Oligomeric Nucleic Acids as Antivirals

Abstract
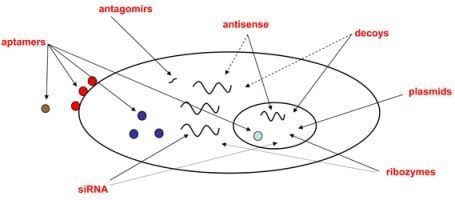
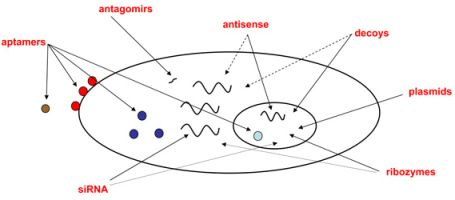
:
1. Introduction
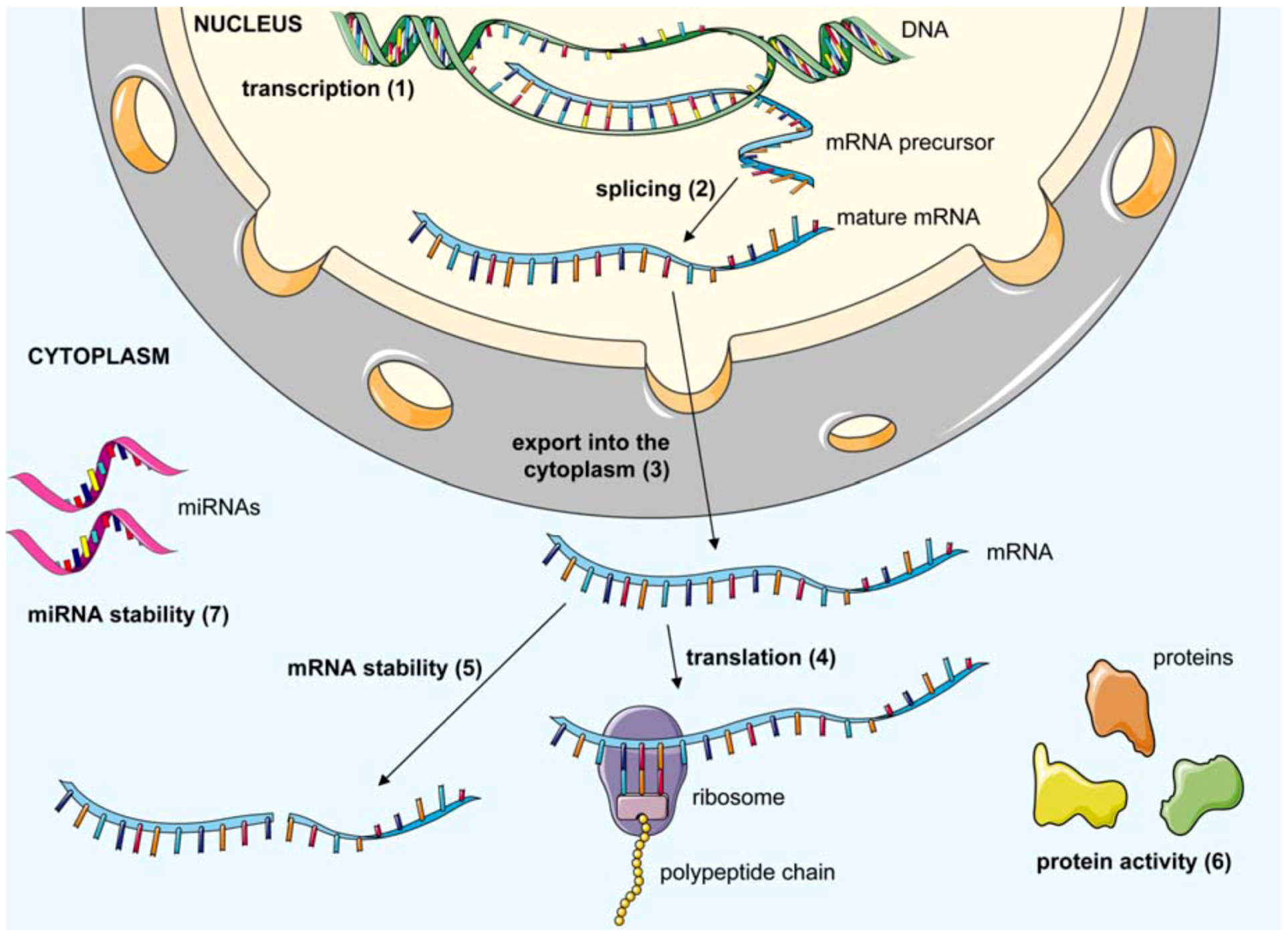
2. Antiviral Oligonucleotide-Based Therapeutics

2.1. Classes of oligonucleotides targeting viral or cellular nucleic acids
2.1.1. AsONs
2.1.2. Ribozymes

2.1.3. MiRNAs and their inhibitors
2.1.4. SiRNAs and siDNAs
2.3. Classes of oligonucleotides targeting viral nucleic acids or viral proteins
2.3.1. Aptamers

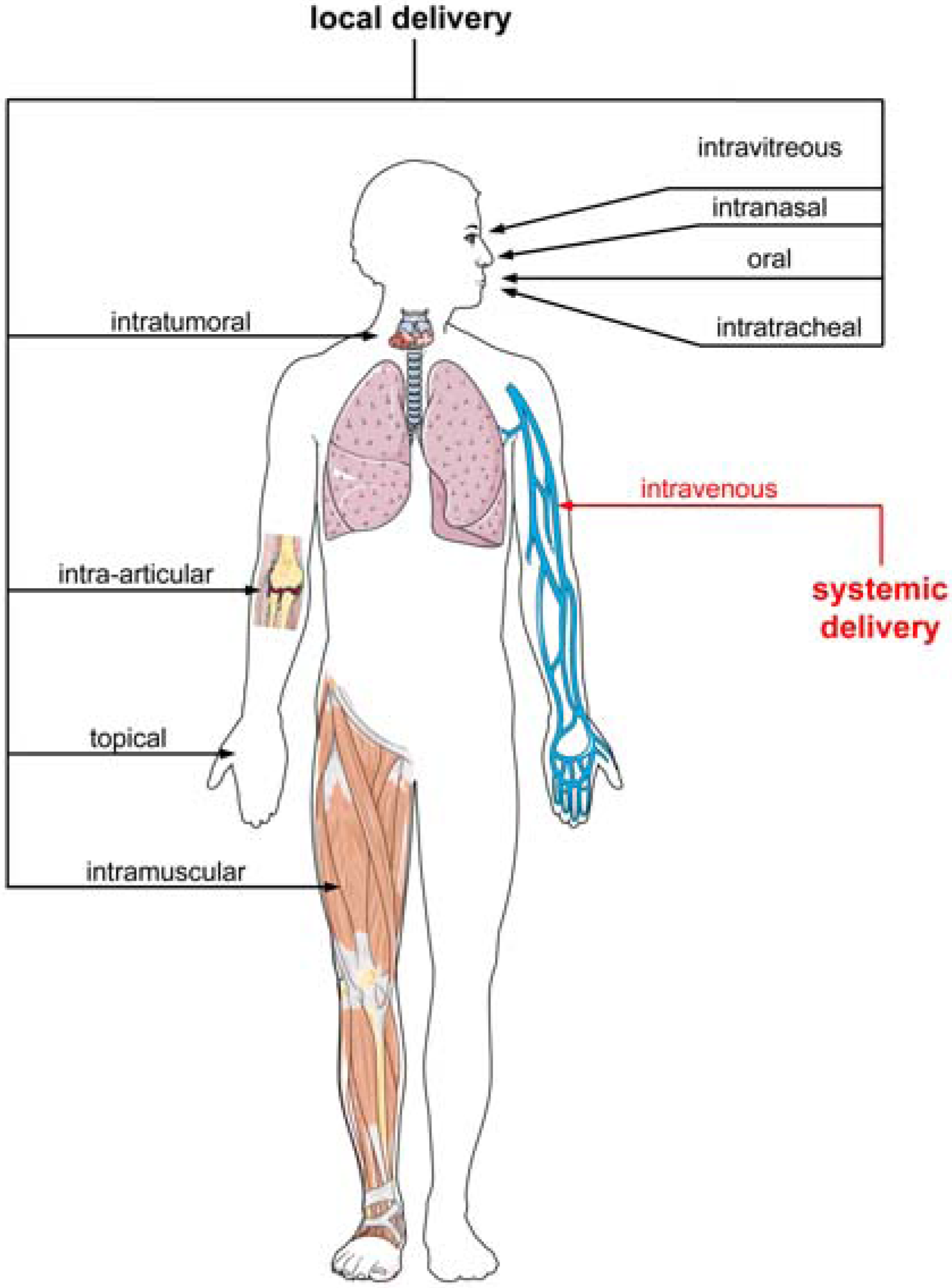
3. Advantages of Oligonucleotide-Based Drugs
4. Limitations of Oligonucleotide-Based Drugs

5. Conclusions and Perspectives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Class | Virus | Target transcript/s | Developer/Reference | Status |
|---|---|---|---|---|---|
| Fomivirsen (VitraveneTM) | asON | CMV | IE2 | Isis Pharmaceuticals | Approved |
| VRX496 | asON | HIV | env | [32] | Phase II |
| Morpholino asON | asON | ZEBOV and MARV | VP24 and VP35 | [35,36] | Entering Phase I |
| OZ1 | ribozyme | HIV | tat and vpr | [50] | Phase II |
| SPC3649 | anti-miRNA | HCV | miR-122 | Santaris Pharma | Phase I |
| ALN-RSV01 | siRNA | RSV | N-protein transcript | Alnylam | Phase II |
| siRNA-SNALP | siRNA | ZEBOV | L-polymerase, VP24 and VP35 | Tekmira Pharmaceuticals | Pre-clinical |
Acknowledgements
References
- Turner, B.G.; Summers, M.F. Structural biology of HIV. J. Mol. Biol. 1999, 285, 1–32. [Google Scholar] [CrossRef]
- Vivet-Boudou, V.; Didierjean, J.; Isel, C.; Marquet, R. Nucleoside and nucleotide inhibitors of HIV-1 replication. Cell Mol. Life Sci. 2006, 63, 163–186. [Google Scholar] [CrossRef]
- Pomerantz, R.J.; Horn, D.L. Twenty years of therapy for HIV-1 infection. Nat. Med. 2003, 9, 867–873. [Google Scholar] [CrossRef]
- Park, N.H.; Pavan-Langston, D.; McLean, S.L. Acylovir in oral and ganglionic herpes simplex virus infections. J. Infect. Dis. 1979, 140, 802–806. [Google Scholar] [CrossRef]
- Reichard, O.; Andersson, J.; Schvarcz, R.; Weiland, O. Ribavirin treatment for chronic hepatitis C. Lancet 1991, 337, 1058–1061. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Targeting HIV: antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002, 23, 381–388. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Das, K.; Hughes, S.H.; Arnold, E. Taking aim at a moving target: designing drugs to inhibit drug-resistant HIV-1 reverse transcriptases. Curr. Opin. Struct. Biol. 2004, 14, 716–730. [Google Scholar] [CrossRef]
- Turner, D.; Wainberg, M.A. HIV transmission and primary drug resistance. AIDS Rev. 2006, 8, 17–23. [Google Scholar]
- Carr, A. Toxicity of antiretroviral therapy and implications for drug development. Nat. Rev. Drug Discov. 2003, 2, 624–634. [Google Scholar] [CrossRef]
- Pinti, M.; Salomoni, P.; Cossarizza, A. Anti-HIV drugs and the mitochondria. Biochim. Biophys. Acta 2006, 1757, 700–707. [Google Scholar]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: an emerging class of therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar]
- Blank, M.; Blind, M. Aptamers as tools for target validation. Curr. Opin. Chem. Biol. 2005, 9, 336–342. [Google Scholar] [CrossRef]
- Gopinath, S.C. Antiviral aptamers. Arch. Virol. 2007, 152, 2137–2157. [Google Scholar] [CrossRef]
- Pan, Q.W.; Henry, S.D.; Scholte, B.J.; Tilanus, H.W.; Janssen, H.L.; van der Laan, L.J. New therapeutic opportunities for hepatitis C based on small RNA. World J. Gastroenterol. 2007, 13, 4431–4436. [Google Scholar] [Green Version]
- Haasnoot, J.; Berkhout, B. Nucleic acids-based therapeutics in the battle against pathogenic viruses. Handb. Exp. Pharmacol. 2009, 189, 243–263. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef]
- Corey, D.R. RNA learns from antisense. Nat. Chem. Biol. 2007, 3, 8–11. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar]
- Sczakiel, G.; Far, R.K. The role of target accessibility for antisense inhibition. Curr. Opin. Mol. Ther. 2002, 4, 149–153. [Google Scholar]
- Pan, W.H.; Clawson, G.A. Identifying accessible sites in RNA: the first step in designing antisense reagents. Curr. Med. Chem. 2006, 13, 3083–3103. [Google Scholar] [CrossRef]
- Sahu, N.K.; Shilakari, G.; Nayak, A.; Kohli, D.V. Antisense technology: a selective tool for gene expression regulation and gene targeting. Curr. Pharm. Biotechnol. 2007, 8, 291–304. [Google Scholar] [CrossRef]
- Patzel, V.; Steidl, U.; Kronenwett, R.; Haas, R.; Sczakiel, G. A theoretical approach to select effective antisense oligodeoxyribonucleotides at high statistical probability. Nucl. Acid. Res. 1999, 27, 4328–4334. [Google Scholar] [CrossRef]
- Far, R.K.; Leppert, J.; Frank, K.; Sczakiel, G. Technical improvements in the computational target search for antisense oligonucleotides. Oligonucleotides 2005, 15, 223–233. [Google Scholar] [Green Version]
- Spurgers, K.B.; Sharkey, C.M.; Warfield, K.L.; Bavari, S. Oligonucleotide antiviral therapeutics: antisense and RNA interference for highly pathogenic RNA viruses. Antivir. Res. 2008, 78, 26–36. [Google Scholar] [Green Version]
- Hnik, P.; Boyer, D.S.; Grillone, L.R.; Clement, J.G.; Henry, S.P.; Green, E.A. Antisense oligonucleotide therapy in diabetic retinopathy. J. Diabetes Sci. Technol. 2009, 3, 924–930. [Google Scholar] [Green Version]
- Seguin, R.M.; Ferrari, N. Emerging oligonucleotide therapies for asthma and chronic obstructive pulmonary disease. Expert Opin. Invest. Drugs 2009, 18, 1505–1517. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. Antisense-mediated modulation of splicing: Therapeutic implications for duchenne muscular dystrophy. RNA Biol. 2010, 7, 453–461. [Google Scholar] [CrossRef]
- Geary, R.S.; Henry, S.P.; Grillone, L.R. Fomivirsen: clinical pharmacology and potential drug interactions. Clin. Pharmacokinet. 2002, 41, 255–260. [Google Scholar] [CrossRef]
- Schreiber, A.; Harter, G.; Schubert, A.; Bunjes, D.; Mertens, T.; Michel, D. Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin. Pharmacother. 2009, 10, 191–209. [Google Scholar] [CrossRef]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R.; Bushman, F.D.; Dropulic, B.; June, C.H. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar]
- Matzen, K.; Elzaouk, L.; Matskevich, A.A.; Nitzsche, A.; Heinrich, J.; Moelling, K. RNase H-mediated retrovirus destruction in vivo triggered by oligodeoxynucleotides. Nat. Biotechnol. 2007, 25, 669–674. [Google Scholar] [CrossRef]
- Witherell, G.W. ISIS-14803 (Isis Pharmaceuticals). Curr. Opin. Invest. Drugs 2001, 2, 1523–1529. [Google Scholar]
- Swenson, D.L.; Warfield, K.L.; Warren, T.K.; Lovejoy, C.; Hassinger, J.N.; Ruthel, G.; Blouch, R.E.; Moulton, H.M.; Weller, D.D.; Iversen, P.L.; Bavari, S. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob. Agents Chemother. 2009, 53, 2089–2099. [Google Scholar]
- Warren, T.K.; Warfield, K.L.; Wells, J.; Swenson, D.L.; Donner, K.S.; Van Tongeren, S.A.; Garza, N.L.; Dong, L.; Mourich, D.V.; Crumley, S.; Nichols, D.K.; Iversen, P.L.; Bavari, S. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat. Med. 2010, 16, 991–994. [Google Scholar] [CrossRef]
- Lilley, D.M. Structure, folding and mechanisms of ribozymes. Curr. Opin. Struct. Biol. 2005, 15, 313–323. [Google Scholar] [CrossRef]
- Walter, N.G. Ribozyme catalysis revisited: is water involved? Mol. Cell 2007, 28, 923–929. [Google Scholar] [CrossRef]
- Serganov, A.; Patel, D.J. Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat. Rev. Genet. 2007, 8, 776–790. [Google Scholar] [CrossRef]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Chen, X.; Li, N.; Ellington, A.D. Ribozyme catalysis of metabolism in the RNA world. Chem. Biodivers. 2007, 4, 633–655. [Google Scholar] [CrossRef]
- Strobel, S.A.; Cochrane, J.C. RNA catalysis: ribozymes, ribosomes, and riboswitche. Curr. Opin. Chem. Biol. 2007, 11, 636–643. [Google Scholar] [CrossRef]
- Peracchi, A. Prospects for antiviral ribozymes and deoxyribozymes. Rev. Med. Virol. 2004, 14, 47–64. [Google Scholar] [CrossRef]
- Opalinska, J.B.; Gewirtz, A.M. Nucleic-acid therapeutics: basic principles and recent applications. Nat. Rev. Drug Discov. 2002, 1, 503–514. [Google Scholar] [CrossRef]
- Sullenger, B.A.; Gilboa, E. Emerging clinical applications of RNA. Nature 2002, 418, 252–258. [Google Scholar] [CrossRef]
- Sakamoto, N.; Wu, C.H.; Wu, G.Y. Intracellular cleavage of hepatitis C virus RNA and inhibition of viral protein translation by hammerhead ribozymes. J. Clin. Invest. 1996, 98, 2720–2728. [Google Scholar] [CrossRef]
- Ryu, K.J.; Lee, S.W. Identification of the most accessible sites to ribozymes on the hepatitis C virus internal ribosome entry site. J. Biochem. Mol. Biol. 2003, 36, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Trepanier, J.B.; Tanner, J.E.; Alfieri, C. Oligonucleotide-based therapeutic options against hepatitis C virus infection. Antivir. Ther. 2006, 11, 273–287. [Google Scholar] [Green Version]
- Levesque, M.V.; Levesque, D.; Briere, F.P.; Perreault, J.P. Investigating a new generation of ribozymes in order to target HCV. PLoS One 2010, 5, e9627. [Google Scholar] [Green Version]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; Akil, B.; Khanlou, H.; Finlayson, R.; McFarlane, R.; Smith, D.E.; Garsia, R.; Ma, D.; Law, M.; Murray, J.M.; von Kalle, C.; Ely, J.A.; Patino, S.M.; Knop, A.E.; Wong, P.; Todd, A.V.; Haughton, M.; Fuery, C.; Macpherson, J.L.; Symonds, G.P.; Evans, L.A.; Pond, S.M.; Cooper, D.A. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [Green Version]
- Burnett, J.C.; Rossi, J.J. Stem cells, ribozymes and HIV. Gene Ther. 2009, 16, 1178–1179. [Google Scholar] [CrossRef]
- Müller-Kuller, T.; Capalbo, G.; Klebba, C.; Engels, J.W.; Klein, S.A. Identification and characterization of a highly efficient anti-HIV pol hammerhead ribozyme. Oligonucleotides 2009, 19, 265–272. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar]
- Cullen, B.R. Five questions about viruses and microRNAs. PLoS Pathog. 2010, 6, e1000787. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- Okamura, K.; Phillips, M.D.; Tyler, D.M.; Duan, H.; Chou, Y.T.; Lai, E.C. The regulatory activity of microRNA* species has substantial influence on microRNA and 3' UTR evolution. Nat. Struct. Mol. Biol. 2008, 15, 354–363. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar]
- Castanotto, D.; Rossi, J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009, 457, 426–433. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Mack, G.S. MicroRNA gets down to business. Nat. Biotechnol. 2007, 25, 631–638. [Google Scholar] [CrossRef]
- Mattes, J.; Yang, M.; Foster, P.S. Regulation of microRNA by antagomirs: a new class of pharmacological antagonists for the specific regulation of gene function? Am. J. Respir. Cell Mol. Biol. 2007, 36, 8–12. [Google Scholar]
- Reshmi, G.; Pillai, M.R. Beyond HPV: oncomirs as new players in cervical cancer. FEBS Lett. 2008, 582, 4113–4116. [Google Scholar] [CrossRef]
- Bala, S.; Marcos, M.; Szabo, G. Emerging role of microRNAs in liver diseases. World J. Gastroenterol. 2009, 15, 5633–5640. [Google Scholar] [CrossRef]
- Esau, C.; Kang, X.; Peralta, E.; Hanson, E.; Marcusson, E.G.; Ravichandran, L.V.; Sun, Y.; Koo, S.; Perera, R.J.; Jain, R.; Dean, N.M.; Freier, S.M.; Bennett, C.F.; Lollo, B.; Griffey, R. MicroRNA-143 regulates adipocyte differentiation. J. Biol. Chem. 2004, 279, 52361–52365. [Google Scholar]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with 'antagomirs'. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Weiler, J.; Hunziker, J.; Hall, J. Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther. 2006, 13, 496–502. [Google Scholar] [CrossRef]
- Stenvang, J.; Kauppinen, S. MicroRNAs as targets for antisense-based therapeutics. Expert Opin. Biol. Ther. 2008, 8, 59–81. [Google Scholar] [CrossRef]
- Mattes, J.; Collison, A.; Foster, P.S. Emerging role of microRNAs in disease pathogenesis and strategies for therapeutic modulation. Curr. Opin. Mol. Ther. 2008, 10, 150–157. [Google Scholar]
- Petri, A.; Lindow, M.; Kauppinen, S. MicroRNA silencing in primates: towards development of novel therapeutics. Cancer Res. 2009, 69, 393–395. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar]
- Moens, U. Silencing viral microRNA as a novel antiviral therapy? J. Biomed. Biotechnol. 2009. Article ID 419539. [Google Scholar]
- He, S.; Yang, Z.; Skogerbo, G.; Ren, F.; Cui, H.; Zhao, H.; Chen, R.; Zhao, Y. The properties and functions of virus encoded microRNA, siRNA, and other small noncoding RNA. Crit. Rev. Microbiol. 2008, 34, 175–188. [Google Scholar] [CrossRef]
- Chang, J.; Guo, J.T.; Jiang, D.; Guo, H.; Taylor, J.M.; Block, T.M. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J. Virol. 2008, 82, 8215–8223. [Google Scholar]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar]
- Randall, G.; Panis, M.; Cooper, J.D.; Tellinghuisen, T.L.; Sukhodolets, K.E.; Pfeffer, S.; Landthaler, M.; Landgraf, P.; Kan, S.; Lindenbach, B.D.; Chien, M.; Weir, D.B.; Russo, J.J.; Ju, J.; Brownstein, M.J.; Sheridan, R.; Sander, C.; Zavolan, M.; Tuschl, T.; Rice, C.M. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. USA 2007, 104, 12884–12889. [Google Scholar]
- Elmen, J.; Lindow, M.; Silahtaroglu, A.; Bak, M.; Christensen, M.; Lind-Thomsen, A.; Hedtjarn, M.; Hansen, J.B.; Hansen, H.F.; Straarup, E.M.; McCullagh, K.; Kearney, P.; Kauppinen, S. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucl. Acid. Res. 2008, 36, 1153–1162. [Google Scholar]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; Gullans, S.; Kearney, P.; Sarnow, P.; Straarup, E.M.; Kauppinen, S. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar]
- Amarzguioui, M.; Rossi, J.J.; Kim, D. Approaches for chemically synthesized siRNA and vector-mediated RNAi. FEBS Lett. 2005, 579, 5974–5981. [Google Scholar] [CrossRef]
- Song, E.; Lee, S.K.; Wang, J.; Ince, N.; Ouyang, N.; Min, J.; Chen, J.; Shankar, P.; Lieberman, J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 2003, 9, 347–351. [Google Scholar]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef]
- Kretschmer-Kazemi, F.R.; Sczakiel, G. The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucl. Acid. Res. 2003, 31, 4417–4424. [Google Scholar] [CrossRef]
- Overhoff, M.; Alken, M.; Far, R.K.; Lemaitre, M.; Lebleu, B.; Sczakiel, G.; Robbins, I. Local RNA target structure influences siRNA efficacy: a systematic global analysis. J. Mol. Biol. 2005, 348, 871–881. [Google Scholar]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar]
- Amarzguioui, M.; Lundberg, P.; Cantin, E.; Hagstrom, J.; Behlke, M.A.; Rossi, J.J. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat. Protoc. 2006, 1, 508–517. [Google Scholar]
- Muhonen, P.; Holthofer, H. Bioinformatic approaches to siRNA selection and optimization. Methods Mol. Biol. 2010, 623, 93–107. [Google Scholar] [CrossRef]
- Mittal, V. Improving the efficiency of RNA interference in mammals. Nat. Rev. Genet. 2004, 5, 355–365. [Google Scholar]
- Moschos, S.A.; Spinks, K.; Williams, A.E.; Lindsay, M.A. Targeting the lung using siRNA and antisense based oligonucleotides. Curr. Pharm. Des. 2008, 14, 3620–3627. [Google Scholar]
- Bitko, V.; Musiyenko, A.; Shulyayeva, O.; Barik, S. Inhibition of respiratory viruses by nasally administered siRNA. Nat. Med. 2005, 11, 50–55. [Google Scholar] [CrossRef]
- DeVincenzo, J.; Cehelsky, J.E.; Alvarez, R.; Elbashir, S.; Harborth, J.; Toudjarska, I.; Nechev, L.; Murugaiah, V.; Van Vliet, A.; Vaishnaw, A.K.; Meyers, R. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antivir. Res. 2008, 77, 225–231. [Google Scholar]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef]
- Westerhout, E.M.; Ooms, M.; Vink, M.; Das, A.T.; Berkhout, B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucl. Acid. Res. 2005, 33, 796–804. [Google Scholar]
- ter Brake, O.; 't Hooft, K.; Liu, Y.P.; Centlivre, M.; von Eije, K.J.; Berkhout, B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol. Ther. 2008, 16, 557–564. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Lee, A.C.; Robbins, M.; Geisbert, J.B.; Honko, A.N.; Sood, V.; Johnson, J.C.; de Jong, S.; Tavakoli, I.; Judge, A.; Hensley, L.E.; Maclachlan, I. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. Lancet 2010, 375, 1896–1905. [Google Scholar]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; Vaish, N.; Zinnen, S.; Vargeese, C.; Bowman, K.; Shaffer, C.S.; Jeffs, L.B.; Judge, A.; Maclachlan, I.; Polisky, B. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar]
- Rossi, J.J. RNAi therapeutics: SNALPing siRNAs in vivo. Gene Ther. 2006, 13, 583–584. [Google Scholar] [CrossRef]
- Nowak, M.; Wyszko, E.; Fedoruk-Wyszomirska, A.; Pospieszny, H.; Barciszewska, M.Z.; Barciszewski, J. A new and efficient method for inhibition of RNA viruses by DNA interference. FEBS J. 2009, 276, 4372–4380. [Google Scholar]
- Sullenger, B.A.; Gallardo, H.F.; Ungers, G.E.; Gilboa, E. Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell 1990, 63, 601–608. [Google Scholar] [CrossRef]
- Sullenger, B.A.; Gallardo, H.F.; Ungers, G.E.; Gilboa, E. Analysis of trans-acting response decoy RNA-mediated inhibition of human immunodeficiency virus type 1 transactivation. J. Virol. 1991, 65, 6811–6816. [Google Scholar]
- Bohjanen, P.R.; Colvin, R.A.; Puttaraju, M.; Been, M.D.; Garcia-Blanco, M.A. A small circular TAR RNA decoy specifically inhibits Tat-activated HIV-1 transcription. Nucl. Acid. Res. 1996, 24, 3733–3738. [Google Scholar] [CrossRef]
- Anderson, J.; Li, M.J.; Palmer, B.; Remling, L.; Li, S.; Yam, P.; Yee, J.K.; Rossi, J.; Zaia, J.; Akkina, R. Safety and efficacy of a lentiviral vector containing three anti-HIV genes (CCR5 ribozyme, tat-rev siRNA, and TAR decoy) in SCID-hu mouse-derived T cells. Mol. Ther. 2007, 15, 1182–1188. [Google Scholar]
- Anderson, J.S.; Javien, J.; Nolta, J.A.; Bauer, G. Preintegration HIV-1 inhibition by a combination lentiviral vector containing a chimeric TRIM5 alpha protein, a CCR5 shRNA, and a TAR decoy. Mol. Ther. 2009, 17, 2103–2114. [Google Scholar] [CrossRef]
- Bahner, I.; Kearns, K.; Hao, Q.L.; Smogorzewska, E.M.; Kohn, D.B. Transduction of human CD34+ hematopoietic progenitor cells by a retroviral vector expressing an RRE decoy inhibits human immunodeficiency virus type 1 replication in myelomonocytic cells produced in long-term culture. J. Virol. 1996, 70, 4352–4360. [Google Scholar]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; Brody, K.; Fox, S.; Haden, E.; Wilson, K.; Salata, C.; Dolan, C.; Wetter, C.; Aguilar-Cordova, E.; Church, J. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar]
- Wyatt, J.R.; Vickers, T.A.; Roberson, J.L.; Buckheit, R.W., Jr.; Klimkait, T.; DeBaets, E.; Davis, P.W.; Rayner, B.; Imbach, J.L.; Ecker, D.J. Combinatorially selected guanosine-quartet structure is a potent inhibitor of human immunodeficiency virus envelope-mediated cell fusion. Proc. Natl. Acad. Sci. USA 1994, 91, 1356–1360. [Google Scholar]
- Pinskaya, M.; Romanova, E.; Volkov, E.; Deprez, E.; Leh, H.; Brochon, J.C.; Mouscadet, J.F.; Gottikh, M. HIV-1 integrase complexes with DNA dissociate in the presence of short oligonucleotides conjugated to acridine. Biochemistry 2004, 43, 8735–8743. [Google Scholar]
- Mescalchin, A.; Wünsche, W.; Laufer, S.D.; Grohmann, D.; Restle, T.; Sczakiel, G. Specific binding of a hexanucleotide to HIV-1 reverse transcriptase: a novel class of bioactive molecules. Nucl. Acid. Res. 2006, 34, 5631–5637. [Google Scholar] [CrossRef]
- Mescalchin, A.; Wünsche, W.; Sczakiel, G. Specific recognition of proteins by array-bound hexanucleotides. Angew. Chem. Int. Ed. Engl. 2011. In Press. [Google Scholar]
- Bunka, D.H.; Stockley, P.G. Aptamers come of age - at last. Nat. Rev. Microbiol. 2006, 4, 588–596. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Morris, K.N.; Jensen, K.B.; Julin, C.M.; Weil, M.; Gold, L. High affinity ligands from in vitro selection: complex targets. Proc. Natl. Acad. Sci. USA 1998, 95, 2902–2907. [Google Scholar]
- Patel, D.J.; Suri, A.K. Structure, recognition and discrimination in RNA aptamer complexes with cofactors, amino acids, drugs and aminoglycoside antibiotics. J. Biotechnol. 2000, 74, 39–60. [Google Scholar]
- Hermann, T.; Patel, D.J. Adaptive recognition by nucleic acid aptamers. Science 2000, 287, 820–825. [Google Scholar] [CrossRef]
- Niazi, J.H.; Lee, S.J.; Gu, M.B. Single-stranded DNA aptamers specific for antibiotics tetracyclines. Bioorg. Med. Chem. 2008, 16, 7245–7253. [Google Scholar] [CrossRef]
- Dausse, E.; Da Rocha, G.S.; Toulme, J.J. Aptamers: a new class of oligonucleotides in the drug discovery pipeline? Curr. Opin. Pharmacol. 2009, 9, 602–607. [Google Scholar] [CrossRef]
- Jayasena, S.D. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar]
- Cerchia, L.; Hamm, J.; Libri, D.; Tavitian, B.; de Franciscis, V. Nucleic acid aptamers in cancer medicine. FEBS Lett. 2002, 528, 12–16. [Google Scholar]
- Proske, D.; Blank, M.; Buhmann, R.; Resch, A. Aptamers--basic research, drug development, and clinical application. Appl. Microbiol. Biotechnol. 2005, 69, 367–374. [Google Scholar]
- Famulok, M. Allosteric aptamers and aptazymes as probes for screening approaches. Curr. Opin. Mol. Ther. 2005, 7, 137–143. [Google Scholar]
- Que-Gewirth, N.S.; Sullenger, B.A. Gene therapy progress and prospects: RNA aptamers. Gene Ther. 2007, 14, 283–291. [Google Scholar] [CrossRef]
- Ulrich, H.; Trujillo, C.A.; Nery, A.A.; Alves, J.M.; Majumder, P.; Resende, R.R.; Martins, A.H. DNA and RNA aptamers: from tools for basic research towards therapeutic applications. Comb. Chem. High Throughput. Screen. 2006, 9, 619–632. [Google Scholar] [CrossRef]
- Gold, L. RNA as the catalyst for drug screening. Nat. Biotechnol. 2002, 20, 671–672. [Google Scholar] [CrossRef]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar]
- Srisawat, C.; Engelke, D.R. Selection of RNA aptamers that bind HIV-1 LTR DNA duplexes: strand invaders. Nucl. Acid. Res. 2010, 38, 8306–8315. [Google Scholar] [CrossRef]
- Duconge, F.; Toulme, J.J. In vitro selection identifies key determinants for loop-loop interactions: RNA aptamers selective for the TAR RNA element of HIV-1. RNA 1999, 5, 1605–1614. [Google Scholar] [CrossRef]
- Boiziau, C.; Dausse, E.; Yurchenko, L.; Toulme, J.J. DNA aptamers selected against the HIV-1 trans-activation-responsive RNA element form RNA-DNA kissing complexes. J. Biol. Chem. 1999, 274, 12730–12737. [Google Scholar]
- Sekkai, D.; Dausse, E.; Di Primo, C.; Darfeuille, F.; Boiziau, C.; Toulme, J.J. In vitro selection of DNA aptamers against the HIV-1 TAR RNA hairpin. Antisense Nucleic Acid Drug Dev. 2002, 12, 265–274. [Google Scholar] [CrossRef]
- Kolb, G.; Reigadas, S.; Castanotto, D.; Faure, A.; Ventura, M.; Rossi, J.J.; Toulme, J.J. Endogenous expression of an anti-TAR aptamer reduces HIV-1 replication. RNA Biol. 2006, 3, 150–156. [Google Scholar]
- Watrin, M.; Von Pelchrzim, F.; Dausse, E.; Schroeder, R.; Toulme, J.J. In vitro selection of RNA aptamers derived from a genomic human library against the TAR RNA element of HIV-1. Biochemistry 2009, 48, 6278–6284. [Google Scholar]
- Kikuchi, K.; Umehara, T.; Fukuda, K.; Kuno, A.; Hasegawa, T.; Nishikawa, S. A hepatitis C virus (HCV) internal ribosome entry site (IRES) domain III-IV-targeted aptamer inhibits translation by binding to an apical loop of domain IIId. Nucl. Acid. Res. 2005, 33, 683–692. [Google Scholar] [CrossRef]
- Kikuchi, K.; Umehara, T.; Nishikawa, F.; Fukuda, K.; Hasegawa, T.; Nishikawa, S. Increased inhibitory ability of conjugated RNA aptamers against the HCV IRES. Biochem. Biophys. Res. Commun. 2009, 386, 118–123. [Google Scholar]
- Konno, K.; Fujita, S.; Iizuka, M.; Nishikawa, S.; Hasegawa, T.; Fukuda, K. Isolation and characterization of RNA aptamers specific for the HCV minus-IRES domain I. Nucl. Acid. Symp. Ser. 2008, 52, 493–494. [Google Scholar]
- Schneider, D.J.; Feigon, J.; Hostomsky, Z.; Gold, L. High-affinity ssDNA inhibitors of the reverse transcriptase of type 1 human immunodeficiency virus. Biochemistry 1995, 34, 9599–9610. [Google Scholar] [CrossRef]
- Burke, D.H.; Scates, L.; Andrews, K.; Gold, L. Bent pseudoknots and novel RNA inhibitors of type 1 human immunodeficiency virus (HIV-1) reverse transcriptase. J. Mol. Biol. 1996, 264, 650–666. [Google Scholar]
- Andreola, M.L.; Pileur, F.; Calmels, C.; Ventura, M.; Tarrago-Litvak, L.; Toulme, J.J.; Litvak, S. DNA aptamers selected against the HIV-1 RNase H display in vitro antiviral activity. Biochemistry 2001, 40, 10087–10094. [Google Scholar]
- Joshi, P.; Prasad, V.R. Potent inhibition of human immunodeficiency virus type 1 replication by template analog reverse transcriptase inhibitors derived by SELEX (systematic evolution of ligands by exponential enrichment). J. Virol. 2002, 76, 6545–6557. [Google Scholar] [CrossRef]
- Hannoush, R.N.; Min, K.L.; Damha, M.J. Diversity-oriented solid-phase synthesis and biological evaluation of oligonucleotide hairpins as HIV-1 RT RNase H inhibitors. Nucl. Acid. Res. 2004, 32, 6164–6175. [Google Scholar] [CrossRef]
- Zhang, Z.; Blank, M.; Schluesener, H.J. Nucleic acid aptamers in human viral disease. Arch. Immunol. Ther. Exp. 2004, 52, 307–315. [Google Scholar] [Green Version]
- Toulmé, J.J.; Di Primo, C.; Boucard, D. Regulating eukaryotic gene expression with aptamers. FEBS Lett. 2004, 567, 55–62. [Google Scholar] [Green Version]
- DeStefano, J.J.; Cristofaro, J.V. Selection of primer-template sequences that bind human immunodeficiency virus reverse transcriptase with high affinity. Nucl. Acid. Res. 2006, 34, 130–139. [Google Scholar] [CrossRef]
- Tarrago-Litvak, L.; Andreola, M.L.; Nevinsky, G.A.; Sarih-Cottin, L.; Litvak, S. The reverse transcriptase of HIV-1: from enzymology to therapeutic intervention. FASEB J. 1994, 8, 497–503. [Google Scholar] [Green Version]
- Tuerk, C.; MacDougal, S.; Gold, L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 1992, 89, 6988–6992. [Google Scholar] [Green Version]
- Jaeger, J.; Restle, T.; Steitz, T.A. The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. EMBO J. 1998, 17, 4535–4542. [Google Scholar] [CrossRef]
- Kensch, O.; Connolly, B.A.; Steinhoff, H.J.; McGregor, A.; Goody, R.S.; Restle, T. HIV-1 reverse transcriptase-pseudoknot RNA aptamer interaction has a binding affinity in the low picomolar range coupled with high specificity. J. Biol. Chem. 2000, 275, 18271–18278. [Google Scholar]
- Chaloin, L.; Lehmann, M.J.; Sczakiel, G.; Restle, T. Endogenous expression of a high-affinity pseudoknot RNA aptamer suppresses replication of HIV-1. Nucl. Acid. Res. 2002, 30, 4001–4008. [Google Scholar]
- Held, D.M.; Kissel, J.D.; Saran, D.; Michalowski, D.; Burke, D.H. Differential susceptibility of HIV-1 reverse transcriptase to inhibition by RNA aptamers in enzymatic reactions monitoring specific steps during genome replication. J. Biol. Chem. 2006, 281, 25712–25722. [Google Scholar]
- Mayer, G. The chemical biology of aptamers. Angew. Chem. Int. Ed. Engl. 2009, 48, 2672–2689. [Google Scholar] [CrossRef]
- White, R.; Rusconi, C.; Scardino, E.; Wolberg, A.; Lawson, J.; Hoffman, M.; Sullenger, B. Generation of species cross-reactive aptamers using ´toggle´ SELEX. Mol. Ther. 2001, 4, 567–573. [Google Scholar] [CrossRef]
- Padilla, R.; Sousa, R. A Y639F/H784A T7 RNA polymerase double mutant displays superior properties for synthesizing RNAs with non-canonical NTPs. Nucl. Acid. Res. 2002, 30, e138. [Google Scholar] [CrossRef]
- Chelliserrykattil, J.; Ellington, A.D. Evolution of a T7 RNA polymerase variant that transcribes 2'-O-methyl RNA. Nat. Biotechnol. 2004, 22, 1155–1160. [Google Scholar] [CrossRef]
- Keefe, A.D.; Cload, S.T. SELEX with modified nucleotides. Curr. Opin. Chem. Biol. 2008, 12, 448–456. [Google Scholar] [CrossRef]
- Beigelman, L.; McSwiggen, J.A.; Draper, K.G.; Gonzalez, C.; Jensen, K.; Karpeisky, A.M.; Modak, A.S.; Matulic-Adamic, J.; DiRenzo, A.B.; Haeberli, P. Chemical modification of hammerhead ribozymes. Catalytic activity and nuclease resistance. J. Biol. Chem. 1995, 270, 25702–25708. [Google Scholar]
- Cox, J.C.; Rudolph, P.; Ellington, A.D. Automated RNA selection. Biotechnol. Prog. 1998, 14, 845–850. [Google Scholar] [CrossRef]
- Cox, J.C.; Hayhurst, A.; Hesselberth, J.; Bayer, T.S.; Georgiou, G.; Ellington, A.D. Automated selection of aptamers against protein targets translated in vitro: From gene to aptamer. Nucl. Acid. Res. 2002, 30, e108. [Google Scholar]
- Vater, A.; Klussmann, S. Toward third-generation aptamers: Spiegelmers and their therapeutic prospects. Curr. Opin. Drug Discov. Dev. 2003, 6, 253–261. [Google Scholar]
- Ryan, S.M.; Mantovani, G.; Wang, X.; Haddleton, D.M.; Brayden, D.J. Advances in PEGylation of important biotech molecules: delivery aspects. Expert Opin. Drug Deliv. 2008, 5, 371–383. [Google Scholar]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar]
- Laufer, S.D.; Restle, T. Peptide-mediated cellular delivery of oligonucleotide-based therapeutics in vitro: Quantitative evaluation of overall efficacy employing easy to handle reporter systems. Curr. Pharm. Des. 2008, 14, 3637–3655. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar]
- Laufer, S.D.; Detzer, A.; Sczakiel, G.; Restle, T. Selected Strategies for the Delivery of siRNA In vitro and In vivo. In RNA Technologies and Their Applications; Erdmann, V.A., Barciszewski, J., Eds.; Springer-Verlag: Berlin Heidelberg, Germany, 2010; pp. 29–58. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mescalchin, A.; Restle, T. Oligomeric Nucleic Acids as Antivirals. Molecules 2011, 16, 1271-1296. https://doi.org/10.3390/molecules16021271
Mescalchin A, Restle T. Oligomeric Nucleic Acids as Antivirals. Molecules. 2011; 16(2):1271-1296. https://doi.org/10.3390/molecules16021271
Chicago/Turabian StyleMescalchin, Alessandra, and Tobias Restle. 2011. "Oligomeric Nucleic Acids as Antivirals" Molecules 16, no. 2: 1271-1296. https://doi.org/10.3390/molecules16021271
APA StyleMescalchin, A., & Restle, T. (2011). Oligomeric Nucleic Acids as Antivirals. Molecules, 16(2), 1271-1296. https://doi.org/10.3390/molecules16021271