General
Diethyl-2,2-(diethoxy)ethylphosphonate, diethyl methylphosphonate and diethylethylphosphonate were purchased from Alfa Aesar (Ward Hill, MA, USA). Diethyl ethylphosphonate can also be prepared in 98% yield by refluxing a mixture of triethyl phosphite and ethyl iodide for 3 h [
26]. THF was purified by distillation from sodium benzophenone ketyl. Lithium diisopropylamide (LDA) was purchased from the Aldrich Chemical Co (Milwaukee, WI, USA) as a 2 M solution in a mixture of heptane/THF/ethylbenzene. Other organic reagents were obtained from Aldrich and were used without further purification. Non-aqueous reactions were performed under an atmosphere of dry argon in oven-dried glassware. Removal of solvent was accomplished by rotary evaporation at water aspirator pressure.
Progress of synthetic reactions was monitored by gas chromatography (GC). All reaction products were identified by GC/MS and the structures were additionally verified by NMR. Yields were corrected for purity. Both 2E and 2Z isomers were considered to be the product for all product compounds; however, the 2E:2Z isomer ratios are reported.
The Hewlett-Packard (HP) 5890 Series II gas chromatograph was equipped with flame ionization detector and split/splitless inlet and was interfaced to an HP ChemStation data system. The column was a DB-5 capillary (30 m × 0.25 mm, 0.25-μm film thickness, J&W Scientific, Folsom, CA, USA). Carrier gas was He. The oven temperature was programmed from 50 to 280 °C at 10 °C/min, and the detector temperature was 280 °C. The inlet temperature was 220 °C, and 1.0 µL sample injections were made in splitless mode. The inlet temperature was 120 °C, and 1.0 µL sample injections were made in splitless mode for analysis for compounds 9 and 10 because considerable degradation of unsaturated aldehyde dimethylhydrazones was observed during GC runs with higher inlet temperatures.
Electron impact mass spectra (70 eV) were obtained with an HP 5973 MSD instrument, interfaced to an HP 6890 GC, equipped with a splitless inlet. Several columns were used, but gave comparable results to that used for GC. The oven temperature was programmed from 50 to 250 °C at 10 °C/min; the inlet temperature was 220 °C and the transfer line temperature was 250 °C. The inlet temperature was 120 °C for compounds 9 and 10.
1H-NMR,
13C-NMR and 2D NMR spectra were collected on a Bruker (Bellerica, MA, USA) Avance 500 spectrometer using a 5 mm broadband probe. Samples were dissolved in either CDCl
3 or C
6D
6 (where indicated) and all spectra (
1H and COSY at 500 MHz,
13C and DEPT at 125 Mhz) were acquired at 300° K. Trisubstituted double-bond “E” or “Z” assignments were based on 1D PFG-NOE experiments and 2D PFG-NOESY experiments. Chemical shifts are reported as parts per million from tetramethylsilane based on the lock solvent. Coupling constants (
J) are in Hertz. Assignments were made with the help of NMR prediction software from Advanced Chemistry Development, Inc. (ACD/Labs) [
27] and by analogy to known compounds. Although compounds
6,
7,
20,
22,
23 and
26 are known, spectral data are given because MS and NMR data are very difficult to find in the older chemical literature. Methyl branches are reported by the point of attachment to the carbon chain (e.g., CH
3 at C-2).
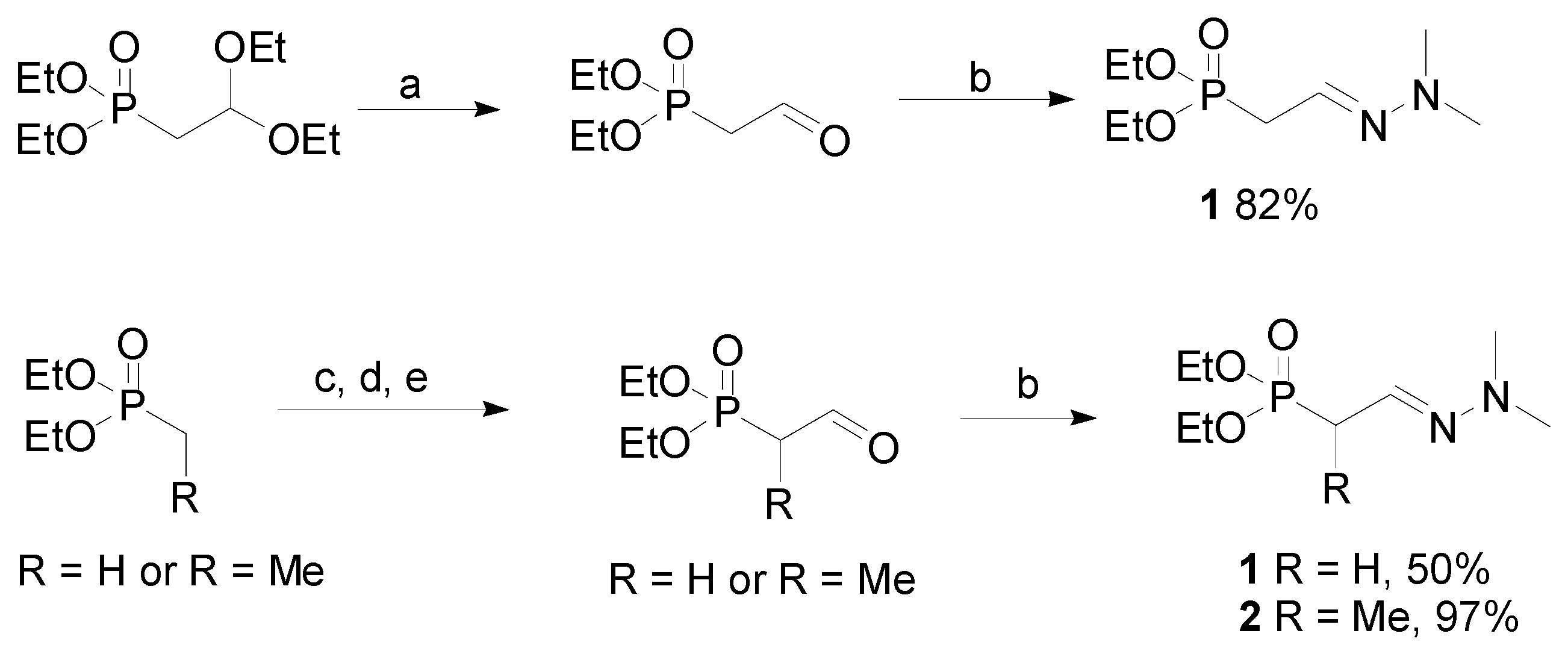
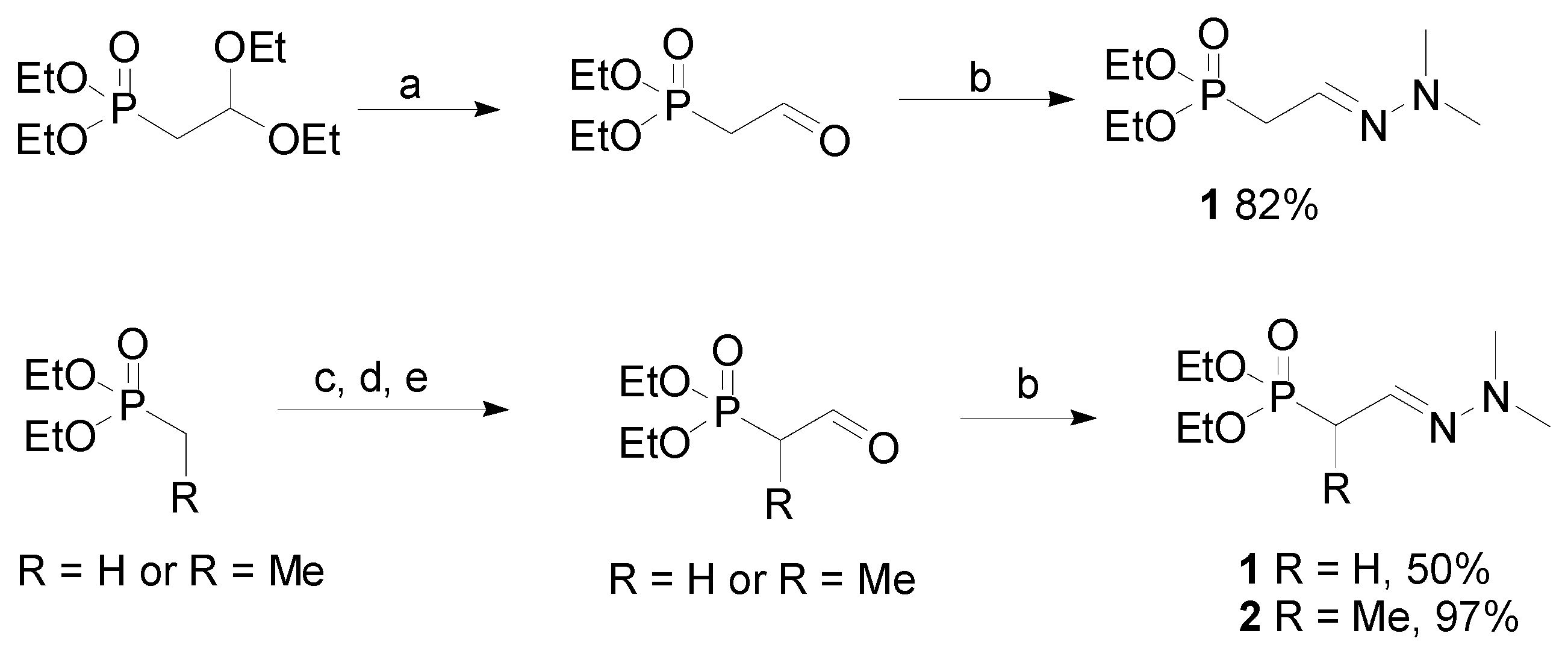
Diethyl methylformylphosphonate dimethylhydrazone (
1). Diethyl-2,2-(diethoxy)ethylphosphonate (5.31 g, 20.9 mmol) was added to acetone (200 mL), followed by water (3 mL) and
p-toluenesulfonic acid (1.3 g, 6.8 mmol). The mixture was stirred at r.t., under an argon atmosphere for 4 days. A solution of NaHCO
3 (570 mg, 6.8 mmol in 6 mL water) was added with stirring to quench the reaction and neutralize acid present. The pH was checked and found to be 7. After removal of solvent, aqueous oil remained. The oil was saturated by the addition of NaCl (25 g). The aqueous oil was repeatedly extracted with CH
2Cl
2 (100 mL then 2 × 50 mL), and the combined CH
2Cl
2 extracts were dried over anhydrous Na
2SO
4 and filtered. Solvent was removed to afford 5.59 g of crude diethyl methylformyl-2-phosphonate. Without further purification, the crude product was converted to the dimethylhydrazone (DMH) derivative by adding it to a mixture of CH
2Cl
2 (75 mL) containing anhydrous MgSO
4 (5.3 g, 44 mmol) then
N,
N-dimethylhydrazine (1.76 g, 2.2 mL, 22 mmol) was added in one portion. The mixture was stirred (48 h) until analysis by GC showed complete protection of the aldehyde as the DMH derivative. Filtration, removal of solvent, and Kugelrohr distillation (oven temperature 62 °C, 0.05 Torr) afforded 4.41 g of oil containing
1 (purity 86%, yield corrected for purity 82%). The oil also contained 8% starting material, diethyl-2,2-(diethoxy)ethylphosphonate. MS of product: (EI)
m/
z (%) 222 (M
+, 32), 180 (9), 152 (91), 125 (100), 122 (74), 108 (27), 97 (37), 85 (85), 71 (15), 58 (8), 44 (91).
1H-NMR δ 1.30 (6H, t,
J = 7.0, C
H3–CH
2–O), 2.77 (6H, s, N–CH
3), 2.81 (1H, dt,
J1-2 = 5.8, H-2), 4.09, (4H, q,
J = 7.0, CH
3–C
H2–O), 6.47 (1H, br d,
J1-2 = 5.8, H-1).
13C-NMR δ 16.4 (
CH
3–CH
2–O), 31.3 (C-2), 42.9 (N–CH
3), 61.9 (CH
3–
CH
2–O), 126.1 (C-1). The MS and NMR spectral data were consistent with the reference spectral data from previous work [
19].
Alternative preparation of diethyl methylformylphosphonate dimethylhydrazone (1). A solution of n-butyllithium (2.5 M, 25 mL, 0.213 mol, slight excess) was added to dry THF (80 mL) and the mixture was cooled in an argon atmosphere to dry ice-ethanol bath temperature (approx −78 °C). Diethyl methylphosphonate (9.13 g, 8.8 mL, 0.06 mol) was added dropwise and the mixture was stirred for an additional hr before dry dimethylformamide (DMF, distilled from CaH2, 4.8 mL, 0.066 mol) was added. After warming to 20 °C, ice cold 3 M aqueous HCl (300 mL) was added and the mixture was stirred 5 min. A fine white precipitate formed initially, and then the solution cleared and separated into two phases (liberation of the free phosphonate aldehyde). The phases were separated, and the organic phase was washed with 10 mL of alkaline brine solution (0.5 % NaHCO3 in saturated aqueous NaCl), dried over anhydrous MgSO4, and filtered. The acidic aqueous phase was repeatedly extracted with CH2Cl2 (5 × 50 mL), and the combined CH2Cl2 extracts were washed with alkaline brine solution (2 × 10 mL). The alkaline brine washes were combined and extracted again with CH2Cl2 (4 × 15 mL). All CH2Cl2 extracts were combined, dried over anhydrous MgSO4, and filtered. Solvent was removed from the dried organic phase and all dried CH2Cl2 extracts to afford 9.8 g of crude diethyl methylformyl-2-phosphonate, which still contained some DMF. Without further purification, the crude product was converted to the dimethylhydrazone (DMH) derivative by adding it to a mixture of CH2Cl2 (200 mL) containing anhydrous MgSO4 (14.4 g, 0.12 mol) then N,N-dimethylhydrazine (4.7 g, 6 mL, 0.08 mol) was added in one portion. The mixture was stirred (48 h) until analysis by GC showed complete protection of the aldehyde as the DMH derivative. Filtration, removal of solvent, and Kugelrohr distillation (oven temperature 44 °C, 0.02 Torr) afforded 7.1 g of 1 (purity 93%, yield corrected for purity 50%). MS (EI) m/z (%) 222 (M+, 32), 180 (9), 152 (91), 125 (100), 122 (74), 108 (27), 97 (37), 85 (85), 71 (15), 58 (8), 44 (91). EI-HRMS C8H19N2O3P calcd. for 222.1133 (obs. 222.1133). 1H-NMR δ 1.30 (6H, t, J = 7.0, CH3–CH2–O), 2.77 (6H, s, N–CH3), 2.81 (1H, dt, J1-2 = 5.8, H-2), 4.09, (4H, q, J = 7.0, CH3–CH2–O), 6.47 (1H, br d, J1-2 = 5.8, H-1). 13C-NMR δ 16.4 (CH3–CH2–O), 31.3 (C-2), 42.9 (N–CH3), 61.9 (CH3–CH2–O), 126.1 (C-1).
Diethyl ethylformyl-2-phosphonate dimethylhydrazone (2). A solution of n-butyllithium (2.5 M, 85 mL, 0.213 mol, slight excess) was added to dry THF (250 mL) and the mixture was cooled in an argon atmosphere to dry ice-ethanol bath temperature (approx −78 °C). Diethyl ethylphosphonate (33.2 g, 32.4 mL, 0.20 mol) was added dropwise and the mixture was stirred for an additional hr before dry DMF (20 mL, 0.26 mol) was added. After warming to 20 °C, ice cold 3 M aqueous HCl (300 mL) was added and the mixture was stirred 5 min. A fine white precipitate formed initially, and then the solution cleared and separated into two phases (liberation of the free phosphonate aldehyde). The phases were separated, and the organic phase was washed with 30 mL of alkaline brine solution (0.5% NaHCO3 in saturated aqueous NaCl), dried over anhydrous MgSO4, and filtered. The acidic aqueous phase was repeatedly extracted with CH2Cl2 (5 × 150 mL), and the combined CH2Cl2 extracts were washed with 30 mL of alkaline brine solution. The alkaline brine washes were combined and extracted again with CH2Cl2 (4 × 15 mL). All CH2Cl2 extracts were combined, dried over anhydrous MgSO4, and filtered. Solvent was removed from the dried organic phase and all dried CH2Cl2 extracts to afford 44 g of crude diethyl ethylformyl-2-phosphonate, which still contained some DMF. Without further purification, the crude product was converted to the dimethylhydrazone (DMH) derivative by adding it to a mixture of CH2Cl2 (600 mL) containing anhydrous MgSO4 (48g, 0.4 mol) then N,N-dimethylhydrazine (15.6 g, 20 mL, 0.26 mol) was added in one portion. The mixture was stirred (48 h) until analysis by GC showed complete protection of the aldehyde as the DMH derivative. Filtration, removal of solvent, and Kugelrohr distillation (oven temperature 50 °C, 0.04 Torr) afforded 47.6 g of 3 (purity 98%, yield corrected for purity 97%). Isolated yields >95% were routinely obtained. MS (EI) m/z (%) 236 (M+, 34), 194 (8), 166 (82), 136 (42), 122 (10), 111 (12), 99 (100), 53 (12), 81 (12), 72 (11), 56 (12), 44 (36). EI-HRMS C9H21N2O3P calcd. for 236.1290 (obs. 236.1293) 1H-NMR δ 1.19 and 1.20 (6H, overlapping t, J = 7.0, CH3–CH2–O), 1.26 (3H, d, J2-3 = 7.3, H-3), 2.76 (1H, dt, J1-2 = 4.1 and J2-3 = 7.3, H-2), 2.66 (6H, s, N–CH3), 3.99 and 4.00, (4H, overlapping q, J = 7.0, CH3–CH2–O), 6.38 (1H, br d, J1-2 = 4.1, H-1). 13C-NMR δ 12.7 (C-3), 16.4 (CH3–CH2–O), 36.0 and 37.1 (C-2, isomers), 43.1 (N–CH3), 62.0 (CH3–CH2–O), 132.6 (C-1).
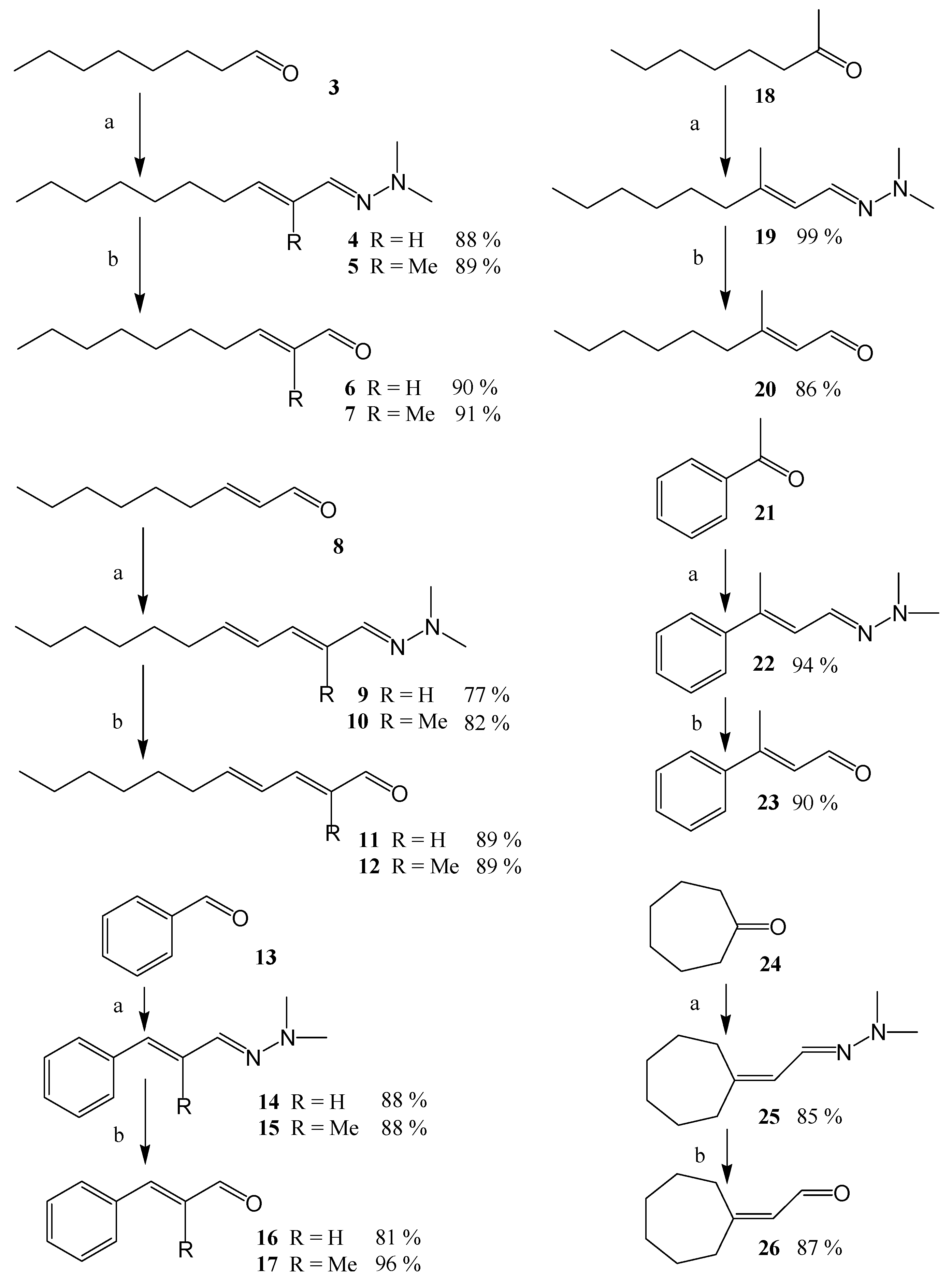
(2E)-N,N-Dimethyl-N’-(dec-2-enylidene)-hydrazine (4). A commercial 2.0 M solution of LDA (5.2 mL, 10.4 mmol, slight excess) was added dropwise (reaction exothermic) to a 250 mL round-bottomed flask containing dry THF (25 mL), 1 (2.22 g, containing 2.06 g of 1, 9.3 mmol) and a few mg of ethyl(triphenyl)phosphonium bromide, as an indicator, and the mixture was stirred in an argon atmosphere at r.t. for 1.5 h to ensure complete formation of the phosphonate ylide. A persistent red color developed in the solution when sufficient base was added to convert the phosphonate to its anion. Then, n-octanal (1.67 g, 13 mmol) was added dropwise; final color was yellow-orange. The mixture was stirred at r.t. for 5 h. Consumption of the phosphonate 1 was monitored by GC. Water (6 mL) was added to quench the reaction (excess water should be avoided because it leads to problematic emulsions during subsequent extraction steps). The solvent was removed by rotary evaporation to give an oily residue, which was extracted with ethyl acetate (5 × 25 mL). The combined extracts were washed with saturated aqueous NaCl solution (15 mL), dried over anhydrous Na2SO4 and filtered. Removal of solvent left a yellow oil. Kugelrohr distillation (oven temperature 59 °C, 0.05 Torr) afforded 1.80 g of 4 (89% purity by GC, yield corrected for purity 88%). By GC and GC-MS, the 2E/2Z isomer ratio was 8.5 to 1. MS (EI) m/z (%) 196 (M+, 72), 181 (8), 167 (2), 152 (19), 140 (7), 125 (14), 111 (100), 82 (40), 59 (21), 44 (32), 42 (19). EI-HRMS C12H24N2 calcd. for 196.1939 (obs. 196.1940). 1H-NMR δ 0.89 (3H, t, J9-10 = 6.9, H-10), 1.27-1.30 (8H, m, H-6 to H-9), 1.42 (2H, m, H-5), 2.15 (2H, m, J3-4 = 7.8, H-4), 2.84 (6H, s, N–CH3), 5.87 (1H, dd, J3-4 = 7.8 and J2-3 = 15.5, =CH, H-3), 6.20 (1H, dd, J1-2 = 9.1 and J2-3 = 15.5, =CH, H-2), 7.11 (1H, d, J1-2 = 9.1, =CH, H-1). 13C-NMR δ 13.3 (C-10), 22.6 (C-9), 29.0 (C-5, C-6 and C-7), 31.7 (C-8), 32.6 (C-4), 43.0 (N–CH3), 128.6 (C-2), 136.4 (C-3), 137.6 (C-1).
(2E)-2-Decenal (
6). Deprotection was initiated by stirring
4 (1.10 g, containing 0.98 g
4, 5.0 mmol) with 1 M HCl (50 mL) at room temperature for 5 min (
4 dissolves as the hydrochloride salt forms), and then petroleum ether (b.p. 35 °C to 60 °C, 50 mL) was added and stirring continued. After 4 h, the phases were separated and the aqueous phase was returned to the reaction vessel. Petroleum ether (30 mL) was added and the mixture was stirred at r.t. for an additional 1 h before the phases were separated. The combined petroleum ether phases were washed with alkaline brine solution (2 × 10 mL), and the brine phase was back-extracted once with petroleum ether (20 mL). The combined organic phases were dried, filtered, and removal of solvent afforded 0.75 g of the free aldehyde
6. Purity by GC was 92%, and yield corrected for purity was 90%. The 2
E:2
Z isomer ratio was 25 to 1. The MS and NMR spectral data were consistent with the reference spectral data from previous work [
9].
(2E)-N,N-Dimethyl-N’-(2-methyl-dec-2-enylidene)-hydrazine (5). Compound 5 was prepared in a way similar to compound 4. The phosphonate anion was prepared from 2 (2.36 g, containing 2.31 g of 2, 9.8 mmol) in THF (25 mL) by treatment with LDA (2.0 M, 5.2 mL, 10.4 mmol, slight excess), and n-octanal (1.92 g, 15 mmol) was subsequently added and the mixture was stirred at r.t. for 20 h. Water (3 mL) was added to quench the reaction and the reaction was worked up as before. Kugelrohr distillation (oven temperature 69 °C, 0.1 Torr) afforded 2.04 g of 5. (90% purity by GC, yield corrected for purity 89%). By GC and GC-MS, the 2E/2Z isomer ratio was 8.2 to 1. MS (EI) m/z (%) 210 (M+, 68), 195 (24), 180 (3), 166 (82), 125 (100), 111 (13), 96 (29), 82 (37), 59 (18), 44 (31), 42 (15). EI-HRMS C13H26N2 calcd. for 210.2096 (obs. 210.2093). 1H-NMR δ 0.90 (3H, t, J9-10 = 7.0, H-10), 1.28-1.31 (8H, m, H-6 to H-9), 1.41 (2H, m, H-5), 1.84 (3H, s, CH3 attached to C-2), 2.20 (2H, m, H-4), 2.83 (6H, s, N–CH3), 5.61 (1H, dd, J3-4 = 6.9, =CH, H-3), 7.15 (1H, s, =CH, H-1). 13C-NMR δ 11.6 (CH3 attached to C-2), 14.1 (C-10), 22.6 (C-9), 28.1 (C-4), 29.3 (C-6 and C-7), 29.9 (C-5), 31.9 (C-8), 43.0 (N–CH3), 132.4 (C-2), 134.2 (C-3), 141.2 (C-1).
(2E)-2-Methyl-2-Decenal (
7). Deprotection procedure was similar to that used for
6. A solution containing
5 (1.15 g, containing 1.04 g
3c, 5.0 mmol) and 1 M HCl (60 mL), to which petroleum ether (b.p. 35 °C to 60 °C, 60 mL) was added after 5 min, was stirred at room temperature for 6 h. The phases were separated and the aqueous phase was returned to the reaction vessel. Petroleum ether (40 mL) was added and the mixture was stirred at r.t. for an additional 1 h before the phases were separated. Workup of the reaction as for
6 resulted in 0.85 g of the free aldehyde
7 (purity by GC showed 90%, yield corrected for purity 91%). The 2
E:2
Z isomer ratio was 24 to 1. The MS and NMR spectral data were consistent with the reference spectral data from previous work [
28].
(2E,4E)-N’-(Undeca-2,4-dienylidene)-N,N-dimethyl-hydrazine (9). Compound 9 was prepared in a way similar to compound 4. The phosphonate anion was prepared from 1 (2.22 g, containing 2.06 g of 1, 9.3 mmol) in THF (25 mL) by treatment with LDA (2.0 M, 5.2 mL, 10.4 mmol, slight excess), and (2E)-2-nonenal (1.68 g, 12 mmol) was subsequently added and the mixture was stirred at r.t. for 3 h. Water (6 mL) was added to quench the reaction and the reaction was worked up as before. Kugelrohr distillation (oven temperature 70 °C, 0.05 Torr) afforded 1.62 g of 9. (92% purity by GC, yield corrected for purity 77%). By GC and GC-MS, the 2E/2Z isomer ratio was 16 to 1. MS (EI) m/z (%) 208 (M+, 18), 137 (3), 123 (100), 108 (2), 94 (8), 80 (11), 67 (5), 44 (7), 42 (3). EI-HRMS C13H24N2 calcd. for 208.1939 (obs. 208.1944). 1H-NMR δ 0.90 (3H, t, J10-11 = 7.2, H-11), 1.29–1.31 (6H, m, H-8 to H-10), 1.41 (2H, m, H-7), 2.12 (2H, m, J5-6 = 7.2, H-6), 2.87 (6H, s, N–CH3), 5.75, (1H, dt, J5-6 = 7.2 and J4-5 = 15.0, =CH, H-5), 6.13 (1H, dd, J3-4 = 10.0 and J4-5 = 15.0, =CH, H-4), 6.28 (1H, m, J3-4 = 10.0 and J2-3 = undeterminable in CDCl3, =CH, H-3), 6.28 (1H, m, J1-2 = 6.0 and J2-3 = undeterminable in CDCl3, =CH, H-2), 7.05 (1H, d, J1-2 = 6.0, =CH, H-1). 1H-NMR in C6D6 δ 0.98 (3H, t, J10-11 = 7.8, H-11), 1.29–1.31 (6H, m, H-8 to H-10), 1.41 (2H, m, H-7), 2.10 (2H, m, J5-6 = 7.2, H-6), 2.63 (6H, s, N–CH3), 5.77, (1H, dt, J5-6 = 7.2 and J4-5 = 15.1, =CH, H-5), 6.22 (1H, dd, J3-4 = 10.4 and J4-5 = 15.1, =CH, H-4), 6.41 (1H, dd, J3-4 = 10.4 and J2-3 = 15.1, =CH, H-3), 6.72 (1H, dd, J1-2 = 8.5 and J2-3 = 15.1, =CH, H-2), 7.14 (1H, d, J1-2 = 8.5, =CH, H-1). 13C-NMR δ 14.0 (C-11), 22.6 (C-10), 28.9 (C-8), 29.2 (C-7), 31.6 (C-9), 32.8 (C-6), 42.9 (N–CH3), 128.6 (C-2), 130.3 (C-4), 133.2 (C-3), 135.8 (C-5), 136.2 (C-1).
(2E,4E)-2,4-undecadienal (11). Deprotection procedure was similar to that used for 6. A solution containing 9 (1.16 g, containing 1.07 g 9, 5.1 mmol) and 1 M HCl (50 mL), to which petroleum ether (b.p. 35 °C to 60 °C, 50 mL) was added after 5 min, was stirred at room temperature for 24 h. The phases were separated and the aqueous phase was returned to the reaction vessel. Petroleum ether (40 mL) was added and the mixture was stirred at r.t. for an additional 1 h before the phases were separated. Workup of the reaction as for 6 resulted in 0.81 g of the free aldehyde 11 (purity by GC showed 93%, yield corrected for purity 89%). The 2E:2Z isomer ratio was 9.5 to 1. MS (EI) m/z (%) 166 (M+, 6), 137 92), 123 (4), 109 (5), 95 (12), 81 (100), 67 (15), 55 (11), 43 (10), 41 (18). 1H-NMR δ 0.91 (3H, t, J10-11 = 6.9, H-11), 1.30-1.32 (6H, m, H-8 to H-10), 1.47 (2H, m, J6-7 = 7.2, H-7), 2.24 (2H, dt, J5-6 = 6.6 and J6-7 = 7.2, H-6), 6.09 (1H, dd, J1-2 = 7.9 and J2-3 = 15.1, =CH, H-2), 5.79 (minor 2Z isomer, 1H, dd, J1-2 = 7.9 and J2-3 = 11.2, =CH, H-2), 6.31, (1H, m, J5-6 = 6.6 and J4-5 = undeterminable in CDCl3, =CH, H-5), 6.31 (1H, d, J3-4 = 9.8 and J4-5 = undeterminable in CDCl3, =CH, H-4), 7.10 (major 2E isomer: 1H, dd, J3-4 = 9.8 and J2-3 = 15.1, =CH, H-3), 6.93 (minor 2Z isomer, 1H, dd, J3-4 = 9.8 and J2-3 = 11.2, =CH, H-3), 9.55 (1H, d, J1-2 = 7.9, O=CH, H-1), 10.19 (minor 2Z isomer, 1H, d, J1-2 = 7.9, O=CH, H-1). The sample also contained about 6% of the 2E,4Z isomer: δ 6.16 (1H, dd, J1-2 = 7.9 and J2-3 = 15.4, =CH, H-2), 6.30 (1H, dd, J4-5 = 11.3, =CH, H-4), 7.45 1H, dd, J3-4 = 9.8 and J2-3 = 15.1, =CH, H-3), 9.63 (1H, d, J1-2 = 7.9, O=CH, H-1). 1H-NMR in C6D6 δ 0.99 (3H, t, J10-11 = 7.1, H-11), 1.27–1.32 (8H, m, H-7 to H-10), 1.91 (2H, m, J5-6 = 7.0, H-6), 5.79, (1H, dt, J5-6 = 7.0 and J4-5 = 15.1, =CH, H-5), 5.90 (1H, dd, J3-4 = 10.7 and J4-5 = 15.1, =CH, H-4), 6.03 (1H, dd, J1-2 = 7.8 and J2-3 = 15.4, =CH, H-2), 6.55 (1H, dd, J3-4 = 10.7 and J2-3 = 15.4, =CH, H-3), 9.54 (1H, d, J1-2 = 7.8, O=CH, H-1). 13C-NMR δ 14.0 (C-11), 22.5 (C-9), 28.5 (C-7), 28.8 (C-8), 31.6 (C-10), 33.2(C-6), 128.6 (C-4), 130.0 (C-2), 147.4 (C-5), 152.8 (C-3), 193.9 (C-1).
(2E, 4E)-N’-(2-Methyl-undeca-2,4-dienylidene)-N,N-dimethyl-hydrazine (10). Compound 10 was prepared in a way similar to compound 4. The phosphonate anion was prepared from 2 (2.36 g, containing 2.31 g of 2, 9.8 mmol) in THF (25 mL) by treatment with LDA (2.0 M, 5.2 mL, 10.4 mmol, slight excess), and (2E)-2-nonenal (1.68 g, 12 mmol) was subsequently added and the mixture was stirred at r.t. for 20 h. Water (3 mL) was added to quench the reaction and the reaction was worked up as before. Kugelrohr distillation (oven temperature 78 °C, 0.06 Torr) afforded 2.09 g of 10. (85% purity by GC, yield corrected for purity 82%). By GC and GC-MS, the 2E/2Z isomer ratio was 39 to 1. MS (EI) m/z (%) 222 (M+, 14), 137 (100), 108 (7), 94 (13), 44 (4), 43 (5). EI-HRMS C14H26N2 calcd. for 222.2096 (obs. 222.2102). 1H-NMR δ 0.91 (3H, t, J10-11 = 7.0, H-11), 1.29-1.31 (6H, m, H-8 to H-10), 1.43 (2H, m, H-7), 1.96 (CH3 attached to C-2), 2.16 (2H, m, J5-6 = 7.2, H-6), 2.87 (6H, s, N–CH3), 5.77, (1H, dt, J5-6 = 7.2 and J4-5 = 15.1, =CH, H-5), 6.12 (1H, d, J3-4 = 11.2, =CH, H-3), 6.44 (1H, dd, J3-4 = 11.2 and J4-5 = 15.1, =CH, H-4), 7.08 (1H, s, =CH, H-1). 13C-NMR δ 11.8 (CH3 attached to C-2), 14.1 (C-11), 22.6 (C-10), 28.9 (C-8), 29.4 (C-7), 31.7 (C-9), 33.2 (C-6), 43.2 (N–CH3), 126.7 (C-4), 131.2 (C-2), 133.2 (C-3), 135.8 (C-5), 140.0 (C-1).
(2E, 4E)-2-Methyl-2,4-undecadienal (12). Deprotection procedure was similar to that used for 6. A solution containing 10 (1.29 g, containing 1.10 g 10, 5.0 mmol) and 1 M HCl (50 mL), to which petroleum ether (b.p. 35 °C to 60 °C, 50 mL) was added after 5 min, was stirred at room temperature for 48 h. The phases were separated and the aqueous phase was returned to the reaction vessel. Petroleum ether (40 mL) was added and the mixture was stirred at rt for an additional 1 h before the phases were separated. Workup of the reaction as for 6 resulted in 0.91 g of the free aldehyde 12 (purity by GC showed 88%, yield corrected for purity 89%). The 2E:2Z isomer ratio was 30 to 1. MS (EI) m/z (%) 180 (M+, 11), 109 (6), 95 (100), 81 (10), 79 (9), 67 (12), 55 (5), 53 (5), 43 (5), 41 (10). 1H-NMR δ 0.91 (3H, t, J10-11 = 6.8, H-11), 1.32–1.34 (6H, m, H-8 to H-10), 1.48 (2H, m, H-7), 1.85 (CH3 attached to C-2), 2.26 (2H, dt, J5-6 = 7.2, H-6), 6.26, (1H, dt, J4-5 = 15.0 and J5-6 = 7.2, =CH, H-5), 6.54 (1H, dd, J3-4 = 11.2 and J4-5 = 15.0, =CH, H-4), 6.84 (1H, d, J3-4 = 11.2, =CH, H-3), 9.44 (major 2E isomer, 1H, s, O=CH, H-1), 10.28 (minor 2Z isomer, 1H, s, O=CH, H-1), The sample also contained a trace amount of the 2E,4Z isomer: δ 6.13 (1H, d, J3-4 = 11.2, =CH, H-3), 6.85 (1H, dd, J3-4 = 11.2 and J4-5 = 11.3, =CH, H-4), 9.51 (1H, s, O=CH, H-1). 13C-NMR δ 14.0 (C-11), 22.5 (C-9), 28.5 (C-7), 28.8 (C-8), 31.6 (C-10), 33.2(C-6), 128.6 (C-4), 130.0 (C-2), 147.4 (C-4), 152.8 (C-3), 193.9 (C-1).
(2E)-N,N-Dimethyl-N’-(3-phenyl-allylidene)-hydrazine (
14). Compound
14 was prepared in a way similar to compound
4. The phosphonate anion was prepared from
1 (2.22 g, containing 2.06 g of
1, 9.3 mmol) in THF (25 mL) by treatment with LDA (2.0 M, 5.2 mL, 10.4 mmol, slight excess), and benzaldehyde (1.11 g, 10.5 mmol) was subsequently added and the mixture was stirred at r.t. for 5 h. Water (3 mL) was added to quench the reaction and the reaction was worked up as before. Kugelrohr distillation (oven temperature 61 °C, 0.1 Torr) afforded 1.62 g of
14. (88% purity by GC, yield corrected for purity 88%). By GC and GC-MS, the 2
E/2
Z isomer ratio was 50 to 1. The MS and NMR spectral data were consistent with the reference spectral data from previous work [
13].
(2E)-Cinnamaldehyde (
16). Deprotection procedure was similar to that used for
6. A solution containing
14 (0.98 g, containing 0.86 g
14, 4.9 mmol) and 1 M HCl (50 mL), to which petroleum ether (b.p. 35 °C to 60 °C, 50 mL) was added after 5 min, was stirred at room temperature for 18 h. The phases were separated and the aqueous phase was returned to the reaction vessel. Petroleum ether (50 mL) was added and the mixture was stirred at r.t. for an additional 1 h before the phases were separated. Workup of the reaction as for
6 resulted in 0.61 g of the free aldehyde
16 (purity by GC showed 86%, yield corrected for purity 81%). The 2
E:2
Z isomer ratio was 170 to 1. The MS and NMR spectral data were consistent with the reference spectral data from previous work [
9].
(2E)-N,N-Dimethyl-N’-(2-methyl-3-phenyl-allylidene)-hydrazine (
15). Compound
15 was prepared in a way similar to compound
4. The phosphonate anion was prepared from
2 (2.36 g, containing 2.31 g of
2, 9.8 mmol) in THF (25 mL) by treatment with LDA (2.0 M, 5.2 mL, 10.4 mmol, slight excess), and benzaldehyde (1.11 g, 10.5 mmol) was subsequently added and the mixture was stirred at r.t. for 20 h. Water (3 mL) was added to quench the reaction and the reaction was worked up as before. Kugelrohr distillation (oven temperature 69 °C, 0.1 Torr) afforded 1.82 g of
15. (89% purity by GC, yield corrected for purity 88%). By GC and GC-MS, the 2
E/2
Z isomer ratio was 25 to 1. The MS and NMR spectral data were consistent with the reference spectral data from previous work [
13].
(2E)-Methyl cinnamaldehyde (
17). Deprotection procedure was similar to that used for
6. A solution containing
15 (1.12 g, containing 1.00 g
15, 5.3 mmol) and 1 M HCl (50 mL), to which petroleum ether (b.p. 35 °C to 60 °C, 50 mL) was added after 5 min, was stirred at r.t. for 48 h. The phases were separated and the aqueous phase was returned to the reaction vessel. Petroleum ether (40 mL) was added and the mixture was stirred at r.t. for an additional 1 hr before the phases were separated. Workup of the reaction as for
6 resulted in 0.78 g of the free aldehyde
17 (purity by GC showed 96%, yield corrected for purity 96%). The 2
E:2
Z isomer ratio was 56 to 1. The MS and NMR spectral data were consistent with the reference spectral data from previous work [
9].
N,N-Dimethyl-N'-(3-methyl-non-2-enylidene)-hydrazine (19). A commercial 2.0 M solution of LDA, (4.4 mL, 8.8 mmol, slight excess) was added dropwise (reaction exothermic) to a 250 mL round-bottomed flask containing dry THF (24 mL), 1 (1.78 g, 8 mmol) and a few mg of ethyl(triphenyl)phosphonium bromide, as an indicator, and the mixture was stirred in an argon atmosphere at r.t. for 1.5 h to ensure complete formation of the phosphonate ylide. A persistent red color developed in the solution when sufficient base was added to convert the phosphonate to its anion. Then, 2-octanone (18, 1.44 g, 11.2 mmol) was added dropwise; final color was yellow-orange. The mixture was stirred at r.t. for 22 h. Consumption of the phosphonate 1 was monitored by GC. Water (6 mL) was added to quench the reaction (excess water should be avoided because it leads to problematic emulsions during subsequent extraction steps). The solvent was removed by rotary evaporation to give an oily residue, which was extracted with ethyl acetate (5 × 25 mL). The combined extracts were washed with saturated aqueous NaCl solution (15 mL), dried over anhydrous Na2SO4 and filtered. Yellow oil remained after removal of solvent. Kugelrohr distillation (oven temperature 60 °C, 0.10 Torr) afforded 1.95 g of 19 (82% purity by GC, yield corrected for purity >99%). By GC and GC-MS, the 2E/2Z isomer ratio was 2.2 to 1. MS (EI) m/z (%) 197, (M+ 1, 197, 14), 196 (M+, 100), 181 (27), 166 (7), 154 (17), 139 (13), 125 (58), 110 (21), 96, (75), 94 ((48), 82 (36), 67 (18), 59 (38), 44 (41). 1H-NMR, major 2E-isomer, δ 0.89 (3H, t, J8-9 = 6.8, H-9), 1.29 (6H, m, H-6, H-7, and H-8) 1.45 (2H, m, H-5), 1.83 (3H, s, H-10 which is the CH3 attached to C-3), 2.09 (2H, t, J4-5 = 7.6, H-4), 2.85 (6H, s, N–CH3, H-11), 5.99 (1H, d, J1-2 = 9.3, H-2), 7.26 (1H, br, H-1). 13C-NMR δ 14.1 (C-9), 17.0 (C-10), 22.6 (C-3), 27.6 (C-5), 28.9 (C-6), 31.7 (C-7), 40.0 (C-4), 43.3 (C-11), 122.5 (C-2), 128.2 (C-1), 141.3 (C-3). 1H-NMR, minor 2Z-isomer, δ 0.89 (3H, t, J8-9 = 6.8, H-9), 1.29 (6H, m, H-6, H-7, and H-8) 1.45 (2H, m, H-5), 1.81 (3H, s, H-10 which is the CH3 attached to C-3), 2.22 (2H, t, J4-5 = 7.5, H-4), 2.84 (6H, s, N-CH3, H-11), 5.99 (1H, d, J1-2 = 9.3, H-2), 7.16 (1H, br, H-1). 13C-NMR δ 14.1 (C-9), 22.6 (C-8), 24.0 (C-10), 28.0 (C-5), 29.1 (C-6), 31.7 (C-7), 32.5 (C-4), 43.3 (C-11), 123.5 (C-2), 128.4 (C-1), 135.2 (C-3).
3-Methyl-non-2-enal (
20). Deprotection was initiated by stirring
19 (1.80 g, containing 1.48 g
19, 7.5 mmol) with 1 M HCl (100 mL) at room temperature for 5 min (
19 dissolves as the hydrochloride salt forms), and then hexane (100 mL) was added and stirring continued. After 3 h, the phases were separated and the aqueous phase was returned to the reaction vessel. Hexane (100 mL) was added and the mixture was stirred at r.t. for an additional 1 h before the phases were separated. The combined hexane phases were washed with alkaline brine solution (2 × 10 mL), and the brine phase was back-extracted once with hexane (20 mL). The combined organic phases were dried, filtered, and removal of solvent afforded 1.04 g of the free aldehyde
20. Purity by GC was 95%, and yield corrected for purity was 86%. The 2
E:2
Z isomer ratio was 2.0 to 1. MS (EI)
m/
z (%) 154 (M
+, 5), 139 (17), 125 (11), 111 (5), 97, (100), 84 (61), 69 (22), 55 (23), 41 (36).
1H-NMR, major 2
E-isomer, δ 0.89 (3H, t,
J8-9 = 6.6, H-9), 1.29 (2H, m, H-7), 1.30 (4H, m, H-6 and H-8), 1.50 (2H, m, H-5), 2.16 (3H, s, H-10 which is the CH
3 attached to C-3), 2.21 (2H, t,
J4-5 = 7.5, H-4), 5.88 (1H, d,
J1-2 = 8.2, H-2), 9.99 (1H, d,
J1-2 = 8.2, H-1).
13C-NMR δ 14.1 (C-9), 17.5 (C-10), 22.5 (C-8), 27.1 (C-5), 28.8 (C-6), 31.5 (C-7), 40.6 (C-4), 127.3 (C-2), 164.4 (C-3), 191.1 (C-1).
1H-NMR, minor 2
Z-isomer, δ 0.89 (3H, t,
J8-9 = 6.6, H-9), 1.29 (2H, m, H-7), 1.30 (2H, m, H-8), 1.32 (2H, m, H-6), 1.54 (2H, m, H-5), 1.97 (3H, s, H-10 which is the CH
3 attached to C-3), 2.57 (2H, t,
J4-5 = 7.6, H-4), 5.87 (1H, d,
J1-2 = 8.2, H-2), 9.96 (1H, d,
J1-2 = 8.2, H-1).
13C-NMR δ 14.0 (C-9), 22.5 (C-8), 25.0 (C-10), 28.8 (C-5), 29.0 (C-6), 31.5 (C-7), 32.6 (C-4), 128.4 (C-2), 164.9 (C-3), 190.7 (C-1). The MS and NMR spectral data were consistent with the reference spectral data from previous work [
22].
N,N-Dimethyl-N'-(3-phenyl-but-2-enylidene)-hydrazine (
22). Compound
22 was prepared in a way similar to compound
19. The phosphonate anion was prepared from
1 (1.78 g, 8 mmol) in THF (25 mL) by treatment with LDA (2.0 M, 4.4 mL, 8.8 mmol, slight excess), and acetophenone (
21, 1.35 g, 11.2 mmol) was subsequently added and the mixture was stirred at r.t. for 22 h. Water (6 mL) was added to quench the reaction and the reaction was worked up as before. Kugelrohr distillation (oven temperature 69 °C, 0.1 Torr) afforded 1.64 g of
22. (86 % purity by GC, yield corrected for purity 94%). By GC and GC-MS, the 2
E/2
Z isomer ratio was 2.1 to 1. MS (EI)
m/
z (%) 189 (M
+ 1, 14), 188 (M
+, 100), 173 (15), 158 (15), 144 (84), 128 (22), 115 (61), 103 (9), 91 (18), 77 (15), 58 (15), 42 (13).
1H-NMR, major 2
E-isomer, δ 2.25 (3H, d,
J2-10 = 1.2, H-10 which is the CH
3 attached to C-3), 2.85 (6H, s, N-CH
3, H-11), 6.68 (1H, dq,
J1-2 = 9.3 and
J2-10 = 1.1, H-2), 7.12 (1H, d,
J1-2 = 9.3, H-1), 7.26 (1H, m, H-7), 7.35 (2H, t,
J5-6 = 8.0, H-6), 7.51 (2H, d,
J5-6 = 8.1, H-5).
13C-NMR δ 16.0 (C-10), 43.0 (C-11), 125.0 (C-2), 125.4 (C-5), 127.1 (C-7), 128.3 (C-6), 135.6 (C-1), 139.4 (C-4), 142.6 (C-3).
1H-NMR, minor 2
Z-isomer, δ 2.18 (3H, d,
J2-10 = 1.3, H-10 which is the CH
3 attached to C-3), 2.78 (6H, s, N–CH
3, H-11), 6.30 (1H, dq,
J1-2 = 9.4 and
J2-10 = 1.3, H-2), 7.26 (1H, m, H-7), 7.35 (2H, t,
J5-6 = 8.0, H-6), 7.41 (1H, d,
J1-2 = 9.3, H-1), 7.51 (2H, d,
J5-6 = 8.1, H-5).
13C-NMR δ 25.6 (C-10), 43.0 (C-11), 125.4 (C-2 and C-5), 127.2 (C-7), 128.3 (C-6), 133.3 (C-1), 139.4 (C-4), 141.4 (C-3). This compound has been prepared previously [
23].
3-Phenyl-but-2-enal (
23). The deprotection procedure was similar to that used for
19. A solution containing
22 (1.49 g, containing 1.28 g
22, 6.8 mmol) and 1 M HCl (100 mL), to which hexane (100 mL) was added after 5 min, was stirred at room temperature for 3 h. The phases were separated and the aqueous phase was returned to the reaction vessel. Hexane (100 mL) was added and the mixture was stirred at r.t. for an additional 1 h before the phases were separated. Workup of the reaction as for
19 resulted in 0.95 g of the free aldehyde
23 (purity by GC showed 93%, yield corrected for purity 90%). The 2
E:2
Z isomer ratio was 2.1 to 1. MS (EI)
m/
z (%) 146 (M
+, 46), 145 (100), 131 (24), 115 (42), 103 (15), 91 (17), 77 (11), 63 (6), 51 (10).
1H-NMR, major 2
E-isomer, δ 2.59 (3H, s, H-10 which is the CH
3 attached to C-3), 6.42 (1H, d,
J1-2 = 7.7, H-2), 7.42 (1H, m, H-7), 7.43 (1H, m, H-6), 7.43 (1H, dd,
J5-6 = 6.0 and
J5-7 = 2.5, H-5), 10.20 (1H, d,
J1-2 = 7.9, H-1).
13C-NMR δ 16.4 (CH
3 attached to C-3), 126.3 (C-5) 127.3 (C-2), 128.8 (C-6), 130.1 (C-7), 140.6 (C-4), 157.6 (C-3), 191.1 (C-1),
1H-NMR, minor 2
Z-isomer, δ 2.33 (3H, s, H-10 which is the CH
3 attached to C-3), 6.15 (1H, d,
J1-2 = 8.1, H-2), 7.31 (1H, dd,
J5-6 = 7.1 and
J5-7 = 3.3, H-5), 7.42 (2H, m, H-6 and H-7), 9.49 (1H, d,
J1-2 = 8.2, H-1).
13C-NMR δ 26.4 (CH
3 attached to C-3), 128.4 (C-5 and C-6), 129.2 (C-2 and C-7), 138.4 (C-4), 162.1 (C-3), 193.4 (C-1). The MS and NMR spectral data were consistent with the reference spectral data from previous work [
24,
25].
N'-(2-Cycloheptylidene-ethylidene)-N,N-dimethyl-hydrazine (25). Compound 25 was prepared in a way similar to compound 19. The phosphonate anion was prepared from 1 (1.78 g, 8 mmol) in THF (25 mL) by treatment with LDA (2.0 M, 4.4 mL, 8.8 mmol, slight excess), cycloheptanone (24, 1.68 g, 11.2 mmol) was subsequently added and the mixture was stirred at r.t. for 22 h. Water (6 mL) was added to quench the reaction and the reaction was worked up as before. Kugelrohr distillation (oven temperature 61 °C, 0.1 Torr) afforded 1.40 g of 25. (88% purity by GC, yield corrected for purity 85%) MS (EI) m/z (%) 181 (M+ 1, 13), 180 (M+, 100), 165 (64), 138 (38), 136 (37), 123 (22), 112 (41), 94 (41), 80 (50), 67 (26), 59 (26), 44 (30). 1H-NMR δ 1.52 (2H, m, H-6), 1.54 (2H, m, H-7), 1.62 (2H, m, H-5), 1.64 (2H,m , H-8), 2.32 (2H, t, J4-5 = 5.8, H-4), 2.46 (2H, td, J8-9 = 6.1 and J2-9 = 1.0, H-9), 2.85 (6H, s, N-CH3, H-10), 5.98 (1H, dp, J1-2 = 6.0 and J2-9 = 1.0, H-2), 7.30 (1H, br, H-1). 13C-NMR δ 27.1 (C-8), 28.8 (C-5), 29.0 (C-6), 29.7 (C-7), 30.6 (C-9), 43.2 (C-10), 123.1 (C-2), 128.2 (C-1), 146.9 (C-3).
Cycloheptylidene-acetaldehyde (
26). The deprotection procedure was similar to that used for
19. A solution containing
25 (1.27 g, containing 1.11 g
25, 5.1 mmol) and 1 M HCl (100 mL), to which hexane (100 mL) was added after 5 min, was stirred at room temperature for 3 h. The phases were separated and the aqueous phase was returned to the reaction vessel. Hexane (100 mL) was added and the mixture was stirred at r.t. for an additional 1 h before the phases were separated. Workup of the reaction as for
19 resulted in 0.76 g of the free aldehyde
26 (purity by GC showed 98%, yield corrected for purity 87%). MS (EI)
m/
z (%) 138 (M
+, 41), 137 (12), 123 (18), 110 (48), 109 (51), 95, (100), 81 (66), 67 (66), 55 (35), 41 (46)..
1H-NMR δ 1.55 (2H, m, H-6), 1.57 (2H, m, H-7), 1.69 (2H, m, H-5), 1.73 (2H,m , H-8), 2.45 (2H, t,
J4-5 = 6.0, H-4), 2.84 (2H, t,
J8,9 = 6.0, H-8), 5.85 (1H, d,
J1-2 = 8.1, H-2), 9.9 (1H, d,
J1-2 = 8.1, H-1).
13C-NMR δ 27.4 (C-5, C-8), 28.6 (C-6), 29.3 (C-7), 30.5 (C-9), 38.9 (C-4), 127.6 (C-2), 170.2 (C-3), 191.1 (C-1), The MS and NMR spectral data were consistent with the reference spectral data from previous work [
20].

{kind=link}
{kind=link}