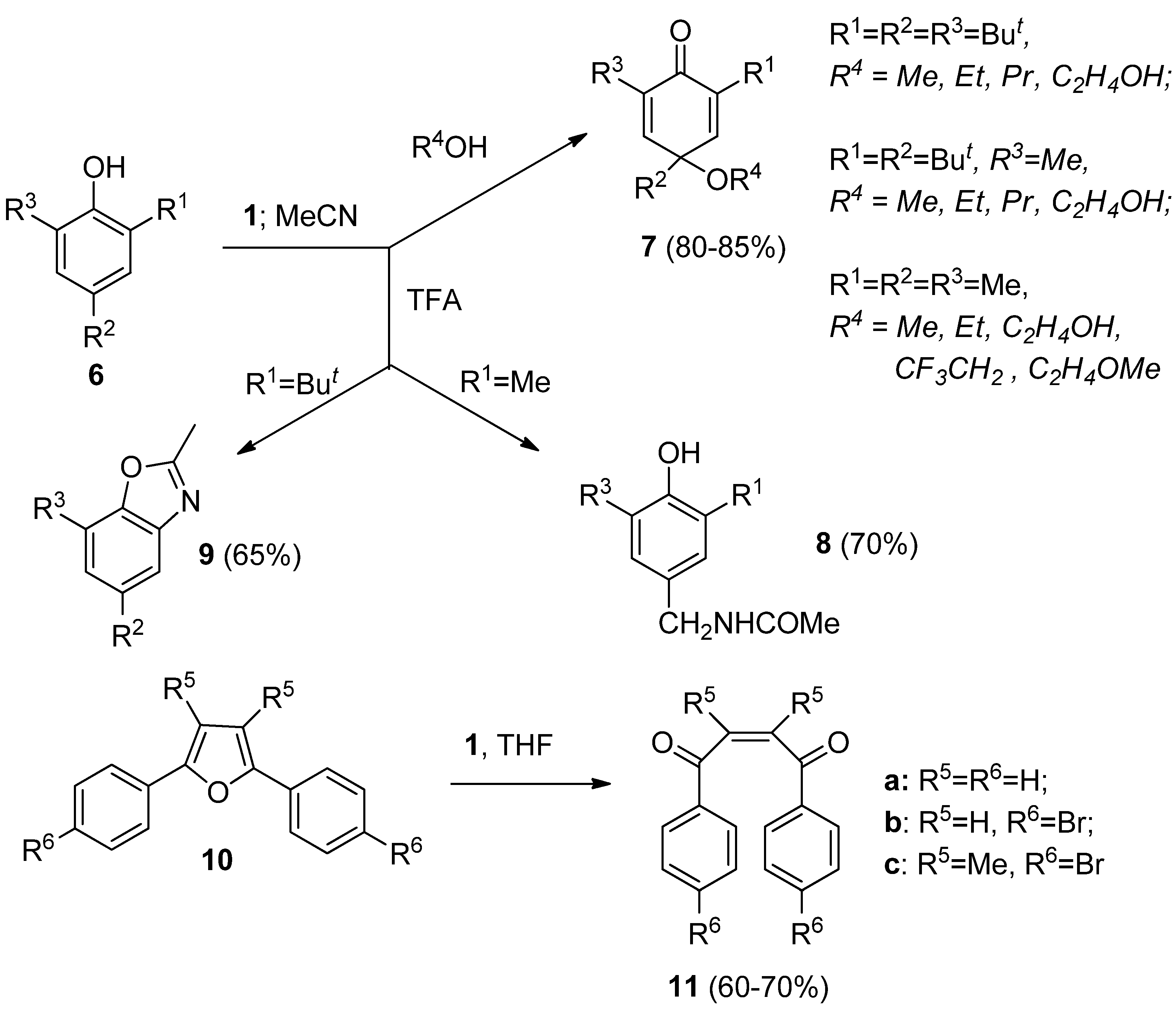
Reactions of phenols with
1 were intensively studied. Phenols substituted by an additional hydroxy substituent at the
ortho or
para position were readily oxidized to the corresponding quinones when treated by
1 in MeCN [
22], while the course of reaction of 2,4,6-trialkyl substituted phenols with
1 was found to be strongly dependent on the structure of the target compounds
6 and the reaction media used (
Scheme 2). Reactions in pure MeCN gave fluorinated products, while in the presence of alcohols or water
para-quinols or
para-quinol ethers
7 were formed in moderate to high yield. The presence of a more acidic nucleophile, such as trifluoroacetic acid (TFA), caused quite different transformations and Ritter-type functionalisation at the 4-benzylic position resulted in the formation of 4-methylacetamido-2,6-dialky substituted phenol derivatives
8, while after
ipso attack at position 2, followed by dealkylation and internal cyclisation, alkyl substituted benzoxazole derivatives
9 were formed [
23,
24]. Another oxidative transformation of oxygen containing functional moieties with
1 was found to be the ring opening of 2,5-diaryl substituted furans
10, resulting in the stereoselective formation of
cis-1,2-dibenzoyldione derivatives
11 [
25].
Scheme 2.
Transformations of 2,4,6-trialkyl substituted phenols and 2,5-diarylfurans with Selectfluor F-TEDA-BF2 1.
Scheme 2.
Transformations of 2,4,6-trialkyl substituted phenols and 2,5-diarylfurans with Selectfluor F-TEDA-BF2 1.
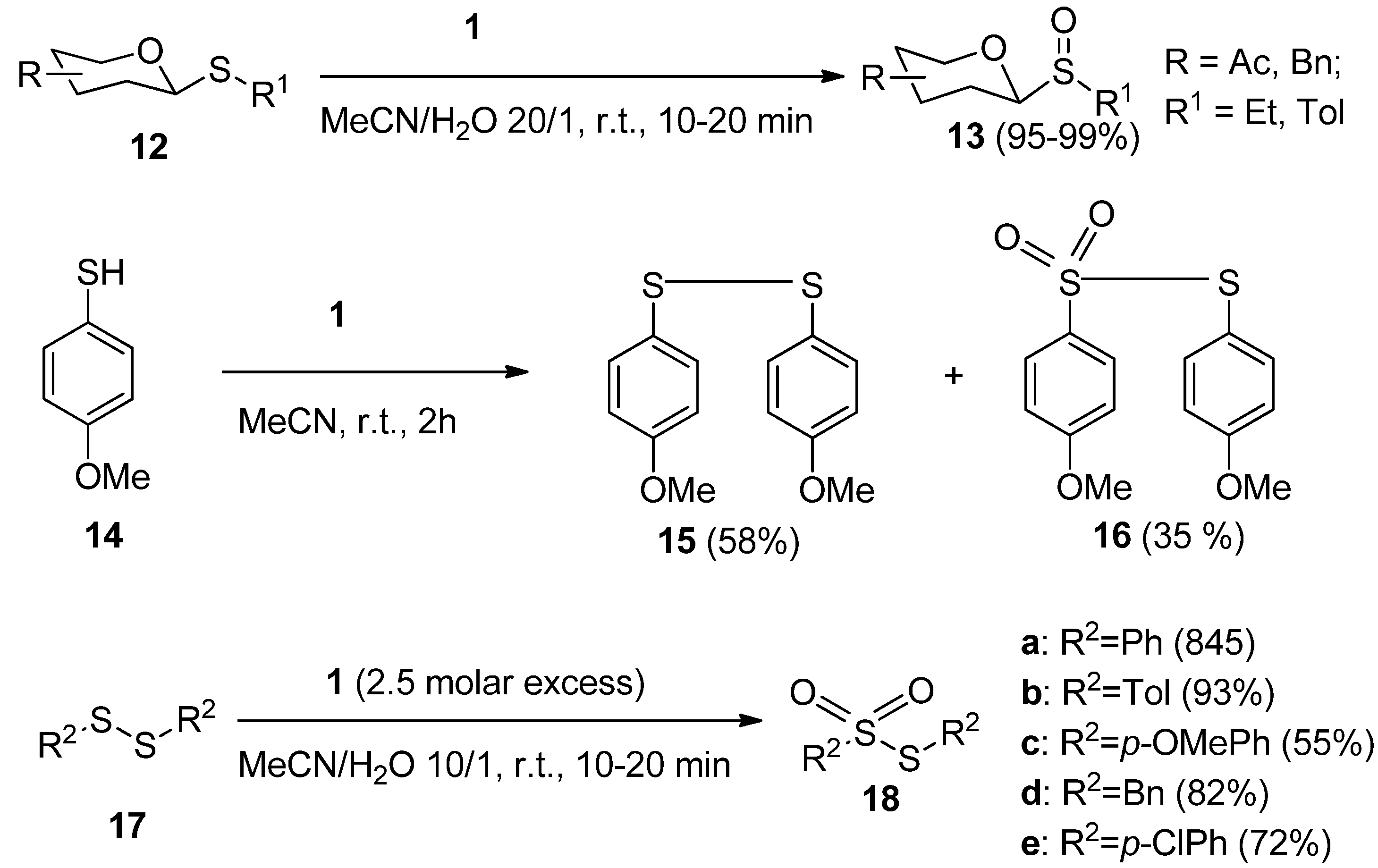
Sulfur-containing functional groups are usually very sensitive to oxidation. The mild oxidative nature of
1 was efficiently used advantageously in glycoside chemistry in the case of the development of a selective and efficient method for the oxidation of thioglycosides to their corresponding sulfoxide derivatives. A variety of thioglycosides (
12,
Scheme 3) were thus readily transformed to their sulfinyl derivatives
13 by treatment with a moderate molar excess of
1 in aqueous MeCN (MeCN/H
20 = 20/1) at room temperature for a few minutes [
26]. The thiophenolic functionality was found to be more unstable towards
1 than its phenolic analogues and could be readily transformed to disulfides and further to sulfonates [
22], and this path was accepted as a methodology for concise synthesis of thiosulfonates. Symmetric aromatic or benzylic disulfides
17 were thus efficiently transformed to thiosulfonates
18 with a 2.5 fold molar excess of
1 in aqueous MeCN [
27], while alkyl phenyl sulfides under these reaction conditions with an equimolar amounts of
1 gave selectively the sulfoxide functionality [
28].
Scheme 3.
Transformations of sulfur-containing functional groups with Selectfluor F-TEDA-BF2 1.
Scheme 3.
Transformations of sulfur-containing functional groups with Selectfluor F-TEDA-BF2 1.
An amino functional group bonded to an aromatic ring usually cannot survive the presence of
1 and demands protection by acetylation, while primary, secondary or tertiary aliphatic amines can be transformed by
1 to N-fluoro-substituted derivatives, often selectively and in moderate to good yield [
14]. On the other hand, amides are relatively stable towards oxidation to imides, and up to now only a few efficient methods for direct preparation of these valuable chemicals are known, but recently the combination of the copper(I) moiety and Selecfluor F-TEDA-BF
4 was introduced as an efficient and selective reagent system for the oxidation of amides to imides [
29]. A variety of amides
19 were thus efficiently transformed to their imide derivatives
20 using the combination of
1 (2.5 equiv)/CuBr (1.2 equiv) in MeCN at room temperature (
Table 1).
Table 1.
Oxidation of amides
19 to imides
20 using
Selectfluor F-TEDA-BF4/CuBr tandem.
a ![Molecules 16 06432 i001]()
Table 1.
Oxidation of amides 19 to imides 20 using Selectfluor F-TEDA-BF4/CuBr tandem. a ![Molecules 16 06432 i001]()
| Entry | R | R 1 | Yield (%) |
|---|
| 1 | Ph | CH2CH(Me)2 | 88 |
| 2 | Ph | Et | 77 |
| 3 | Ph | C2H4COOMe | 82 |
| 4 | Ph | Ph | 84 |
| 5 | Ph | (CH2)5OCOPh | 84 |
| 6 | 4-F-Ph | Et | 80 |
| 7 | 4-F-Ph | c-C6H11 | 50 |
| 8 | Me | Ph | 83 |
| 9 | n-C6H13 | CH2CH(Me)2 | 79 |
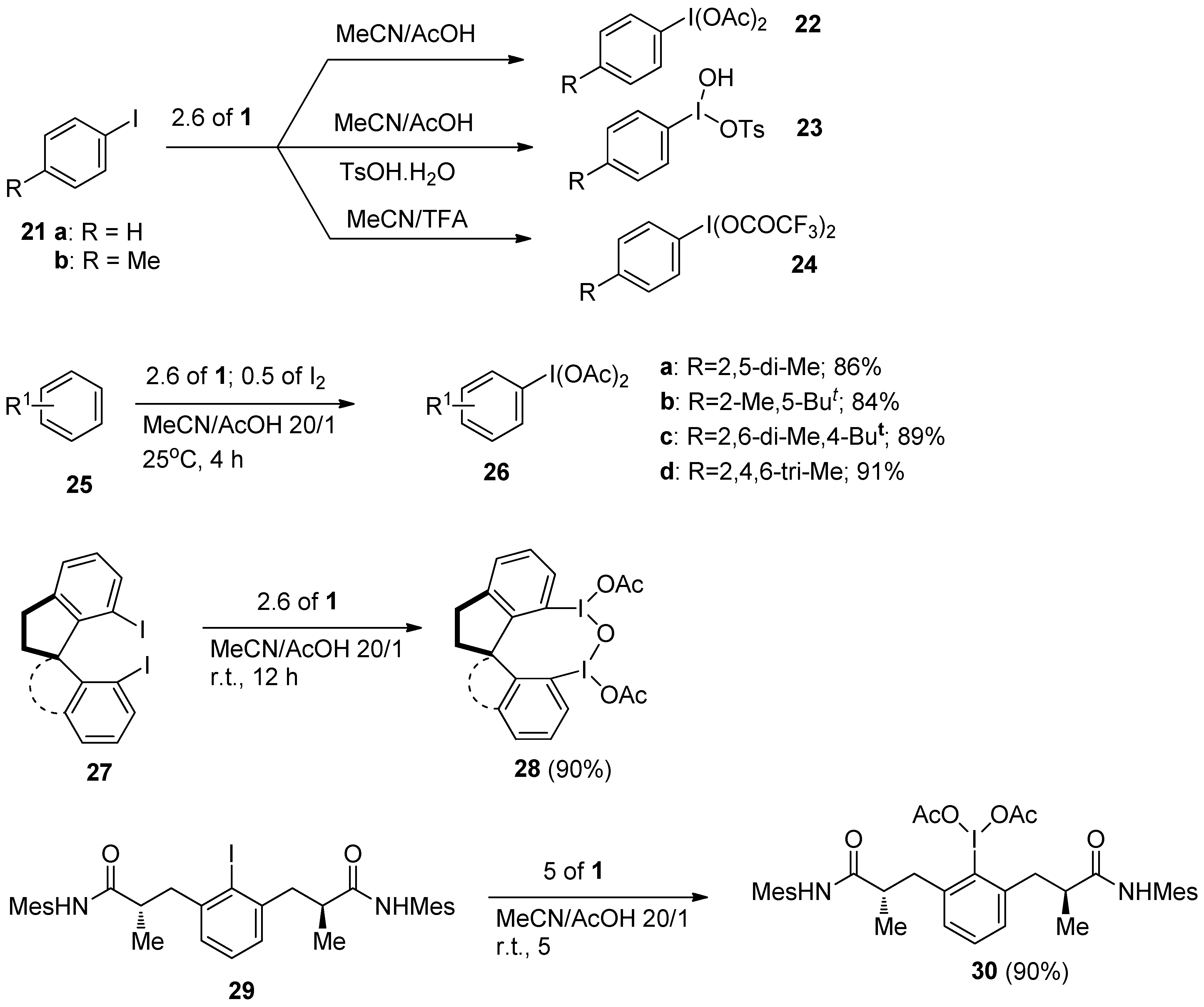
Hypervalent iodine(III) compounds are valuable and versatile reagents in organic synthesis. It has been demonstrated that various types of aryl hypervalent iodine(III) compounds could be efficiently prepared using
Selectfluor F-TED-BF4 starting from the corresponding aryl iodides (
21,
Scheme 4), or even straightforwardly from arenes
25 following
1 mediated oxidative iodination and further in situ functionalization of aryl iodides. Using one or other approach, a variety of phenyliodine(III)diacetates
22,
26 or phenyliodine(III)ditrifluoromethylacetates
24 were prepared with a 2.6 fold molar excess of
1 in MeCN solution in the presence of acetic or trifluoromethyl acetic acid, while in the presence of TsOH.H
2O, Koser`s reagents
23 were synthesized [
30]. The same methodology was applied for the synthesis of chiral hypervalent iodine(III) reagents
28 [
31] and
30 [
32], and further used for various enantioslective transformations.
Scheme 4.
Synthesis of hypervalent iodine(III) compounds using Selectfluor F-TEDA-BF4.
Scheme 4.
Synthesis of hypervalent iodine(III) compounds using Selectfluor F-TEDA-BF4.
2.2. Oxidative Halogenation
Halogenation of organic compounds using the oxidative approach mediated by Selectfluor F-TEDA-BF
4 has been introduced in our laboratory [
33] and the methodology originally applied for the regioselective iodination of aromatic ethers using molecular iodine. Regioselective iodination at the
para position took place, while when this position was occupied, regioselective
ortho iodofunctionalization took place. Acetonitrile was found to be the best medium for these transformations and 50 mol% of molecular iodine was found to be enough for complete transformation of starting the material. This methodology has been intensively used for efficient and selective iodination of alkyl-substituted benzene derivatives [
34], also those sterically hindered [
35], as well for iodofunctinalization of arenes in ionic liquids as the reaction media [
36].
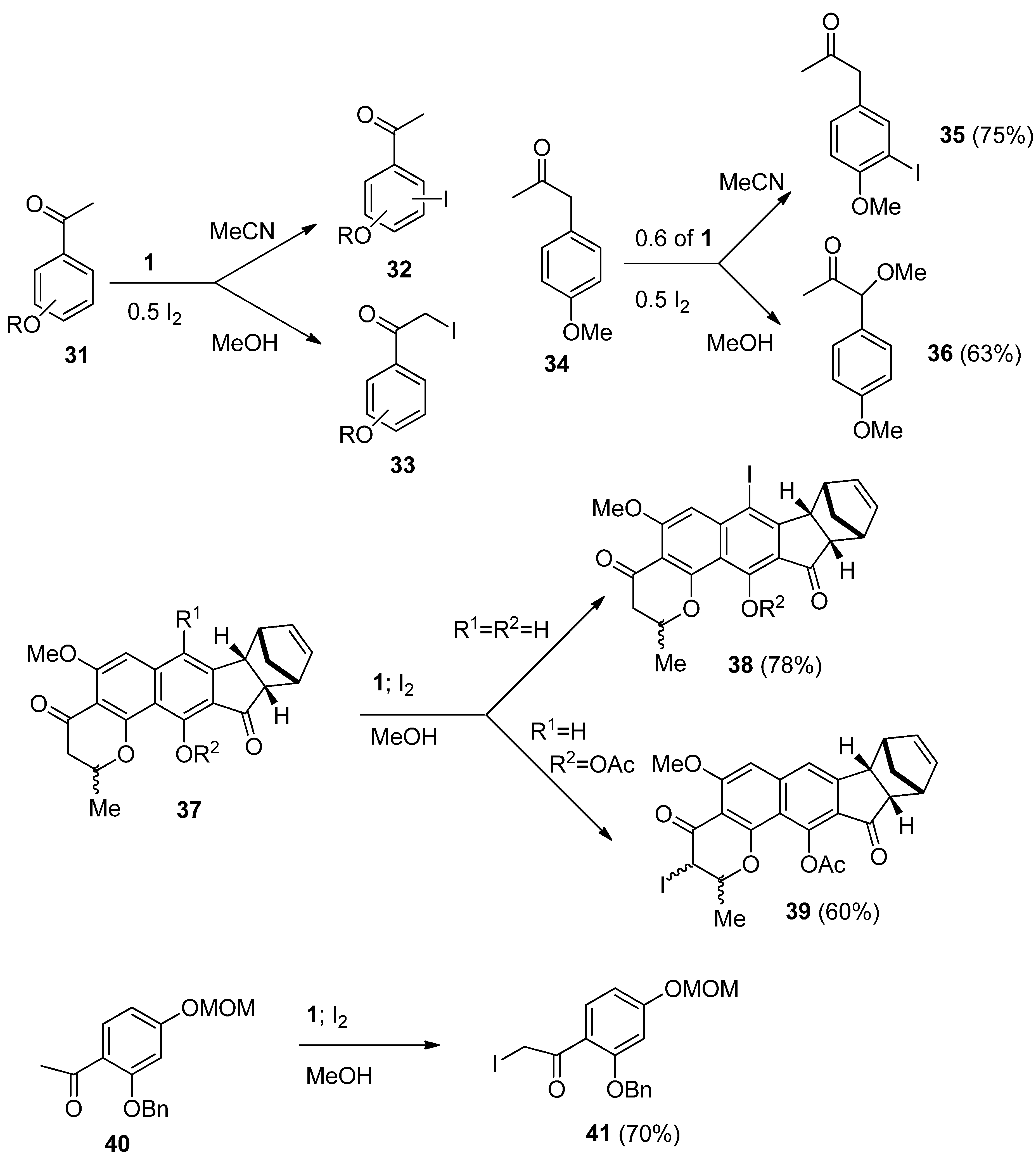
We have also demonstrated that the regioselectivity of iodination could be regulated by the solvent used. In the case of iodination of substituted aryl-alkyl ketones regioselective functionalization of the aromatic ring took place (
32,
Scheme 5) when the reactions were performed in MeCN, while regioselective iodination of the side chain (
eg.
33) has been found in reactions performed in MeOH [
37]. It has been established that the stoichiometry of the process for substrate/I
2/F-TEDA-BF
4 is 1/0.5/0.6. This MeOH directed and F-TEDA-BF
4 mediated iodination methodology was applied for side chain iodination of a variety of acetyl substituted aromatic compounds [
38], and indanone and tetralone derivatives [
39] bearing a strongly activated aromatic ring; these achievements have been reviewed in our previous account [
19]. 1-(4-Methoxyphenyl)propan-2-one (
34) was further chosen as a model substrate; in MeCN ring iodination forming
35 was established, in MeOH exclusive side-chain methoxy functionalization at the benzylic position took place (
36), while in water regioselectivity was lost and a mixture of ring and side-chain functionalized products were observed in the crude reaction mixture [
21]. Recently application of the method was successfully demonstrated for the synthesis of euplectin, where by varying the substituents on the euplectin precursor
37, the regioselectivity of the F-TEDA-BF
4 mediated iodination could be directed towards aryl ring iodofuctionalization resulting in
38, or to the α-to carbonyl position resulting in
39 [
40], and for side chain iodination of the protected 2,4-dihydroxy acetophenone derivative
40 to
41, one of precursors in total synthesis of glyceollin I [
41].
Selectfluor F-TEDA-BF4 mediated iodination of dimethoxybenzenes (
42,
Table 2) was studied and the role of reaction media and the relative ratio of reactants on the course of the transformation evaluated. In the case of 1,2- (
42a) and 1,4-dimethoxybenzene (
42c) equimolar amounts of all three reactants (B) were found to be necessary for high conversion of starting material (entries 1-3 and 8,9 in
Table 2), while for the iodofunctionalization of 1,3-dimethoxybenzene
42b to
43b a 0.5 molar amount of iodine and 0.6 molar amount of F-TEDA-BF
4 (A) was enough for high yield iodination in all three solvents (entries 4-6). This result was explained by the different nature of the reaction path and a predominantly ionic process was proposed for case A, where iodine has the role of activator of the system and F-TEDA-BF
4 the role of activator and regenerator of iodide liberated during the iodination process, while in the case of B, a reaction course through single electron transfer was proposed [
21].
Bromination and chlorination of various unsaturated organic compounds mediated by F-TEDA-BF
4 have also been demonstrated. Electrophilic bromination or chlorination of benzene derivatives was reported at room temperature using the anionic precursors of bromide or chloride transformed
in situ into their electrophilic species by
1 [
42]. Acetonitrile was found to be the best choice for the reaction medium, while reactions did not proceed in MeOH. A number of olefins were oxidative brominated using the F-TEDA-BF
4/KBr tandem and for different types of substrates, addition, monobromine-substituted, or Hunsdiecker-Borodin reaction products were readily obtained [
43].
Scheme 5.
Oxidative iodination of organic compounds mediated by Selectfluor F-TEDA-BF4. The original idea and recent applications.
Scheme 5.
Oxidative iodination of organic compounds mediated by Selectfluor F-TEDA-BF4. The original idea and recent applications.
Table 2.
Iodination of dimethoxy benzenes with elemental iodine mediated by F-TEDA-BF
4.
![Molecules 16 06432 i002]()
Table 2.
Iodination of dimethoxy benzenes with elemental iodine mediated by F-TEDA-BF4. ![Molecules 16 06432 i002]()
| Entry | Substrate | Solvent | T/t (°C/h) | Reactants ratio a | Product | Yield (%) b |
|---|
| 1 | | MeCN | 20/4 | B | | 100(46) |
| 2 | 42a | MeOH | 20/18 | B | 43a | 100(96) |
| 3 | | H2O | 20/22 | B | | 32(5) |
| 4 | | MeCN | 20/2 | A | | 100(89) |
| 5 | 42b | MeOH | 20/3 | A | 43b | 100(71) |
| 6 | | H2O | 20/3 | A | | 88(68) |
| 7 | | MeCN | | B | | 0 |
| 8 | 42c | MeOH | | B | 43c | 60(38) |
| 9 | | H2O | | B | | 93(17) |
2.4. Functionalisation at a Benzylic Carbon Atom
In the transformations described in sections 2.2 and 2.3 F-TEDA-BF4 acts as an oxidant forming electrophilic species from various unreactive sources which afterwards collapse with the electron-rich part of the organic substrates. In this section the opposite situation is described and a variety of examples reviewed where 1 acted as oxidant for the chosen substrates, thus forming an electron deficient reactive intermediate which reacted with an external nucleophile.
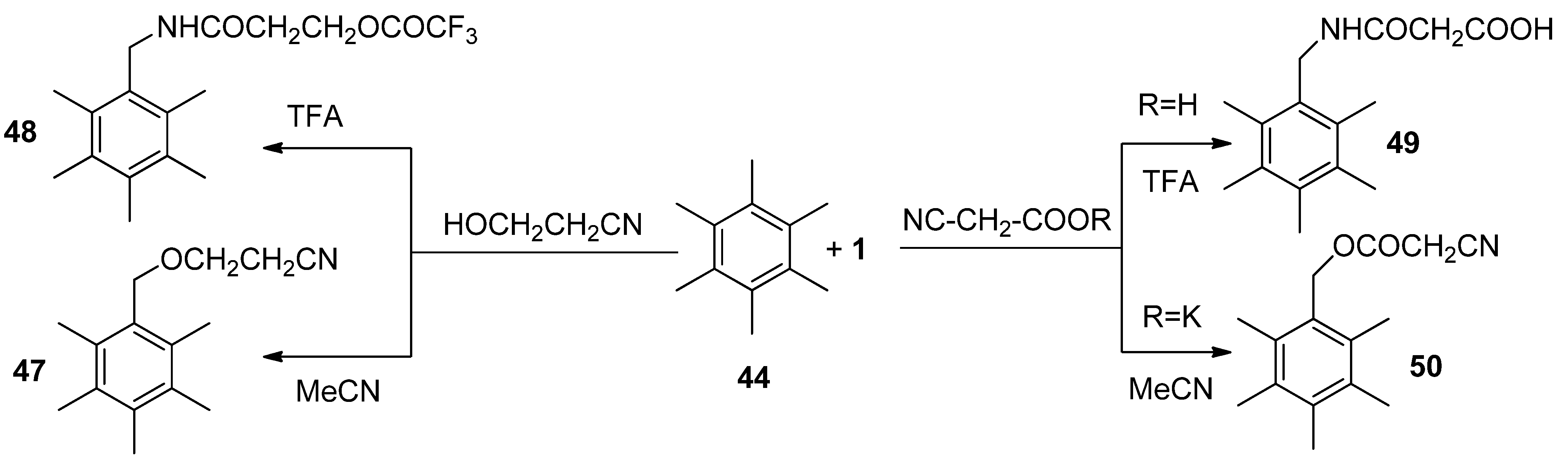
An example of this kind is the versatile derivatisation of a benzylic carbon atom in hexamethylbenzene (HMB,
44).
Table 3 summarizes reactions of HMB with F-TEDA-BF
4 in the presence of alcohols or potassium salts of perfluoroalkanoic acids in MeCN media. Pentamethyl-benzylalkyl ethers (entries 1-9) or esters (entries 10-15) were readily obtained in high to excellent yields. When this reaction was performed in TFA in the presence of various nitriles, Ritter-type benzylic amidation took place and the corresponding pentamethylbenzyl amides (
46,
Table 4) were formed in high yield [
44].
Using appropriate reaction conditions, selective functionalisation of HMB can be obtained in the presence of compounds bearing two different nucleophilic active sites. Reaction in MeCN in the presence of 2-cyanoethanol gave the benzylic ether derivative (
47,
Scheme 6), while in TFA Ritter transformation took place and benzyl amide derivative
48 was formed.
Table 3.
Reactions of hexamethyl benzene
44 with F-TEDA-BF
41 in the presence of alcohols or potassium salts of carboxylic acids.
a ![Molecules 16 06432 i003]()
Table 3.
Reactions of hexamethyl benzene 44 with F-TEDA-BF41 in the presence of alcohols or potassium salts of carboxylic acids.a ![Molecules 16 06432 i003]()
| Entry | R | Y | R 1 | Yield (%) | Reference |
|---|
| 1 | H | O | i-Pr | 88 | [44] |
| 2 | H | O | n-hexyl | 90 | [44] |
| 3 | H | O | c-pentyl | 98 | [44] |
| 4 | H | O | Bn | 75 | [44] |
| 5 | H | O | MeOCH2CH2 | 93 | [44] |
| 6 | H | O | CF3CH2 | 75 | [44] |
| 7 | H | O | CF3CF2CH2 | 70 | [45] |
| 8 | H | O | CF3(CF2)2CH2 | 70 | [45] |
| 9 | H | O | (CF3)2CH | 71 | [45] |
| 10 | H | OCO | Me b | 97 | [44] |
| 11 | H | OCO | CF3 b | 97 | [45] |
| 12 | K | OCO | CF3CF2 | 97 | [45] |
| 13 | K | OCO | CF3CF2CF2 | 72 | [45] |
| 14 | K | OCO | CF2(CF2)3CF2 | 96 | [45] |
| 15 | K | OCO | CF3(CF2)5CF2 | 90 | [45] |
Table 4.
Ritter-type functionalization of the benzylic position in hexamethylbenzene mediated by F-TEDA-BF
4 [
44].
a ![Molecules 16 06432 i004]()
Table 4.
Ritter-type functionalization of the benzylic position in hexamethylbenzene mediated by F-TEDA-BF4 [44].a ![Molecules 16 06432 i004]()
| Entry | R | Time (h) | Yield |
|---|
| 1 | Et | 2 | 82 |
| 2 | n-pentyl | 3 | 65 |
| 3 | i-Pr | 2 | 75 |
| 4 | c-Pr | 1 | 86 |
| 5 | MeOCH2 | 1 | 95 |
| 6 | MeOCOCH2 | 1 | 98 |
| 7 | EtOCOCH2 | 1 | 84 |
| 8 | Ph | 1 | 75 |
| 9 | p-COOMe-Ph | 1 | 71 |
| 10 | Bn | 1 | 90 |
| 11 | C6F5 | 1 | 81 |
Similarly, cyanoacetic acid as a source of an external nucleophile was activated at its cyanide moiety if TFA was used as solvent and the corresponding benzyl amide
49 was formed, while in MeCN, potassium cyanoacetate acted as a carboxy nucleophile and pentamethylbenzyl cyanoacetate
50 was formed [
44].
Scheme 6.
F-TEDA-BF4 mediated benzylic functionalisation of hexamethyl benzene in the presence of compounds bearing two different nucleophilic centres.
Scheme 6.
F-TEDA-BF4 mediated benzylic functionalisation of hexamethyl benzene in the presence of compounds bearing two different nucleophilic centres.
A quite different course of reaction of HMB with
1 was established in the case when water was used as the external nucleophile. In aqueous MeCN phenyl ring transformation took place, starting with
ipso attack of water and further rearrangement of the methyl group as the main process. Primarily formed rearranged 2,3,4,5,6,6-hexamethylcyclohexa-2,4-dienone (
52,
Scheme 7) was further transformed to 5-hydroxy-2,3,5,6,6-pentamethyl-4-methylenecyclohex-2-en-1-one
53 or 5-fluoro-2,3,5,6,6-pentamethyl-4-methylenecyclohex-2-en-1-one
54; the relative yield of these final products was found to be dependent on the concentration of water in the reaction mixture (
Scheme 7). Product
52 was independently obtained in excellent yield by treating hexamethyl Dewar benzene
51 with an aqueous MeCN solution of
1. In the presence of water and alcohol as the second external nucleophile, competition between ring and benzylic functionalisation was observed. In the case of MeOH or EtOH up to 40% of benzylic functionalisation took place thus forming benzyl alkyl ethers, while in the presence of trifluoroethanole or hexafluoro i-propanole product
54 was selectively formed in excellent yield [
46].
The reaction of 1,2,4,5-tetramethyl benzene (
55,
Table 5) with
1 was also studied and the role of solvent and external nucleophile on the course of the transformation established. In MeOH benzylic functionalisation forming benzyl methyl ether derivative
57a (entry 1,
Table 5) was the exclusive process, in acetic acid ring attack of the nucleophile forming 2,3,5,6-tetramethylphenyl acetate (
58a, entry 2) was found to be predominant process, while in TFA exclusive ring esterification thus forming 2,3,5,6-tetramethylphenyl trifluoroacetate
58b (entry 3) was observed. In reactions performed in MeCN, the nature of the external nucleophile regulated the course of reaction. In the presence of TFA (entry 4) Ritter-type benzylic functionalization to
N-(2,4,5-trimethylbenzyl)acetamide
56 took place exclusively, in the presence of acetic acid benzylic amidation, benzylic and ring acetoxylation competed, while in the presence of water (entry 6)
ipso attack of water followed by methyl group rearrangement and further fluorination or fluoro amidation forming equal amounts of products
59 and
60 was observed [
46]. Other isomeric tetra- and trimethyl benzene derivatives were also tested in the presence of
1 and an external nucleophile; the kinetics of the reactions of polymethyl-substituted benzene derivatives with
1 studied and the results obtained supported the assumption that single electron transfer (SET) is the dominant process in these transformations [
46].
Scheme 7.
Transformation of hexamethyl benzene with F-TEDA-BF4 in the presence of water.
Scheme 7.
Transformation of hexamethyl benzene with F-TEDA-BF4 in the presence of water.
Table 5.
Effect of solvent and external nucleophile on the transformation of 1,2,4,5-tetramethyl benzene with F-TEDA-BF
4.
a ![Molecules 16 06432 i005]()
Table 5.
Effect of solvent and external nucleophile on the transformation of 1,2,4,5-tetramethyl benzene with F-TEDA-BF4. a ![Molecules 16 06432 i005]()
| Relative ratio of products (%) | |
|---|
| Entry | Solvent/nucleophile | 56 | 57 | 58 | 59 | 60 | Yield (%) b |
|---|
| 1 | MeOH / - | - | 100 | - | - | - | 93 |
| 2 | AcOH / - | - | 29 | 71 | - | - | 85 |
| 3 | TFA / - | - | - | 100 | - | - | 95 |
| 4 | MeCN / TFA c | 100 | - | - | - | - | 82 |
| 5 | MeCN / AcOH | 27 | 21 | 52 | - | - | 80 |
| 6 | MeCN / H2O d | - | - | - | 50 | 50 | 95 |
2.5. Lewis Acid-Type Mediation of Condensation Reactions and Ring Opening of Epoxides
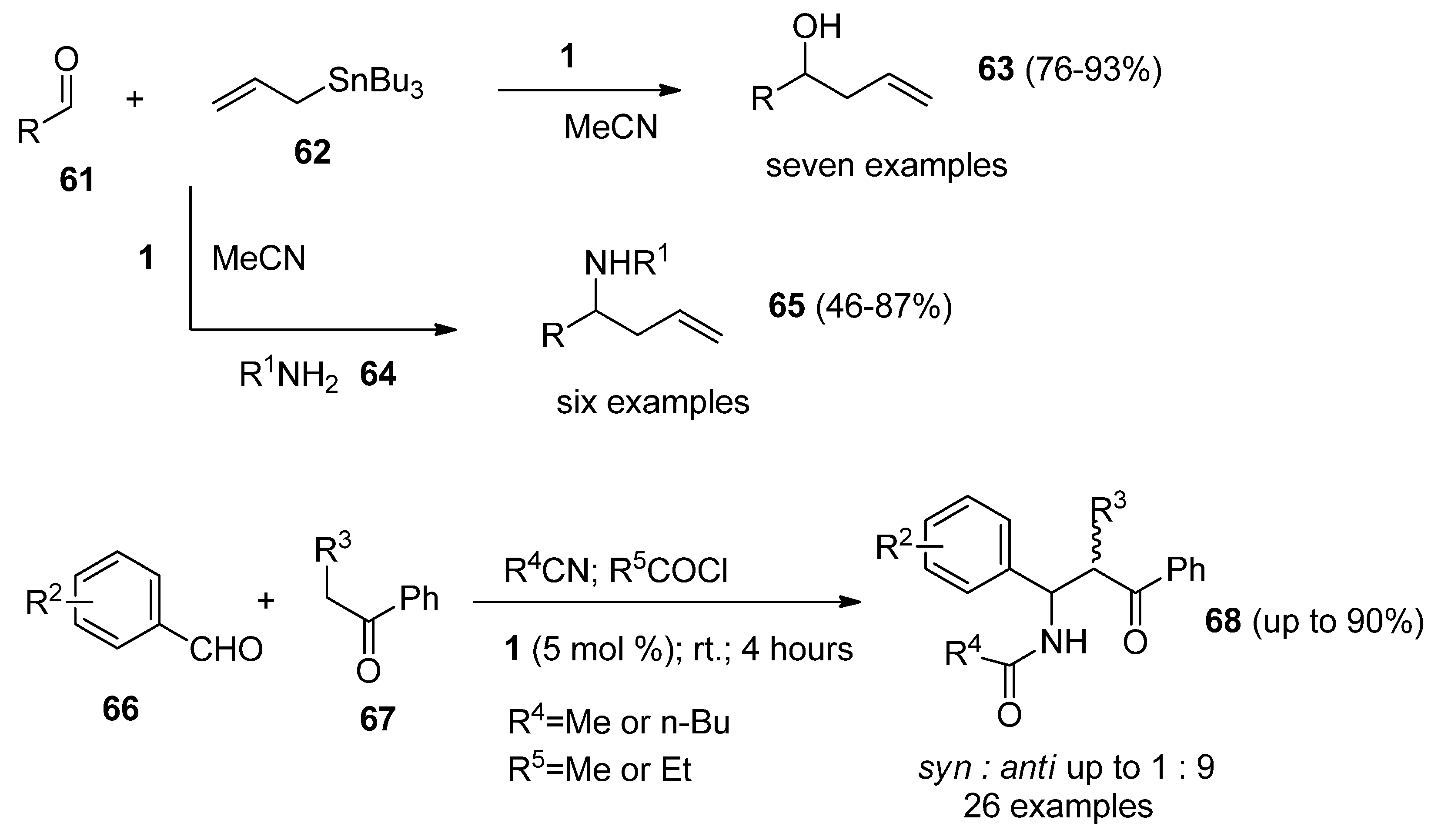
Selectfluor F-TEDA-BF4 can act as a Lewis acid and this fact was used to advantage in a variety of condensation reactions. Reactions of aryl or alkyl adehydes (
61,
Scheme 8) with allylbutyltin mediated
62 by
1 in MeCN resulted in the formation of homoallylic alcohols
63, and the analogous reactions in the presence of amines
64 lead to homoallylic amines
65 in good yields with excellent moisture and air tolerance [
47].
Scheme 8.
Synthesis of homoallylic alcohols or amines and β-acetamido ketones mediated by F-TEDA-BF4.
Scheme 8.
Synthesis of homoallylic alcohols or amines and β-acetamido ketones mediated by F-TEDA-BF4.
An efficient, room temperature process for the stereoselective synthesis of β-amido ketones (
68,
Scheme 8) employing a one-pot multi-component reaction of benzaldehyde derivatives
66, alkyl phenyl ketone
67, an acid chloride, and a nitrile in the presence of catalytic amounts of F-TEDA-BF
4 was reported [
48]. The method offers advantages such as high yield, short reaction time and energy efficiency, high
anti-stereoselectivity and a simple work-up protocol.
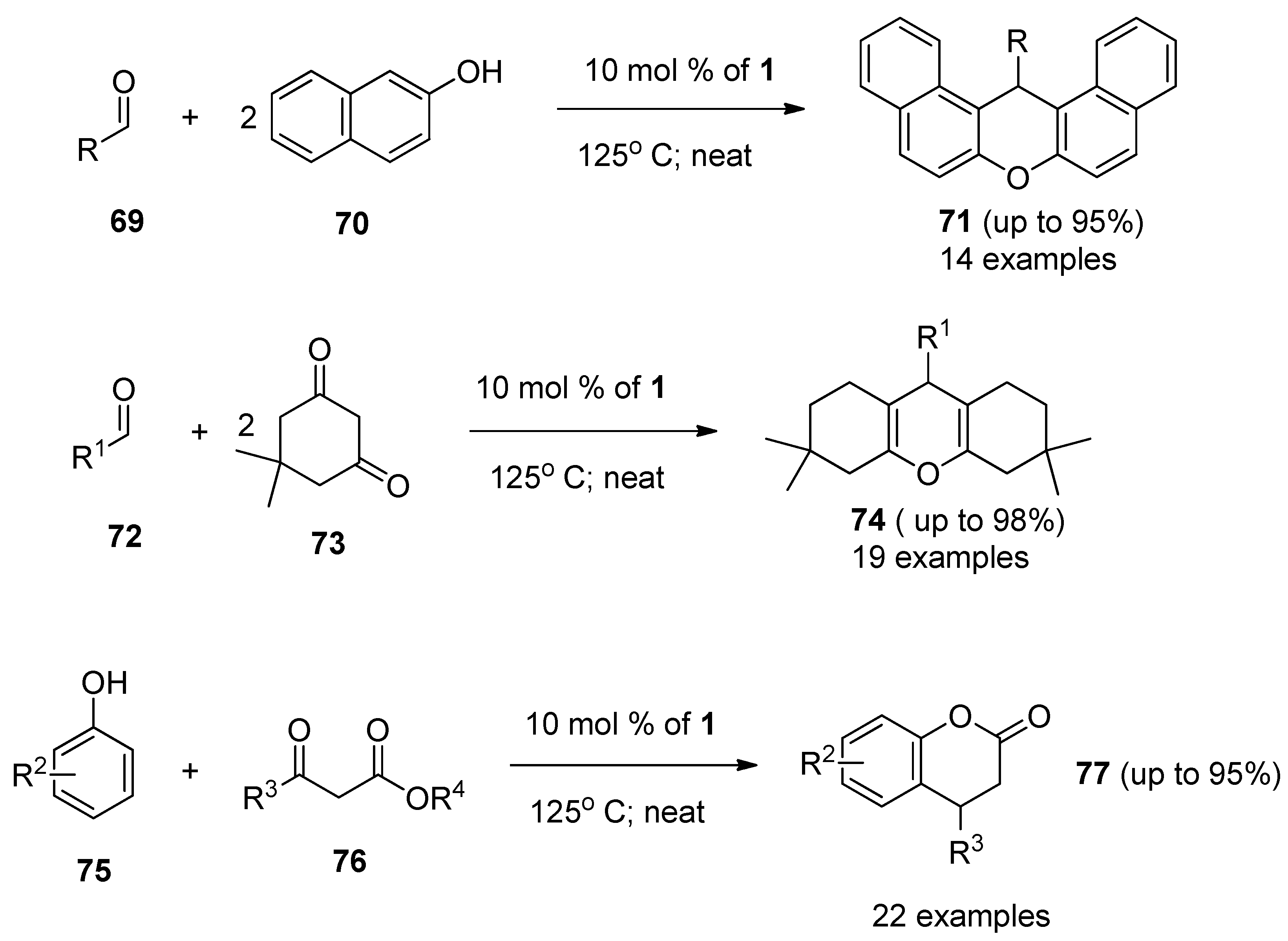
A synthetic protocol for the preparation of aryl-
14H-dibenzo[
a,j]xanthene derivatives (
71,
Scheme 9) through the F-TEDA-BF
4 catalyzed one-pot condensation of substituted benzaldehydes
69 with 2-naphthole
70 under solvent-free conditions was devised and methodology efficiently demonstrated by 14 examples [
49]. An efficient procedure for the synthesis of 1,8-dioxo-octahydro-xanthenes
74 through one-pot condensation of 5,5-dimethyl-1,3-cyclohexadione
73 with aryl aldehyde derivatives
72 in the presence of catalytic amounts of
1 was developed and efficiently demonstrated with 19 examples [
50]. One-pot condensation of β-ketoesters
76 and substituted phenols
75 catalyzed by
1 resulted in the efficient formation of 2H-chromen-2-one derivatives 77 [
51]. Reactions were performed under solvent-free conditions and application of ultrasonic irradiation improved the yields and reduced the reaction times [
52].
Scheme 9.
F-TEDA-BF4 catalyzed condensation reactions forming oxygen heterocycles.
Scheme 9.
F-TEDA-BF4 catalyzed condensation reactions forming oxygen heterocycles.
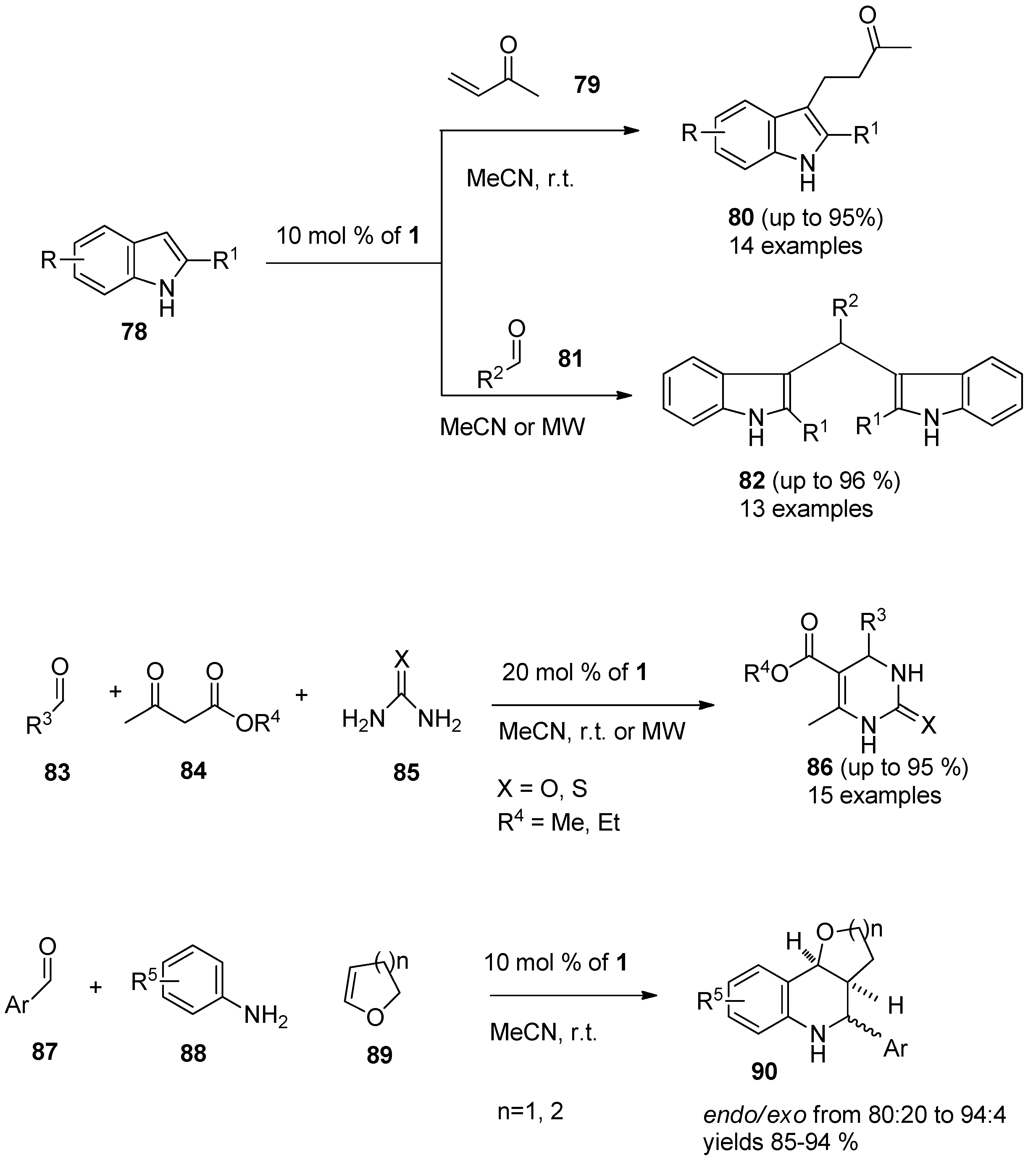
It was also found that F-TEDA-BF
4 efficiently catalyzed the conjugate addition of indoles (
78,
Scheme 10) with α,β-unsaturated ketones
79 thus forming Michael adducts
80 under extremely mild reaction conditions and the methodology was confirmed with 14 examples [
53]. The same approach was used in the case of reactions of indoles 78 with different aldehydes
81, resulting in the formation of bis(indolyl)methane derivatives
82 and the efficiency of the reaction was improved by MW irradiation under solvent-free conditions [
54]. The Biginelli reaction,
i.e., one-pot multi-component condensation of aldehyde
83, β-ketoester
84 and urea or thiourea
85 forming dihydropyrimidinones
86, was considerably improved when 1 was used as the catalyst [
55]. Aryl imines formed
in situ from aryl aldehydes
87 and aromatic amines
88 underwent smooth [
4+2] cycloaddition reactions with cyclic enol ethers
89 such as 3,4-dihydro-2H-pyran or 2,3-dihydrofuran in the presence of 10 mol %
1 in MeCN at room temperature to afford pyrano- and furanotetrahydroquinoline derivatives
90 with high
endo-stereoselectivity and high yield [
56].
A variety of epoxides (
91,
93,
Scheme 11) could be efficiently opened regio and stereoselectively with ammonium thiocyanate in the presence of 10 mol% of F-TEDA-BF
4 in MeCN at room temperature, affording the corresponding β-hydroxy thiocyanates
92, in the case of cyclic epoxides with
trans stereochemistry
94 [
57].
2.6. Deprotection of Functional Groups
An efficient method for cleavage of
p-methoxybenzylidene (PMP), tetrahydropyranyl (THP) and 1,3 dithiane protecting groups with F-TEDA-BF
4 was reported. PMP and THP are very useful protecting groups for diols, but their deprotection usually demands strong acidic or oxidative conditions, and 1,3-dithiane deprotection usually requires harsh conditions, too, which is inconvenient in the case of multifunctionally derivatized target molecules. It has been shown that
1 can smoothly and efficiently cleave PMP (
95,
Scheme 12), THP
97 or 1,3-dithiane protected compounds under mild reaction conditions [
58].
Scheme 10.
F-TEDA-BF4 catalyzed condensation reactions forming nitrogen heterocycles.
Scheme 10.
F-TEDA-BF4 catalyzed condensation reactions forming nitrogen heterocycles.
Scheme 11.
Regio and stereoselective ring opening of epoxides catalysed by F-TEDA-BF4.
Scheme 11.
Regio and stereoselective ring opening of epoxides catalysed by F-TEDA-BF4.
Scheme 12.
Cleavage of PMP, THP, and 1,3-dithiane protecting groups by F-TEDA-BF4.
Scheme 12.
Cleavage of PMP, THP, and 1,3-dithiane protecting groups by F-TEDA-BF4.
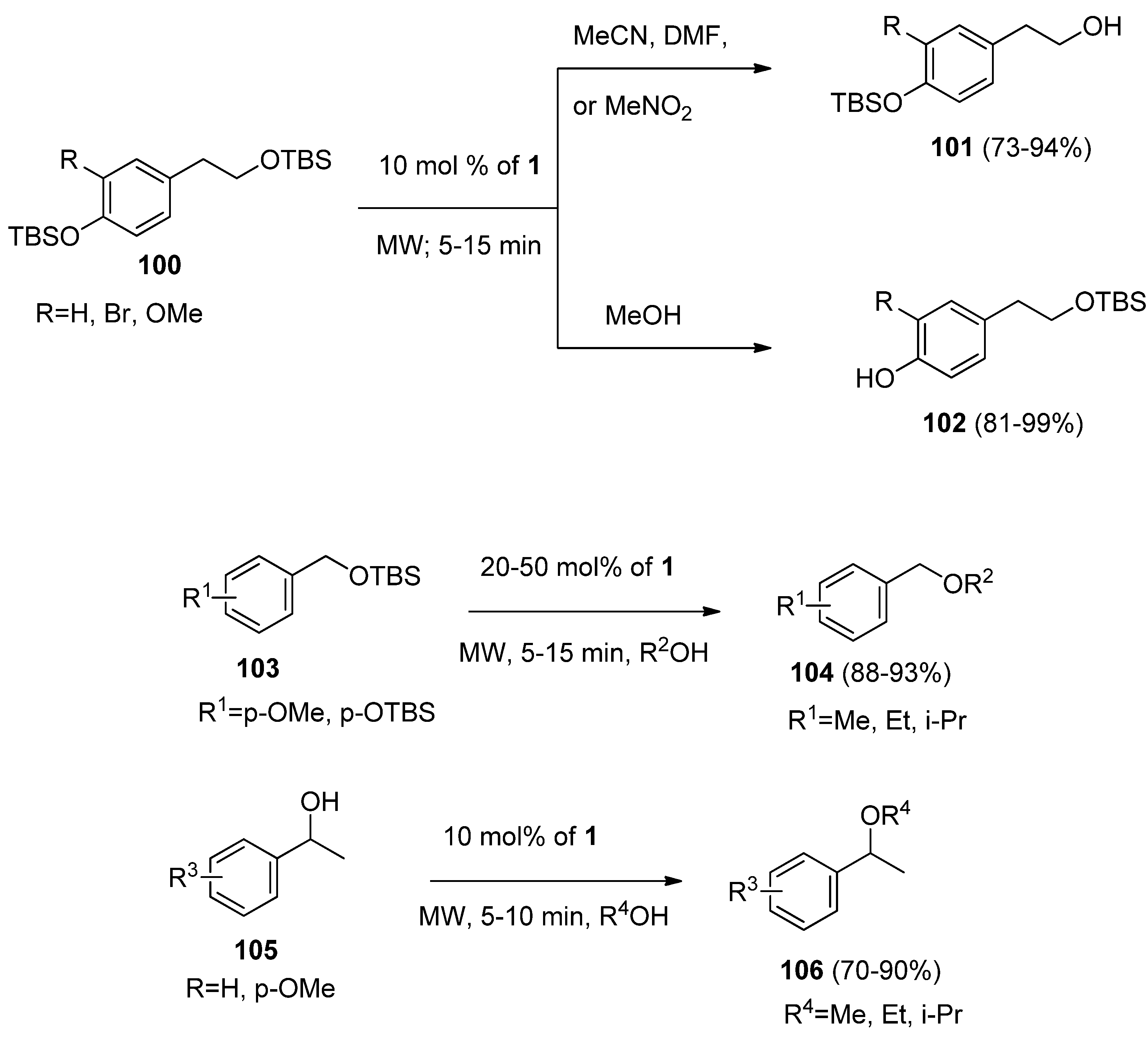
A novel microwave-assisted, chemoselective and efficient method for the cleavage of aliphatic and aromatic silyl ethers catalyzed by F-TEDA-BF
4 was reported. A wide range of aliphatic and aromatic
tert-butyldimethyl (TBS) protected silyl ethers (
100,
Scheme 13) were chemoselectively cleaved. In MeCN, MeNO
2 or DMF alkyl silyl ether was deprotected (
101), while in MeOH phenolic silyl ether was cleaved (
102). In addition, the transetherification of benzylic TBS-protected ethers
103 and etherification of benzyl alcohols
105 in alcoholic solvents resulting in the formation of
104 or
106 was observed [
59].
Scheme 13.
Chemoselective microwave-assisted deprotection of alkyl and aryl silyl ethers, transetherification and etherification of benzylic hydroxyl groups catalyzed by F-TEDA-BF4.
Scheme 13.
Chemoselective microwave-assisted deprotection of alkyl and aryl silyl ethers, transetherification and etherification of benzylic hydroxyl groups catalyzed by F-TEDA-BF4.
2.8. Functionalization of N-Heterocycles
The direct thiolation of indoles (
111,
Table 8) with a variety of thiols
112 has been achieved in the presence of F-TEDA-BF
4. This versatile and efficient method works for thiolation of 5- or 7-substituted indoles, as well as for 1-substituted (entries 6 and 9) and 2-substituted (entries 5, 12, and 13) indole derivatives with aromatic thiols (entries 1–17), alkyl thiols (entries 18 and 19) and benzyl thiol (entry 20). The reaction protocol is simple; the transformation goes to completion at room temperature within 20–30 minutes, efficiently and selectively forming 3-sulfenylindoles
113 [
62].
Table 8.
F-TEDA-BF
4 mediated synthesis of 3-sulfenylindoles.
![Molecules 16 06432 i008]()
Table 8.
F-TEDA-BF4 mediated synthesis of 3-sulfenylindoles. ![Molecules 16 06432 i008]()
| Entry | R | R 1 | R 2 | R 3 | Yield [%] |
|---|
| 1 | H | H | H | Ph | 96 |
| 2 | 5-Br | H | H | Ph | 85 |
| 3 | 5-OMe | H | H | Ph | 96 |
| 4 | 7-Et | H | H | Ph | 89 |
| 5 | H | H | Me | Ph | 89 |
| 6 | H | Bn | H | Ph | 87 |
| 7 | H | H | H | 4-Cl-Ph | 92 |
| 8 | 5-OMe | H | H | 4-Cl-Ph | 97 |
| 9 | H | Bn | H | 4-Cl-Ph | 93 |
| 10 | 7-Et | H | H | 4-Cl-Ph | 90 |
| 11 | 7-Et | H | H | 4-Me-Ph | 89 |
| 12 | H | H | Me | 4-Cl-Ph | 94 |
| 13 | H | H | H | 4-Cl-Ph | 94 |
| 14 | 5-Br | H | H | 4-Me-Ph | 90 |
| 15 | H | H | H | 4-NO2-Ph | 78 |
| 16 | H | H | H | 4-Br-Ph | 87 |
| 17 | H | H | H | 2-naphthyl | 85 |
| 18 | H | H | H | Et | 87 |
| 19 | 5-Br | H | H | n-Bu | 78 |
| 20 | H | H | H | Bn | 82 |
Various substituted indoles
111 have been efficiently thiocyanated under mild and neutral conditions to selectively produce 3-indoylthiocyanates
114 (
Table 9) in excellent yield following the reaction of indole derivatives with ammonium thiocyanate in the presence of F-TEDA-BF
4. Mechanistically, the reaction was declared to be the electrophilic substitution of indole derivatives by in situ generated thiocyanogen electrophilic species from
1 and ammonium thiocyanate. Following the same protocol was also successful for thiocyanation of azaindole, carbazole and pyrrole [
63].
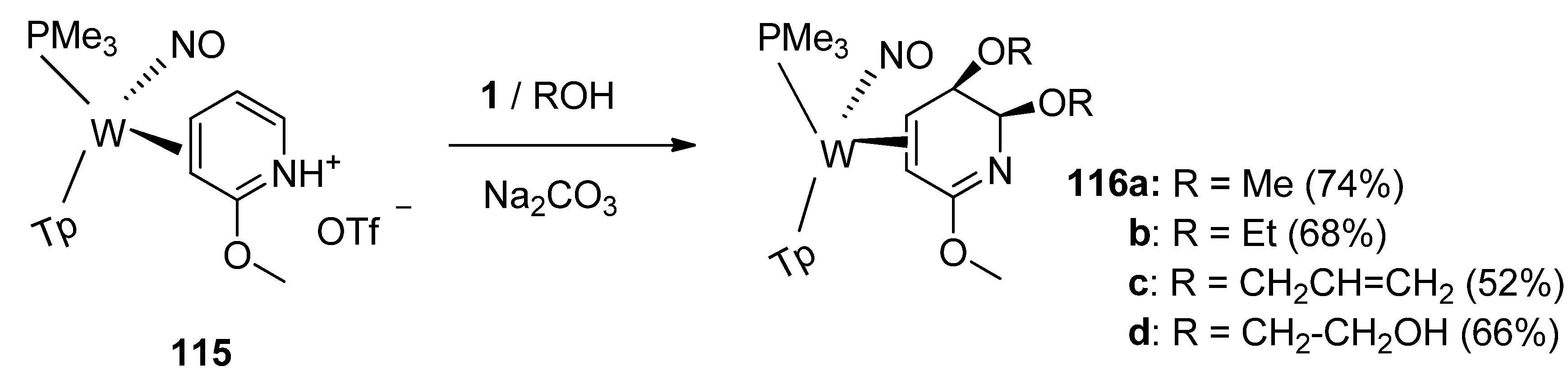
The tungsten η
2-coordinated pyridinium complex
115 (
Scheme 14) undergoes a stereoselective dialkoxylation when treated with F-TEDA-BF
4 in alcohol. The alkoxy groups add to the 5-and 6-positions of TpW(NO)(PMe
3)(3,4-η
2-methoxypyridine
115 in a
syn fashion. The reaction pathway has been not completely investigated but apparent stabilization by tungsten of the allyl cation intermediate resulting from the electrophilic attack of
1 to the 5,6-double bond on
115, captured by alkoxy anion and further fluorine atom replacement by the alkoxide in a subsequent substitution reaction resulting in the final formation of
116 seems to be a reasonable explanation of the reaction route [
64].
Table 9.
Thiocyanation of indole derivatives with ammonium thiocyanate using F-TEDA-BF
4.
![Molecules 16 06432 i009]()
Table 9.
Thiocyanation of indole derivatives with ammonium thiocyanate using F-TEDA-BF4. ![Molecules 16 06432 i009]()
| Entry | R | R 1 | R 2 | Yield [%] |
|---|
| 1 | H | H | H | 95 |
| 2 | H | H | Me | 92 |
| 3 | 7-Et | H | H | 94 |
| 4 | 5-NO2 | H | H | 93 |
| 5 | 5-CN | H | H | 92 |
| 6 | 5-Br | H | H | 93 |
| 7 | 5-OMe | H | H | 96 |
| 8 | H | H | Ph | 89 |
| 9 | H | Bn | H | 94 |
| 10 | H | Bn | Ph | 86 |
Scheme 14.
Dimethoxylation of η2-pyridinium complex mediated by F-TEDA-BF4.
Scheme 14.
Dimethoxylation of η2-pyridinium complex mediated by F-TEDA-BF4.
2.9. Gold-Catalyzed and Palladium-Catalyzed Oxidative C-C or C-Heteroatom Bond Formation
Cross-coupling reactions are powerful tools for the rapid construction of organic molecules and one of the most important and valuable approaches in organic synthesis. Various transition metals catalyze these valuable transformations and gold was introduced for this purpose recently [
65]. The gold/Selectfluor F-TEDA-BF
4 tandem was recognized as a valuable combination in numerous cross-coupling C-C or C-heteroatom bond formations.
The pioneer work on this area has been done by Zhang and co-workers with the discovery that under oxidative conditions gold catalyzes the coupling of propargyl acetates (
117,
Table 10) with boronic acids
118 resulting in the formation of α-aryl α,β-enones
119 in moderate to good yields and total
E-stereoselectivity [
66]. Following the proposed mechanism, reactions start by gold mediated 3,3-rearangement of propargyl acetates to allenyl acetates and their hydrolysation into the vinyl-Au(I) species which is subsequently oxidized by F-TEDA-BF
4 to furnish Au(III) intermediates; later these undergo transmetallation with boronic acids to give diorganogold derivatives, which after reductive elimination, regenerate the active Au(I) species and deliver the final cross-coupled products
119. Without the presence of boronic acid derivatives, oxidative dimerization of propargylic acetates was observed [
67].
Table 10.
Gold-catalyzed oxidative cross-coupling of propargyl acetates with boronic acids.
![Molecules 16 06432 i010]()
Table 10.
Gold-catalyzed oxidative cross-coupling of propargyl acetates with boronic acids. ![Molecules 16 06432 i010]()
| Entry | R | R 1 | R 2 | Yield (%) |
|---|
| 1 | Ph | n-butyl | H | 62 |
| 2 | iPr | n-butyl | H | 65 |
| 3 | Me | Ph | H | 59 |
| 4 | Me | MeOCH2CH2 | H | 60 |
| 5 | Me | cyclohexyl | H | 68 |
| 6 | cyclohexyl | cyclohexyl | H | 70 |
| 7 | PhCH2CH2 | n-butyl | H | 70 |
| 8 | 4-Br-Ph | n-butyl | H | 59 |
| 9 | AcOCH2CH2 | n-butyl | H | 61 |
| 10 | H | cyclohexyl | H | 61 |
| 11 | cyclohexyl | n-butyl | 4-Me-Ph | 72 |
| 12 | cyclohexyl | n-butyl | 4-CO2Me-Ph | 57 |
| 13 | cyclohexyl | n-butyl | 4-Cl-Ph | 58 |
| 14 | cyclohexyl | n-butyl | 3-CO2Me-Ph | 45 |
Analogous reactions were observed when propargyl benzoates (
120,
Table 11) were treated under similar reaction conditions and 1-benzoylvinyl ketones
121 were isolated [
68]. Intramolecular cross-coupling resulting in carboamination, carboalkoxylation or carbolactonization processes and formation of N- or O-heterocycles (
123,
Scheme 15) were reported when alkenes bearing a terminal hydroxyl, tosylamido or carboxy group (
122) were treated with the gold
cat /F-TEDA-BF
4 tandem in the presence of boronic acid [
69]. The scope of this reaction was considerably extended using bimetallic gold complexes as catalysts. The best results were obtained in the case of [dppm(AuBr)
2] catalyst where bis(diphenylphosphine)methane (dppm) was the ligand part of the bimetallic Au catalyst and a variety of alkenes and boronic acid reactants cross-coupled forming N-heterocycle derivatives [
70].
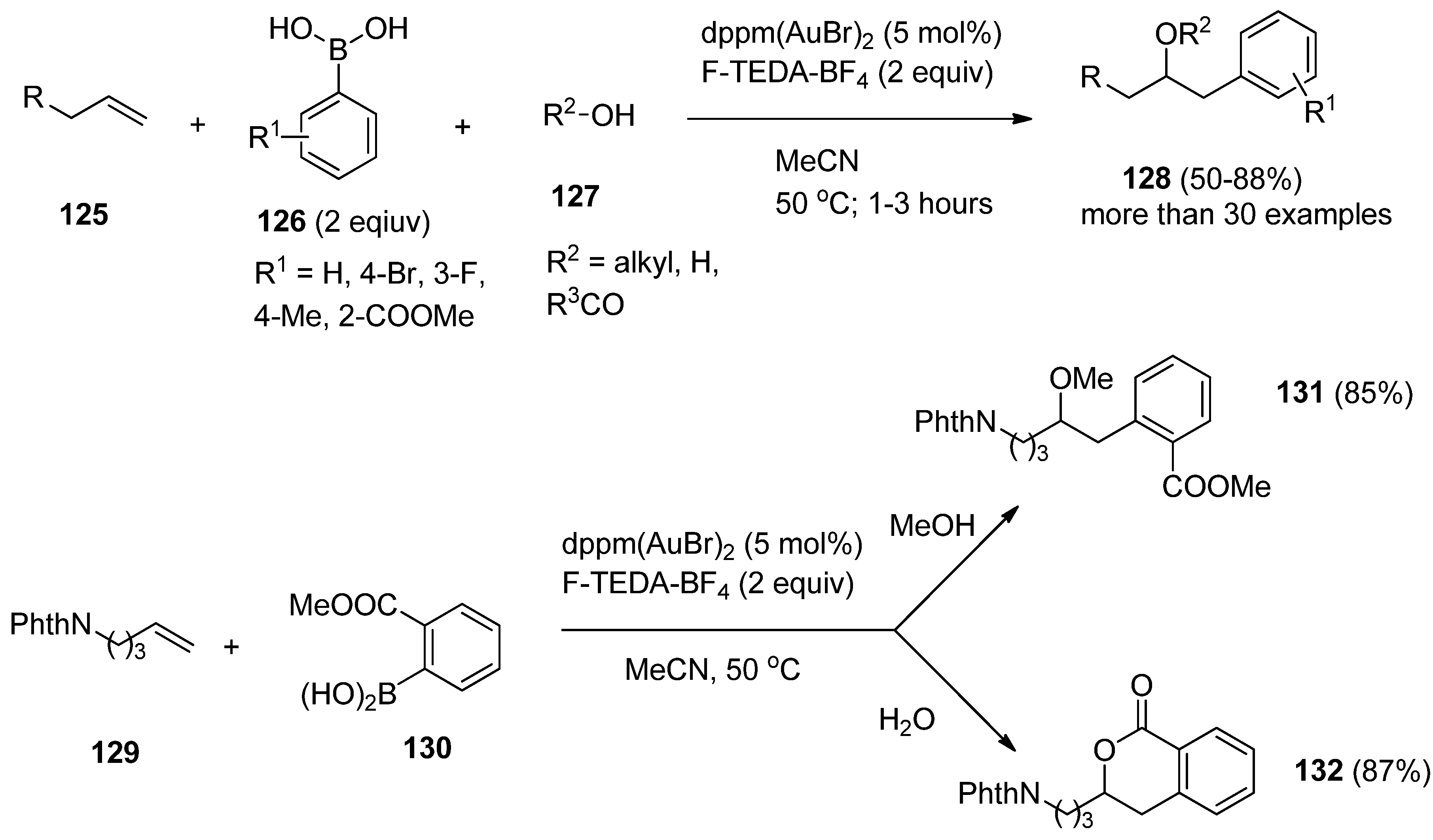
The same group of authors further reported three-component coupling reactions using this valuable methodology. Various combinations of alkenes (
125,
Scheme 16), boronic acid derivatives
126, and alcohols, carbocyclic acids or even water (
127) were treated with catalytic amounts of dppm(AuBr)
2 bimetallic complex in the presence of F-TEDA-BF
4 and oxyarylation of the double bond took place resulting in compounds
128. The ability to use either alcohols or water as nucleophiles in this gold-catalyzed three-component coupling provided access to a greater diversity of products. In the case of alkene
129 and 2-carboxymethyl boronic acid
130, methoxyarylation producing
131 took place when methanol was used as nucleophile, while in the presence of water, hydroxyarylation, followed by
in situ lactone formation
131 was the result of the reaction [
71].
Table 11.
Gold-catalyzed synthesis of 1-benzoylvinyl ketones from propargylbenzoates.
![Molecules 16 06432 i011]()
Table 11.
Gold-catalyzed synthesis of 1-benzoylvinyl ketones from propargylbenzoates. ![Molecules 16 06432 i011]()
| Entry | R | Yield (%) |
|---|
| 1 | cyclohexyl | 76 |
| 2 | Ph | 66 |
| 3 | cyclopropyl | 56 |
| 4 | BnOCH2CH2 | 71 |
| 5 | BzOCH2CH2 | 78 |
| 6 | BzCH2CH2CH2 | 70 |
Scheme 15.
Gold-catalyzed oxidative carboheterofunctionalization of alkenes.
Scheme 15.
Gold-catalyzed oxidative carboheterofunctionalization of alkenes.
Scheme 16.
Gold-catalyzed F-TEDA-BF4 mediated oxyarylation of alkenes.
Scheme 16.
Gold-catalyzed F-TEDA-BF4 mediated oxyarylation of alkenes.
The versatility of this methodology was expanded and arylsilicon compounds were taken as transmetallation components. The best results were obtained with phenyltrimethylsilane (
133,
Table 12) and efficient three-component coupling was accomplished when alkene
129, various alcohols and
133 were treated with the dppm(AuBr)
2 / F-TEDA-BF
4 tandem, resulting in oxyarylated products
134. As in the case of boronic acid in the presence of methanol, 2-carboxymethyl-trimethylphenylsilane was methoxyarylated to product
131, while the water mediated reaction yielded lactone product
132. In the case when a side chain bearing terminal alkene functionality is bonded at the
ortho position of phenyltrimethylsilane reagent (
135,
Table 13), intramolecular coupling reaction took place resulting in products
136 [
72].
Table 12.
Gold-catalyzed and F-TEDA-BF
4 mediated three-component oxyarylation of C-C double bond.
![Molecules 16 06432 i012]()
Table 12.
Gold-catalyzed and F-TEDA-BF4 mediated three-component oxyarylation of C-C double bond. ![Molecules 16 06432 i012]()
| Entry | R | R 1 | Yield [%] |
|---|
| 1 | 4-OAc | Me | 83 |
| 2 | 4-OTf | Me | 53 |
| 3 | 4-N(Me)Ts | Me | 66 |
| 4 | 4-Me | Me | 73 |
| 5 | 4-Br | Me | 82 |
| 6 | 4-CHO | Me | 77 |
| 7 | 4-CO2Me | Me | 68 |
| 8 | 3-CO2Me | Me | 83 |
| 9 | 2-CH2CH2OH | Me | 69 |
| 10 | H | Me | 87 |
| 11 | H | Et | 83 |
| 12 | H | i-Pr | 81 |
| 13 | H | t-Bu | 37 |
| 14 | H | neopentyl | 64 |
| 15 | H | cyclopentyl | 68 |
| 16 | H | 2-methoxyethyl | 86 |
| 17 | H | H | 77 |
| 18 | 2-CH2CH2OH | H | 55 |
A comparison of gold-catalyzed oxyarylation of terminal alkenes (
137,
Table 14) using arylsilanes
138a or arylboronic acids
138b as transmetallating reactants was reported. The results collected in
Table 14 demonstrate some advantages of the application of arylboronic acids in these reactions but the differences are not so remarkable. The commercially available gold catalyst Ph
3PAuCl was used, making this valuable and versatile transformation even more attractive [
73].
Table 13.
Gold-catalyzed and F-TEDA-BF
4 mediated intramolecular coupling reactions.
![Molecules 16 06432 i013]()
Table 13.
Gold-catalyzed and F-TEDA-BF4 mediated intramolecular coupling reactions. ![Molecules 16 06432 i013]()
| Entry | R | R 1 | n | Yield (%) |
|---|
| 1 | H | H | 1 | 66 |
| 2 | H | Me | 1 | 73 |
| 3 | H | Et | 1 | 70 |
| 4 | H | H | 0 | 15 |
| 5 | F | H | 1 | 47 |
| 6 | F | Et | 1 | 68 |
| 7 | Cl | H | 1 | 62 |
| 8 | Cl | Me | 1 | 65 |
| 9 | CF3 | H | 1 | 51 |
| 10 | CF3 | Me | 1 | 59 |
| 11 | Ph | Me | 1 | 74 |
Another valuable application of the Au(catalyst)/F-TEDA-BF
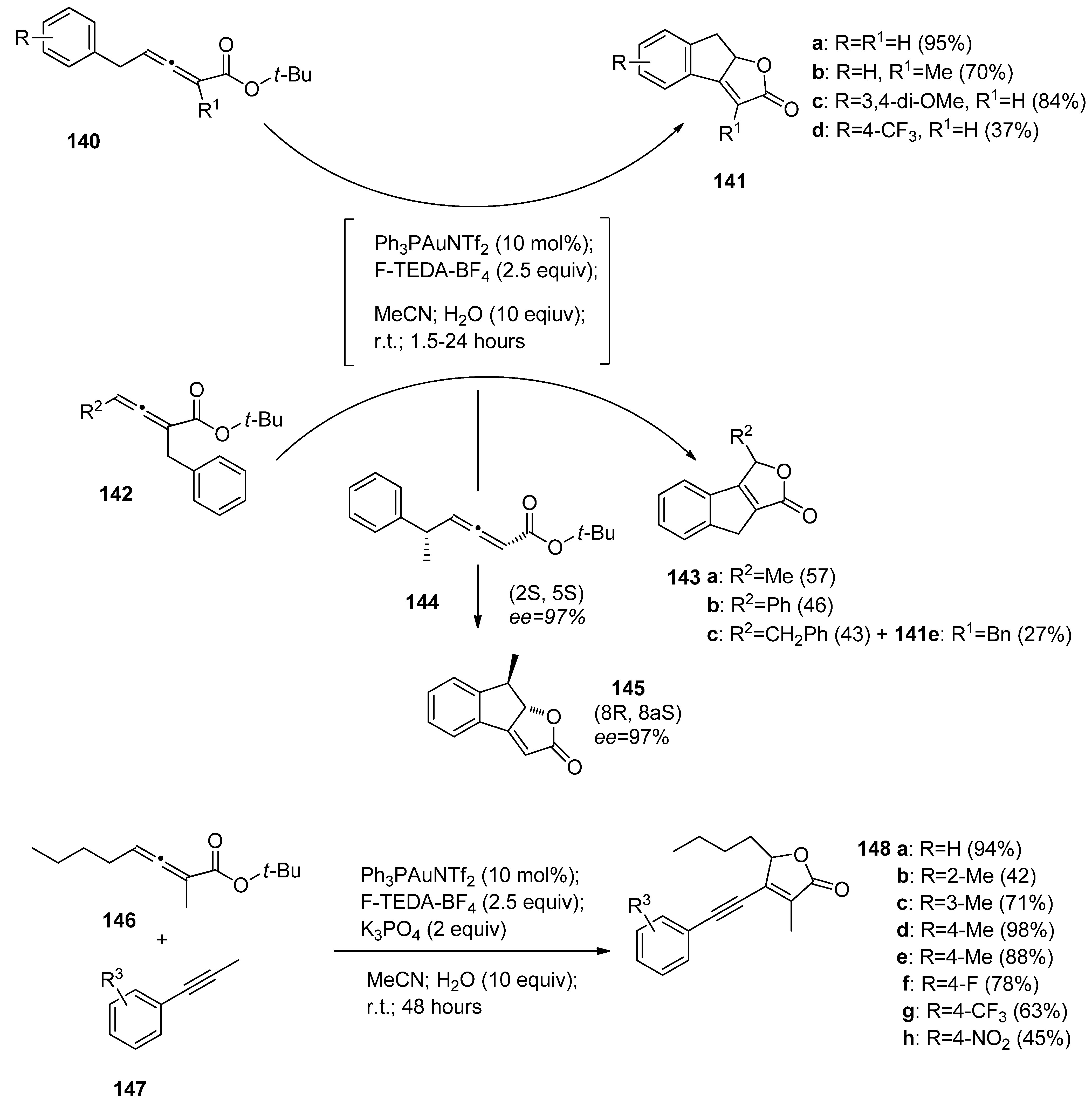
4(oxidant) tandem was reported by Gouverneur and co-authors. They developed a novel cascade cyclization cross-coupling process leading to tricyclic dihydroindenofurane-type compounds (
141a-e,
143a-c, and
145,
Scheme 17) following the Ph
3PAuNTf
2 catalyzed and F-TEDA-BF
4 mediated transformations of t-butyl ester substituted allenoates bearing a benzyl functional group on the opposite side of an allenoate moiety (
140), or vicinal to a
tert-butyl ester group (
142). The substrates
140 readily gave products
141a-e, while starting materials
142 gave products
143 a–b. In the case when both relevant allenoate carbon atoms were substituted by a benzyl group, the formation of product
143c was found to be preferential. It has also been established that the transformation is stereospecific, since pure enantiomer
144 gave only enantiomer
145 [
74].
The same group of authors developed efficient cascade cyclization-oxidative alkynyliation of allenoates (
146,
Scheme 17) with phenyl acetylenes
147, resulting in the formation of 5-butynyl-3-methyl-4-(phenylenthynyl)furan-2(5
H)-one derivatives
148. The selectivity as well as the efficiency of the transformation decreased if other than a n-butyl group was bonded to alleonate
146, or an alkyl group bonded to the alkynyl substrate
147 [
75].
Various arylgold(I) and alkynylgold(I) triphenylphosphane complexes (
149,
Table 15) were subjected to electrophilic halogenations reagents. Iodo, bromo and chloro reagents gave halogenated products, while reactions with F-TEDA-BF
4 followed exclusively the homocoupling process and corresponding dimeric products
150 were isolated in high yield [
76].
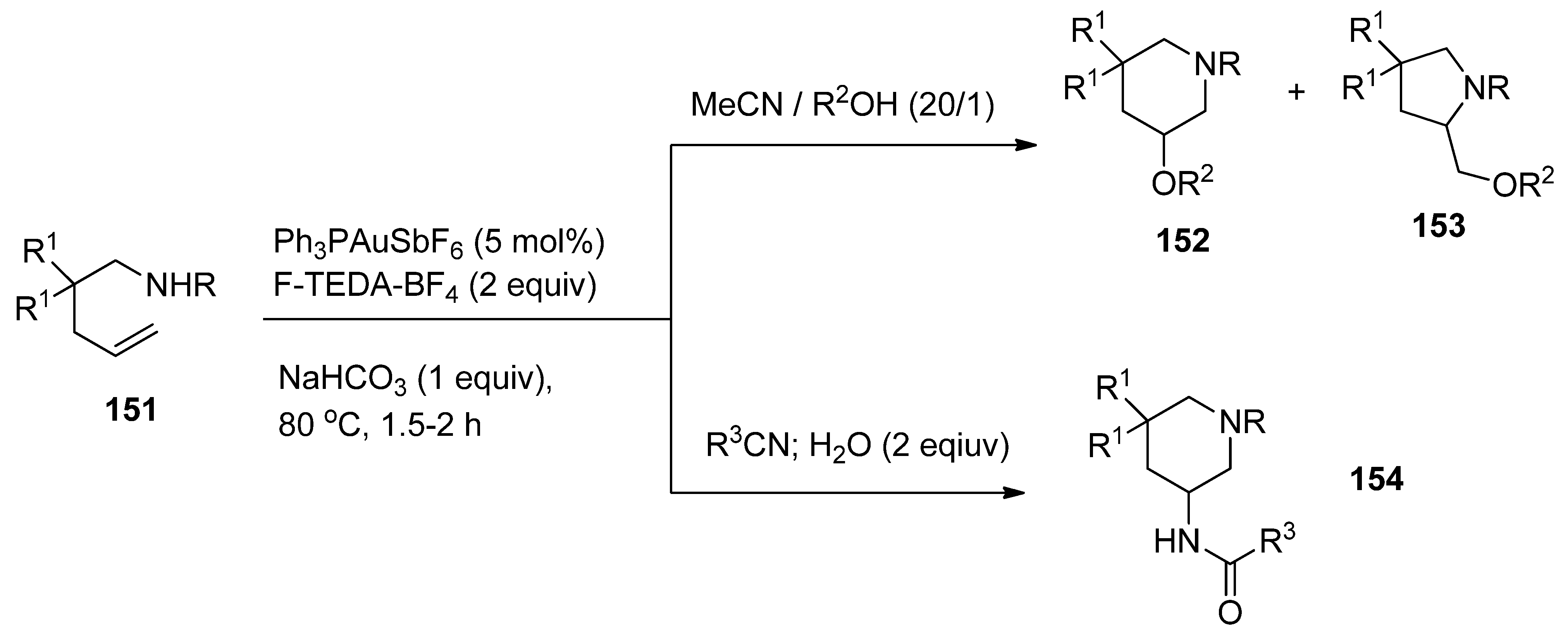
Aminooxygenation of unactivated alkenes (
151,
Scheme 18) were achieved by gold catalysis assisted by F-TEDA-BF
4 as an oxidant. In the case when the solvent was 20/1 mixture of MeCN and water (R
2 = H), methanol (R
2 = Me), or ethanol (R
2 = Et) mixtures of piperidine
152 and pyrrolidine derivatives
153 were formed. The formation of piperidine derivatives prevailed. On the other hand, by reducing the amount of water in the reaction mixture to only 2 equivalents and using nitriles as the reaction media, the aminoamidation process took place and 3-amido substituted piperidine derivatives
154 were selectively formed [
77].
Table 14.
Gold-catalyzed and F-TEDA-BF
4 mediated oxyarylation of terminal alkenes using arylsilanes [
73] or arylboronic acids [
71].
![Molecules 16 06432 i014]()
Table 14.
Gold-catalyzed and F-TEDA-BF4 mediated oxyarylation of terminal alkenes using arylsilanes [73] or arylboronic acids [71]. ![Molecules 16 06432 i014]()
| | | | | Yield [%] |
|---|
| Entry | Alkene | R 1 | R 2 | Z = SiMe3 | Z = B(OH)2 |
|---|
| 1 | 137 a | H | Me | 71 | 79 |
| 2 | 137 a | H | Et | 69 | 85 |
| 3 | 137 a | H | i-Pr | 70 | 90 |
| 4 | 137 a | H | t-Bu | - | 33 |
| 5 | 137 a | H | neopentyl | 80 | 91 |
| 6 | 137 a | H | c-pentyl | 57 | 85 |
| 7 | 137 a | H | Ac | 79 | 62 |
| 8 | 137 a | 4-Me | Me | 55 | 88 |
| 9 | 137 a | 2-Me | Me | 20 | - |
| 10 | 137 a | 4-Br | Me | 80 | 90 |
| 11 | 137 a | 3-F | Me | 63 | 79 |
| 12 | 137 a | 4-CO2Me | Me | 80 | 83 |
| 13 | 137 b | 4-Br | c-pentyl | 51 | 69 |
| 14 | 137 b | 4-Br | Ac | 51 | 51 |
| 15 | 137 b | H | H | 76 | 76 |
| 16 | 137 c | 4-Br | c-pentyl | 38 | 76 |
| 17 | 137 c | 4-Br | neopentyl | 85 | 73 |
| 18 | 137 c | H | H | 78 | 73 |
| 19 | 137 d | H | H | 75 | 67 |
Scheme 17.
Gold-catalyzed F-TEDA-BF4 mediated oxidative transformations of allenoates.
Scheme 17.
Gold-catalyzed F-TEDA-BF4 mediated oxidative transformations of allenoates.
Recently the Zhang group reported the first oxidative cross-coupling reaction between an aryl C-H bond and an alkyl gold compound generated
in situ, combining Au(I)/Au(III) catalysis with C-H functionalization. They have chosen
N,N-diallyl-
N´-phenylurea derivatives (
155d-k,
Table 16) as a substrates, (4-CF
3-C
6H
4)
3P-Au-NTf
2 as the catalyst, and F-TEDA-BF
4 as the oxidant and following an initial aminoauration and subsequent intramolecular [
3+2] annulation process isolated tricyclic indoline derivatives
156 in high yield. The efficiency of the reaction was significantly improved by the addition of 30 eqiuvalents of water in TFH as the optimal reaction media and the transformation was successful in the case when the additional allyl group in
155 was replaced by benzyl (entry 1), alkyl (entry 2) or phenyl group (entry 3). On the basis of performed deuterium labeling and kinetic isotope effect studies along with the isolation of alkyl gold intermediates the reaction mechanism anticipating an electrophilic aromatic substitution for the C-H functionalization and a subsequent inner-sphere concerted reductive elimination for the C
sp2-C
sp3 bond formation were strongly supported [
78].
Table 15.
Homocoupling reactions of organogold(I) triphenylphosphane compounds induced by F-TEDA-BF
4.
![Molecules 16 06432 i015]()
Table 15.
Homocoupling reactions of organogold(I) triphenylphosphane compounds induced by F-TEDA-BF4. ![Molecules 16 06432 i015]()
| Entry | R | Yield (%) |
|---|
| 1 | Ph | 90 |
| 2 | 3-nitrophenyl | 91 |
| 3 | 3-methoxyphenyl | 85 |
| 4 | 4-methoxyphenyl | 94 |
| 5 | 2-formylfuran-5-yl | 82 |
| 6 | 3-formylfuran-5-yl | 81 |
| 7 | phenyletynyl | 94 |
| 8 | ![Molecules 16 06432 i016]() | 71 |
Scheme 18.
Gold-catalyzed and F-TEDA-BF4 assisted aminooxygenation or aminoamidation of unactivated alkenes.
Scheme 18.
Gold-catalyzed and F-TEDA-BF4 assisted aminooxygenation or aminoamidation of unactivated alkenes.
Table 16.
Gold-catalyzed and F-TEDA-BF
4 mediated C-C coupling through C-H functionalization.
![Molecules 16 06432 i017]()
Table 16.
Gold-catalyzed and F-TEDA-BF4 mediated C-C coupling through C-H functionalization. ![Molecules 16 06432 i017]()
| Entry | 155 | R | R 1 | Yield [%] of 156 |
|---|
| 1 | a | H | Bn | 75 |
| 2 | b | H | n-hexyl | 69 |
| 3 | c | H | Ph | 70 |
| 4 | d | 4-Me | allyl | 72 |
| 5 | e | 2-Me | allyl | 43 |
| 6 | f | 3-Me | allyl | 79 |
| 7 | g | 4-F | allyl | 70 |
| 8 | h | 4-OTs | allyl | 67 |
| 9 | i | 4-CF3 | allyl | 64 |
| 10 | j | 4-COOEt | allyl | 84 |
| 11 | k | 4-Ac | allyl | 75 |
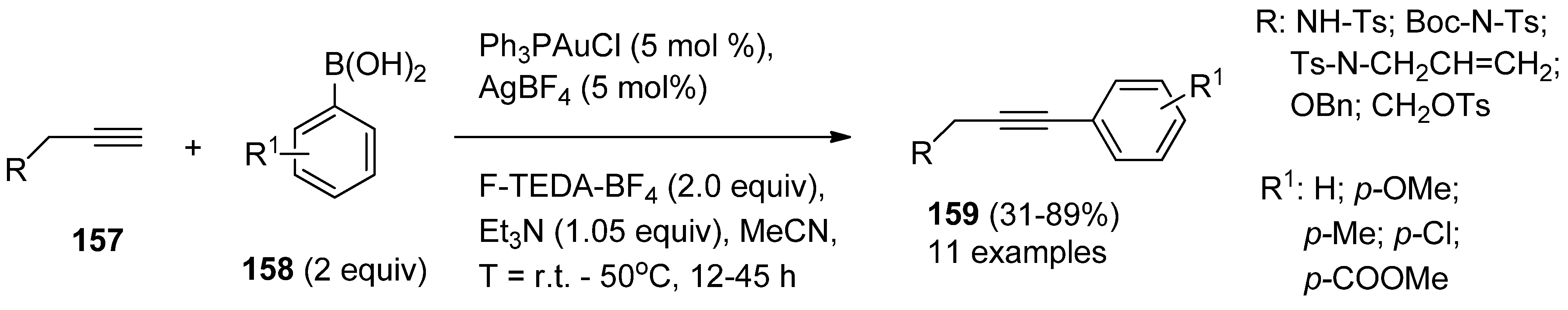
In the same laboratory a straightforward, efficient, and reliable catalyst system for the Sonogashira cross-coupling reaction of terminal alkyne derivatives (
157,
Scheme 19) with arylboronic acids
158 was developed very recently. The catalyst consisting Ph
3PAuCl and AgBF
4 gave the best results in the presence of F-TEDA-BF
4 as the oxidant and Et
3N as the base and the scope of the method was illustrated by eleven examples of cross-coupling yielding aryl functionalized alkyne derivatives
159 [
79].
Scheme 19.
Gold-catalyzed F-TEDA-BF4 mediated Sonogashira-type cross-coupling reactions of terminal alkynes with arylboronic acids.
Scheme 19.
Gold-catalyzed F-TEDA-BF4 mediated Sonogashira-type cross-coupling reactions of terminal alkynes with arylboronic acids.
Palladium-catalyzed directed
ortho amidation of aromatic ketones (
160,
Scheme 20) with both sulfoanamides
161a and amides
161b has been accomplished using different oxidants, including N-F compounds. The efficiency of the formation of the corresponding sulfonamides
162a or amides
162b was moderate to good when F-TEDA-BF
4 mediated the reactions. It has been proposed and supported by X-ray crystallography that the formation of cyclopalladation complexes of aryl ketones and amides are the key intermediates for this valuable transformation. The palladium(II) complex is oxidized to the Pd(IV) moiety, which following reductive elimination, ends in the final ortho amido derivatized product [
79].
Scheme 20.
Palladium-catalyzed F-TEDA-BF4 mediated ortho amidation of aromatic ketones.
Scheme 20.
Palladium-catalyzed F-TEDA-BF4 mediated ortho amidation of aromatic ketones.






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
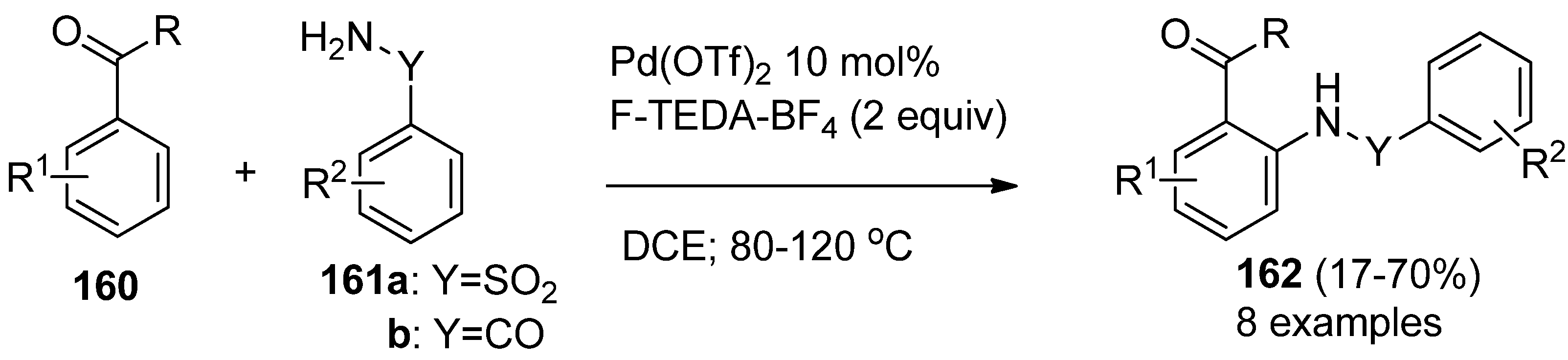{kind=link}

