Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers



Abstract
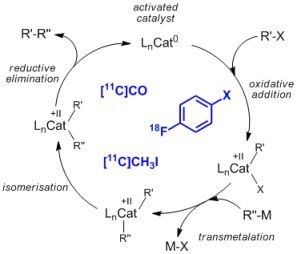
:
1. Introduction
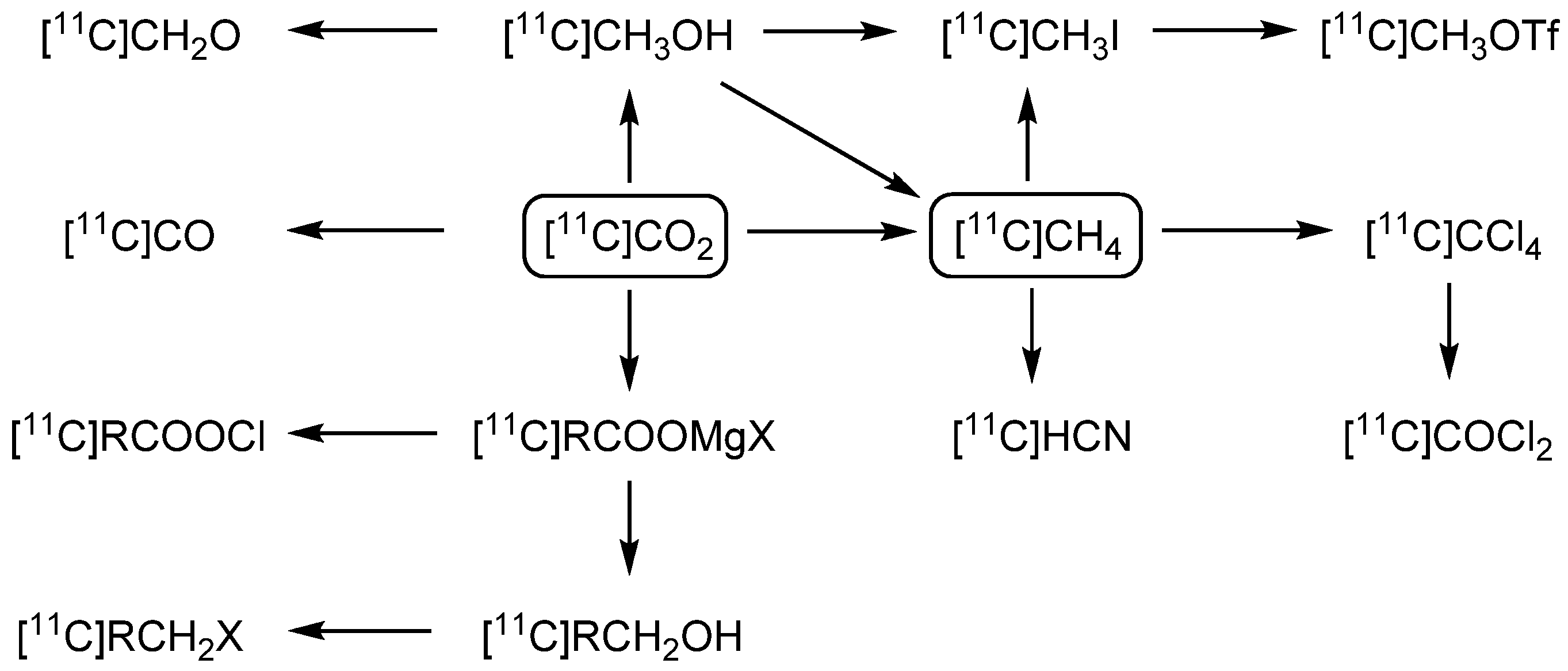
2. Carbon-11 Labeling




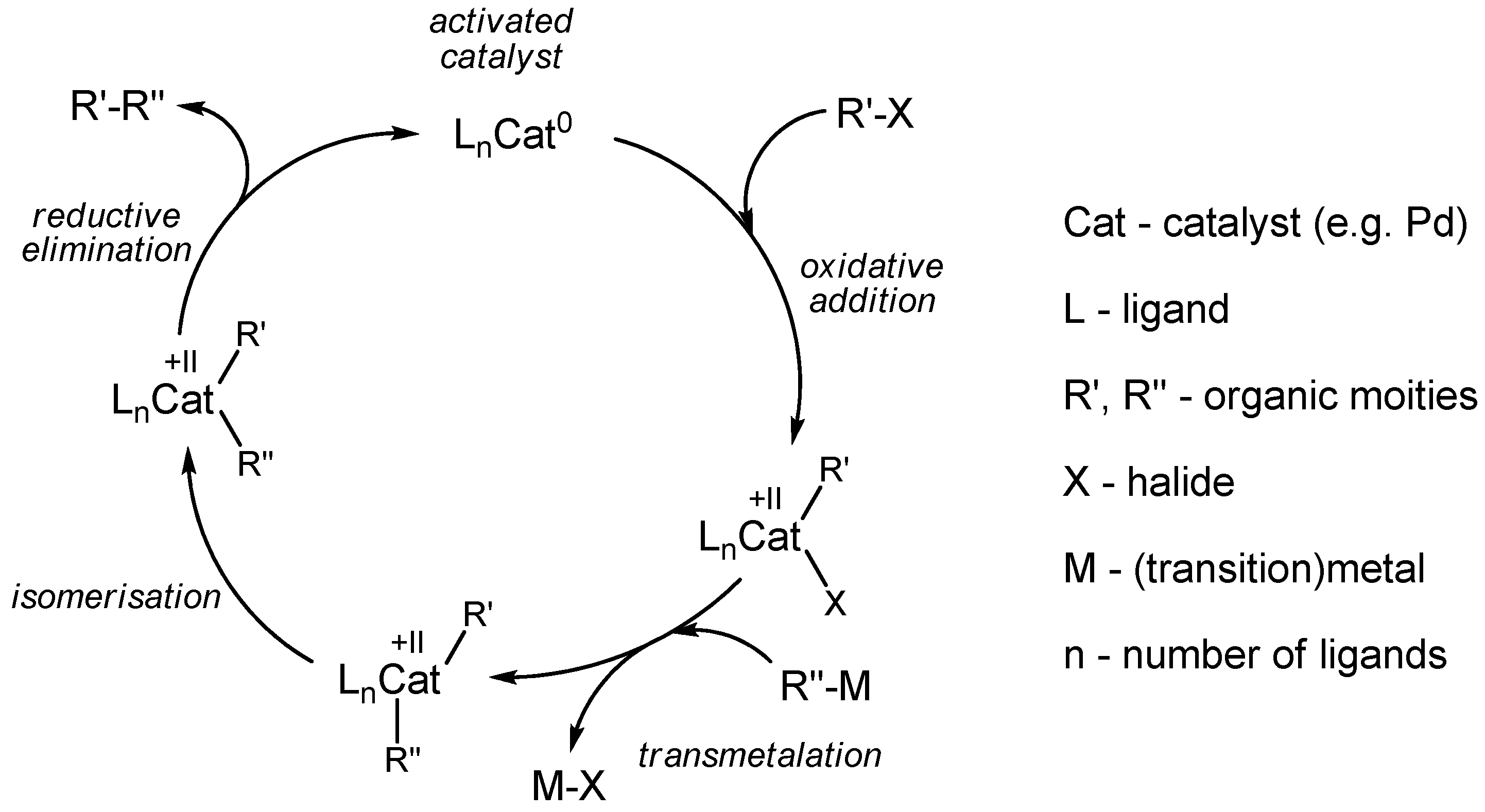
2.1. Cross-coupling reactions with palladium
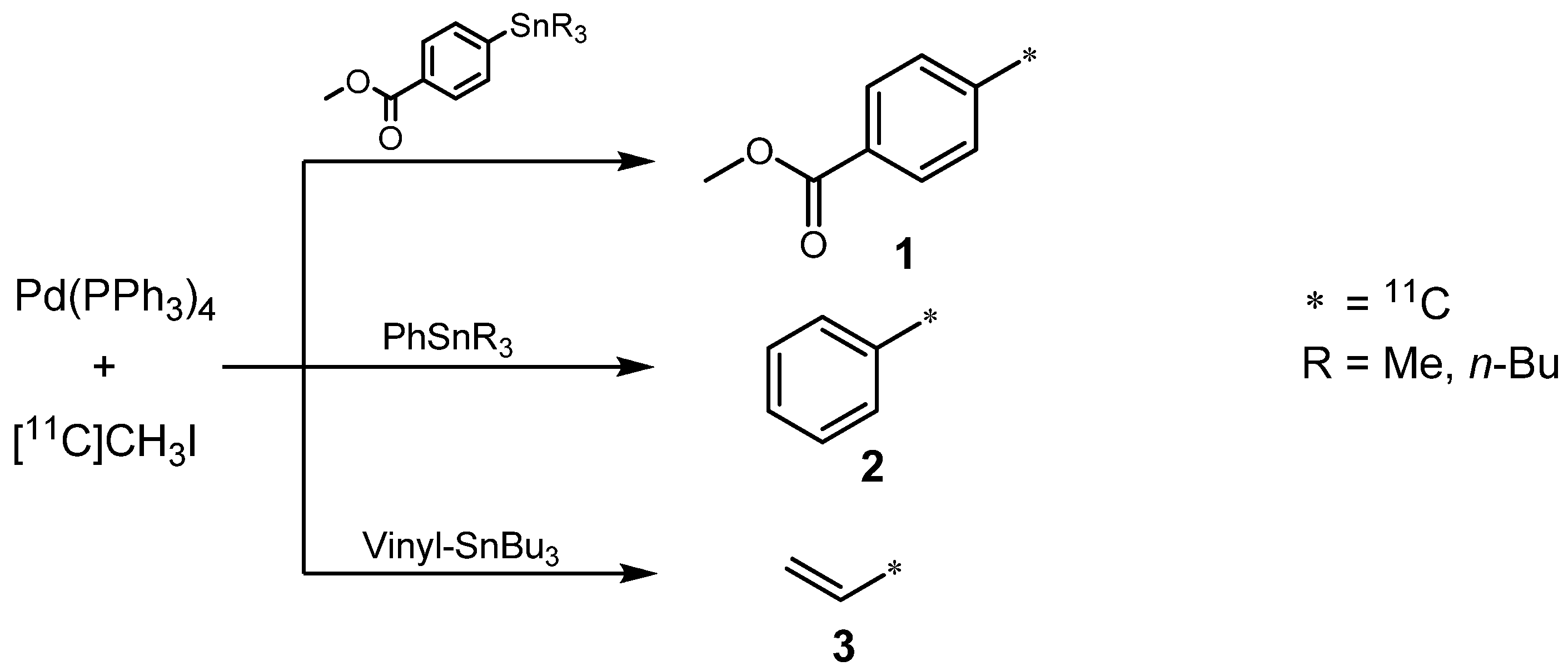
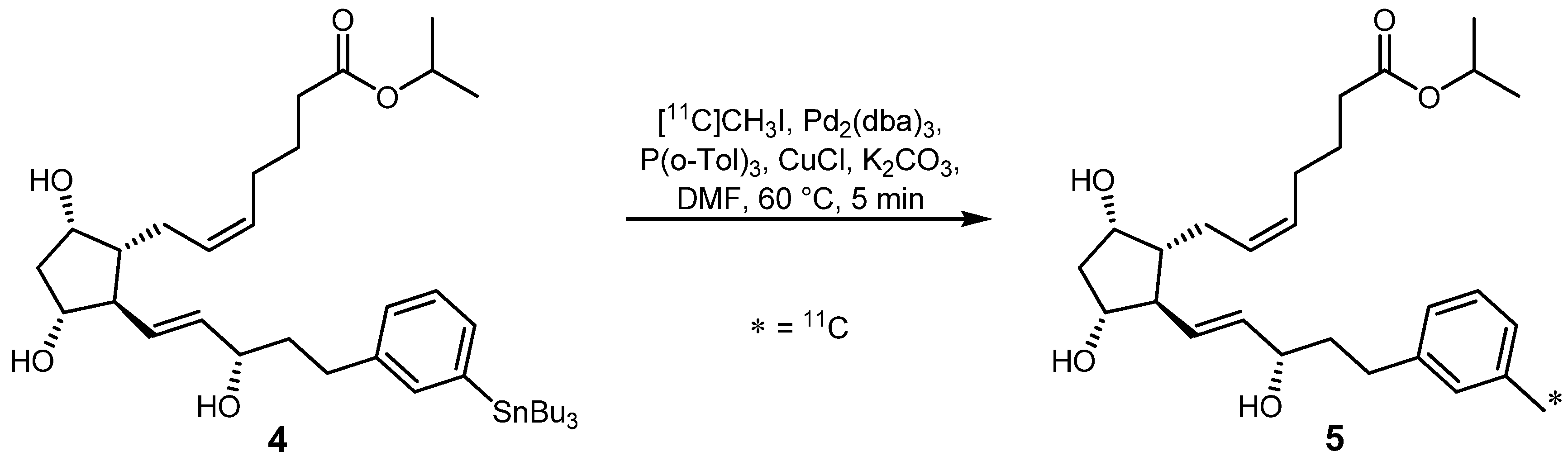
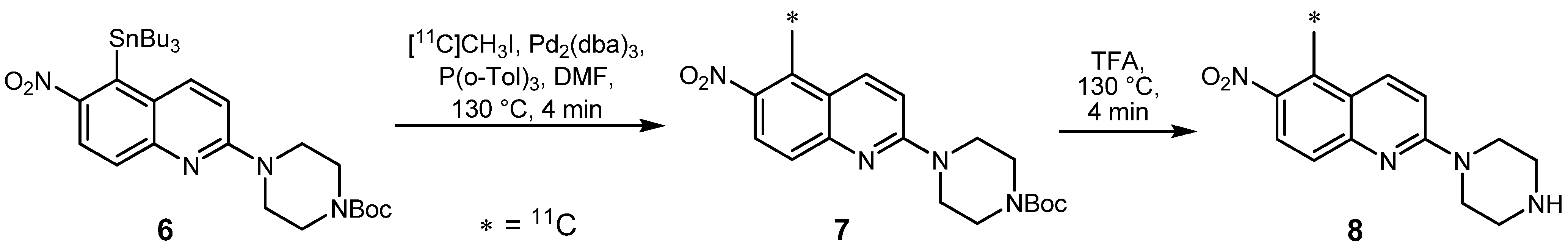
2.1.1. Stille reaction








2.1.2. Suzuki reaction




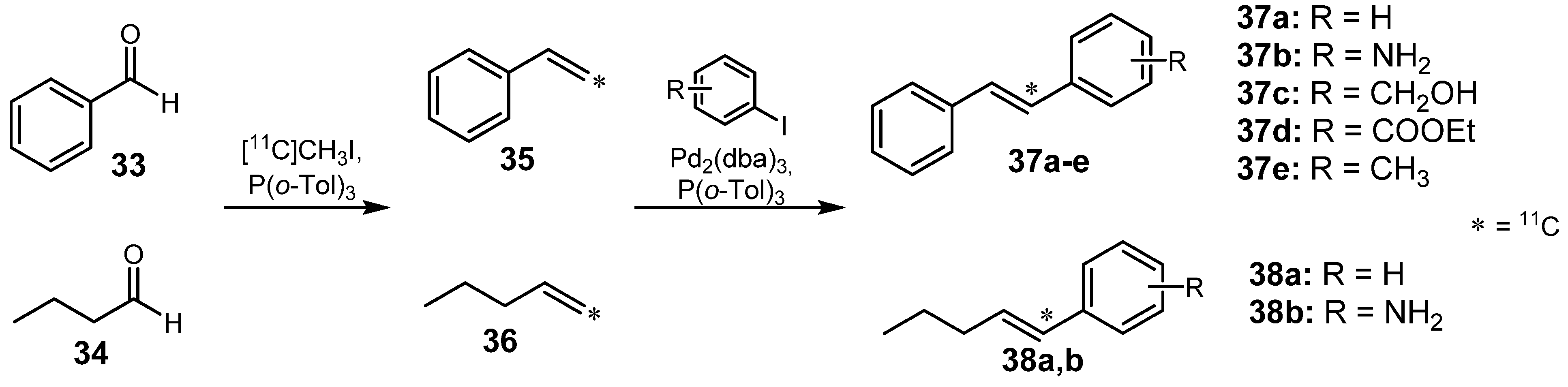
2.1.3. Heck reaction

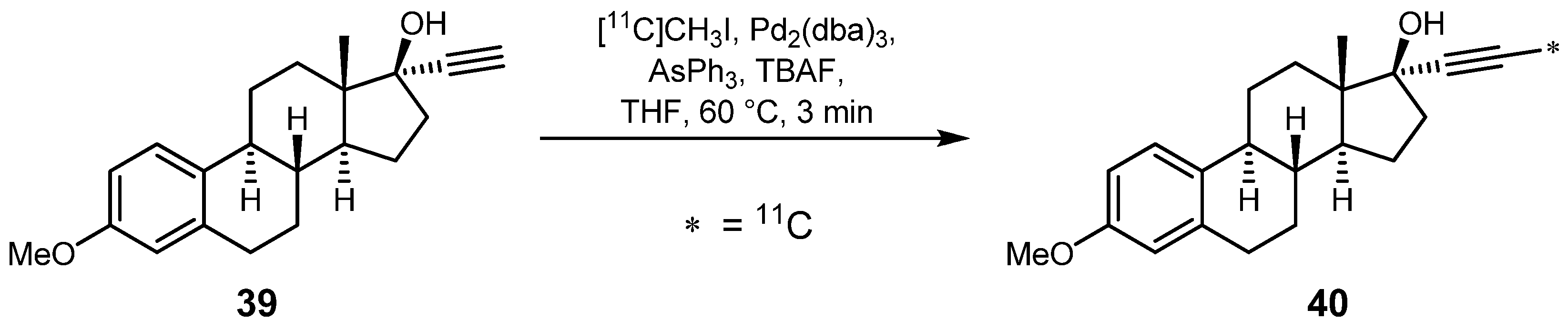
2.1.4. Sonogashira reaction

2.1.5. Miscellaneous cross-coupling reactions






2.2. Cross-coupling reactions with cuprates


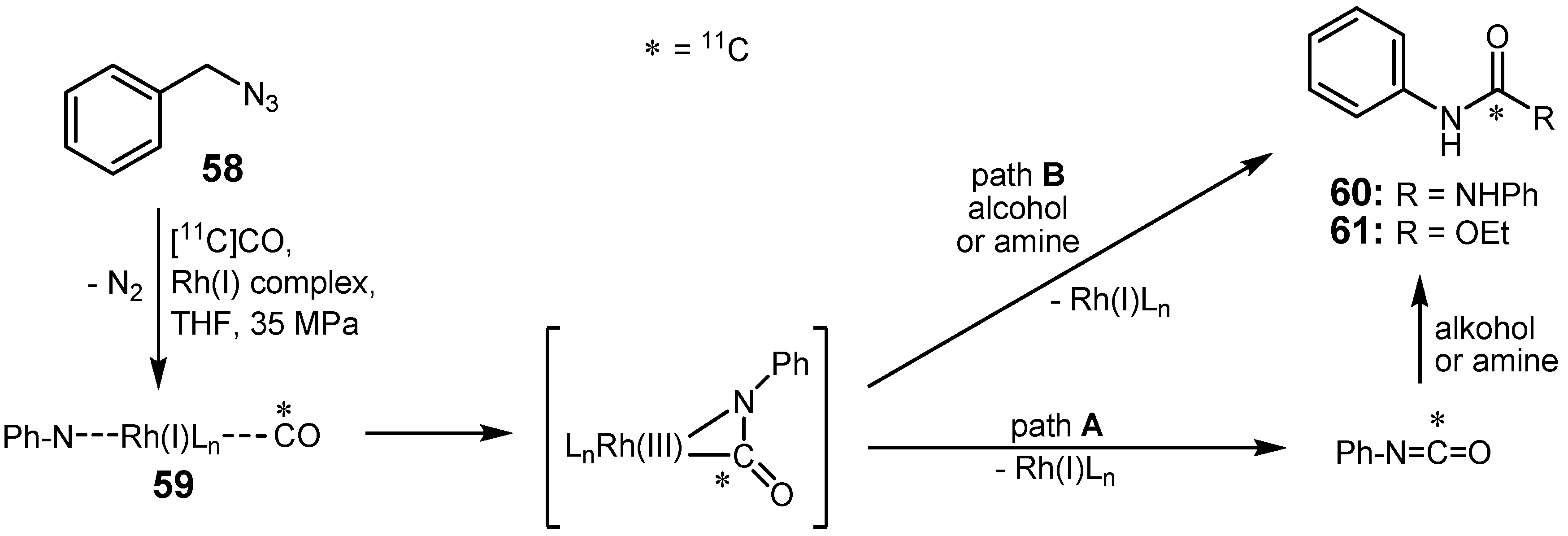
2.3. Cross-coupling reactions with rhodium


3. Fluorine-18 Labeling
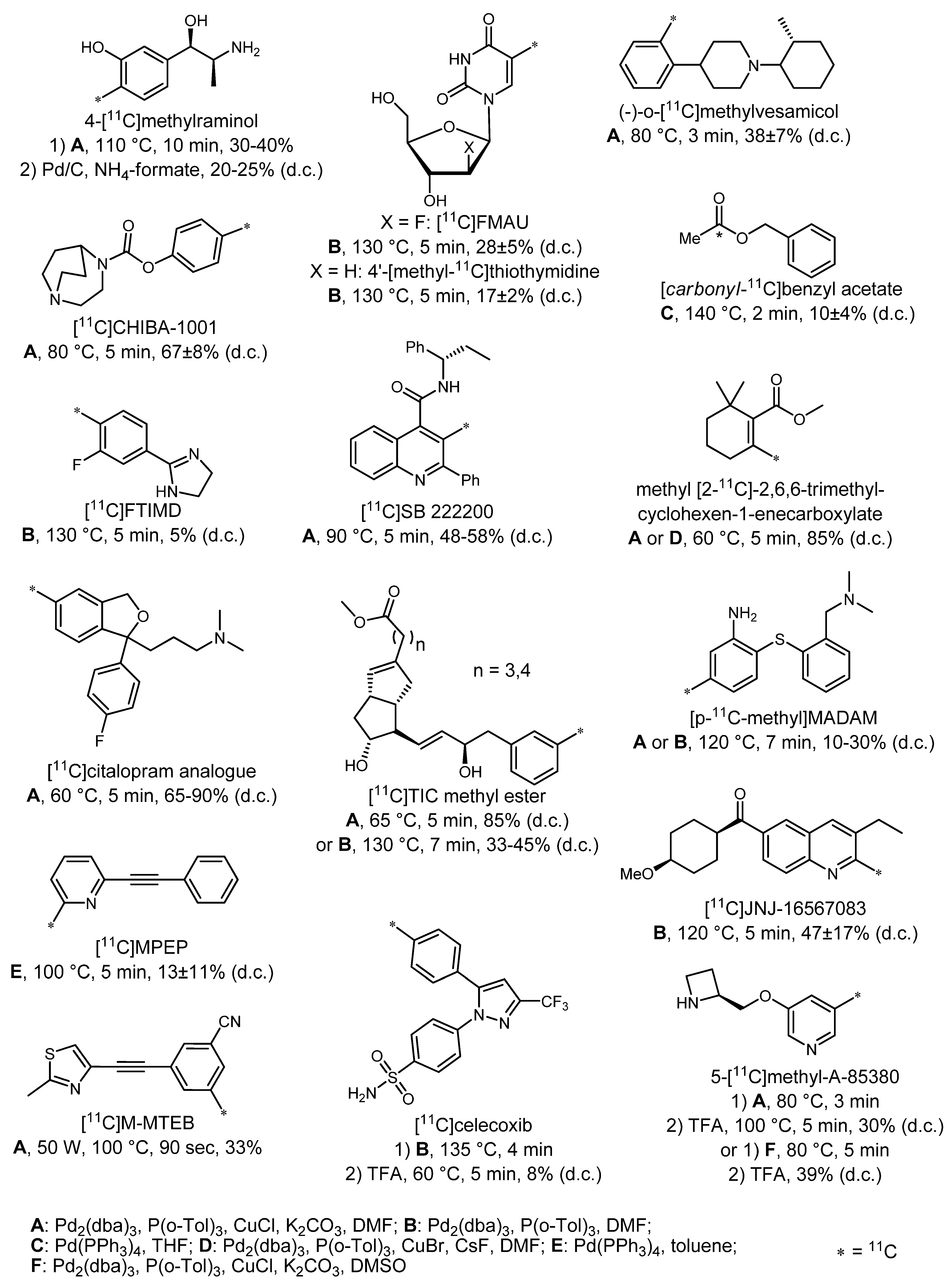
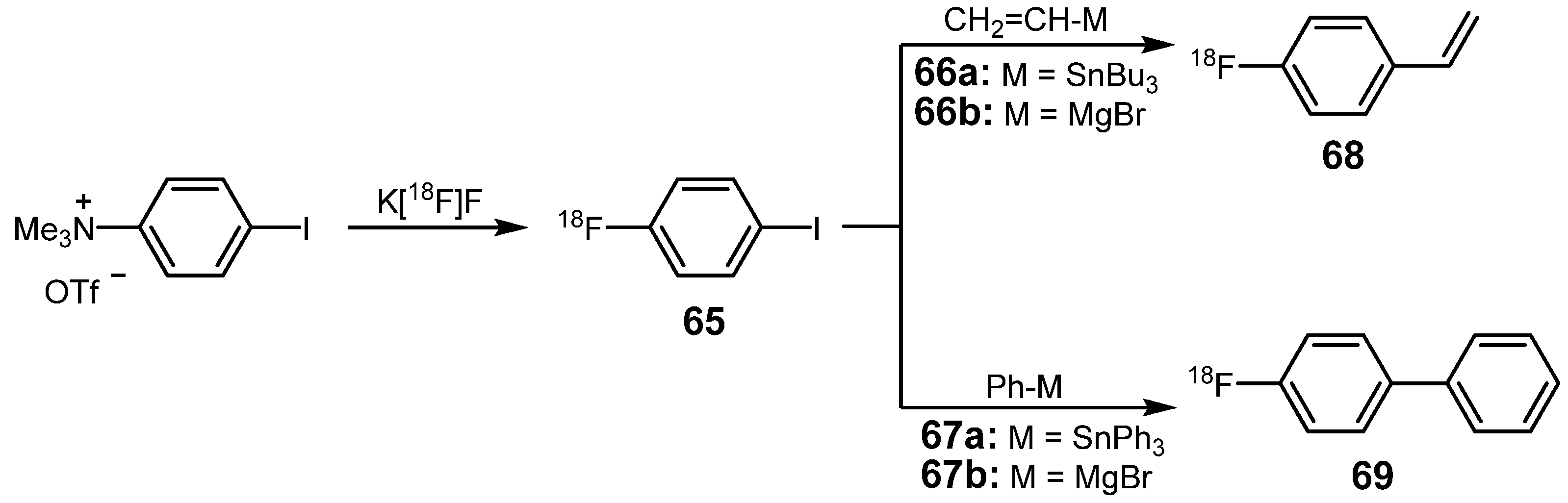
3.1. 18F-labeled compounds prepared via carbon-carbon cross-coupling reactions



{kind=link}
{kind=link}
{kind=link}
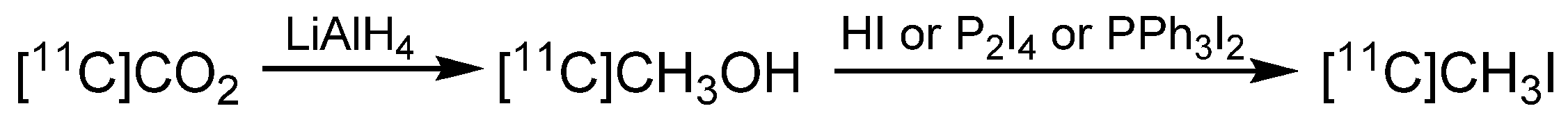{kind=link}
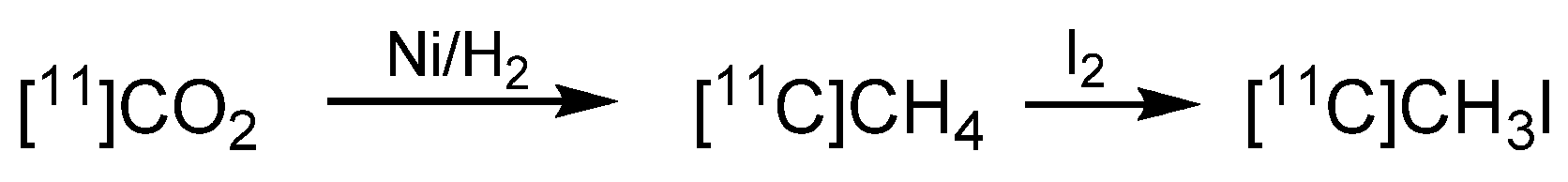{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
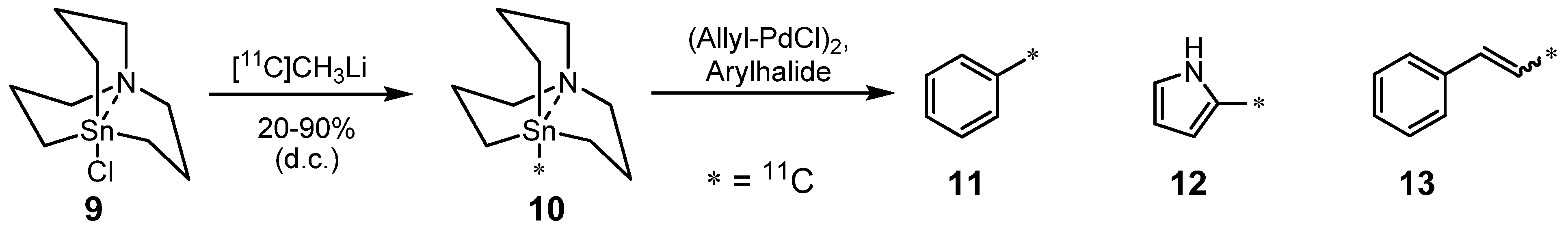{kind=link}
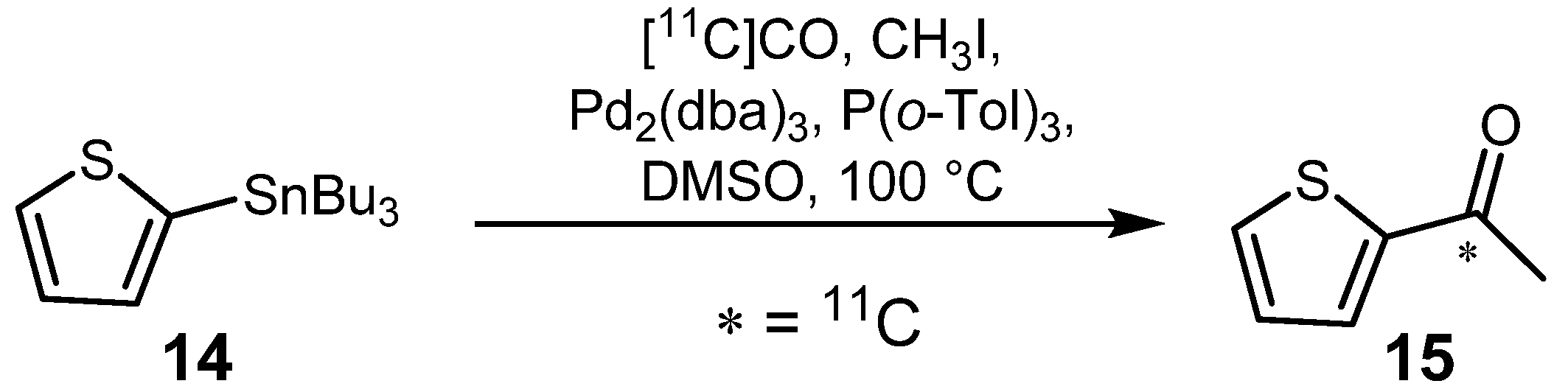{kind=link}
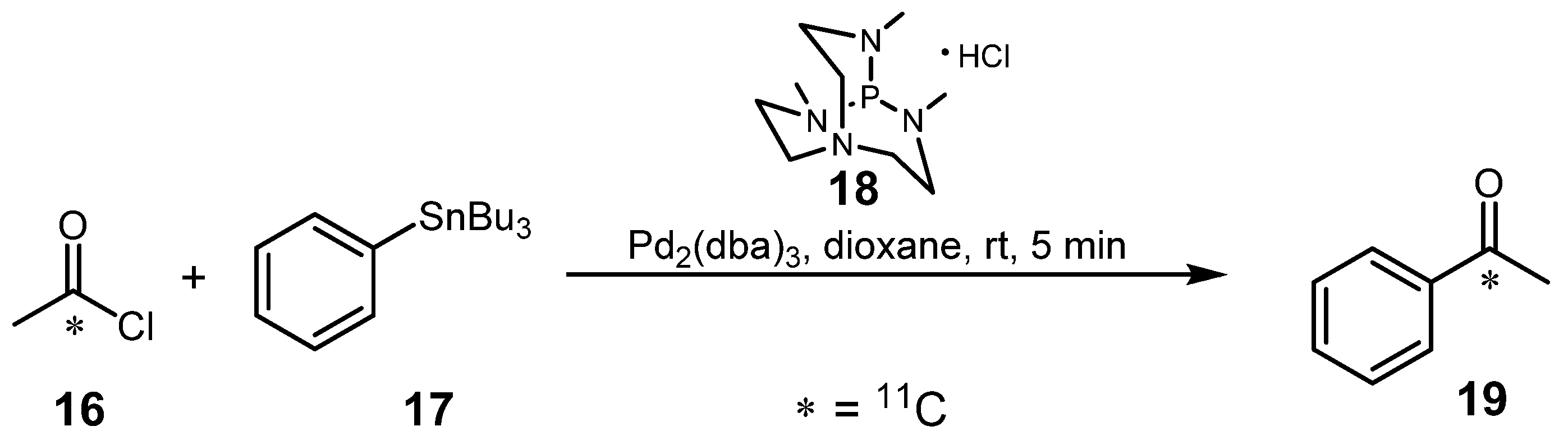{kind=link}
{kind=link}
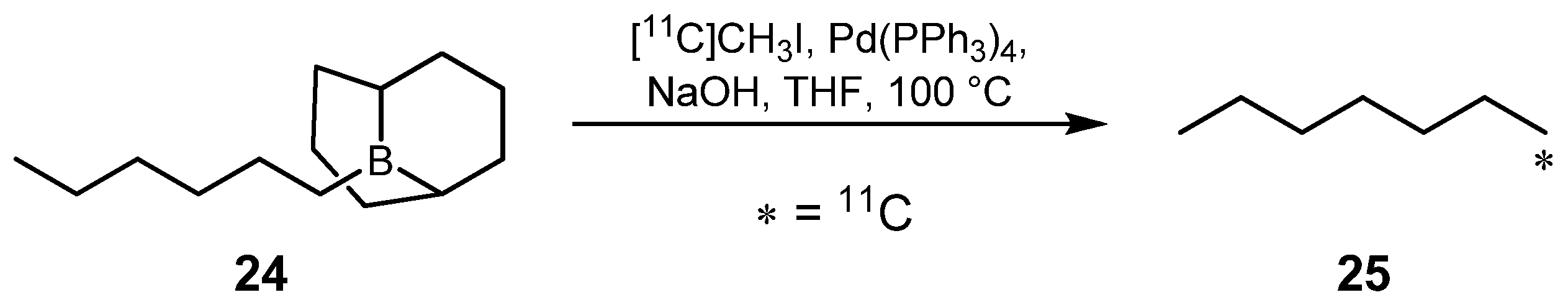{kind=link}
{kind=link}
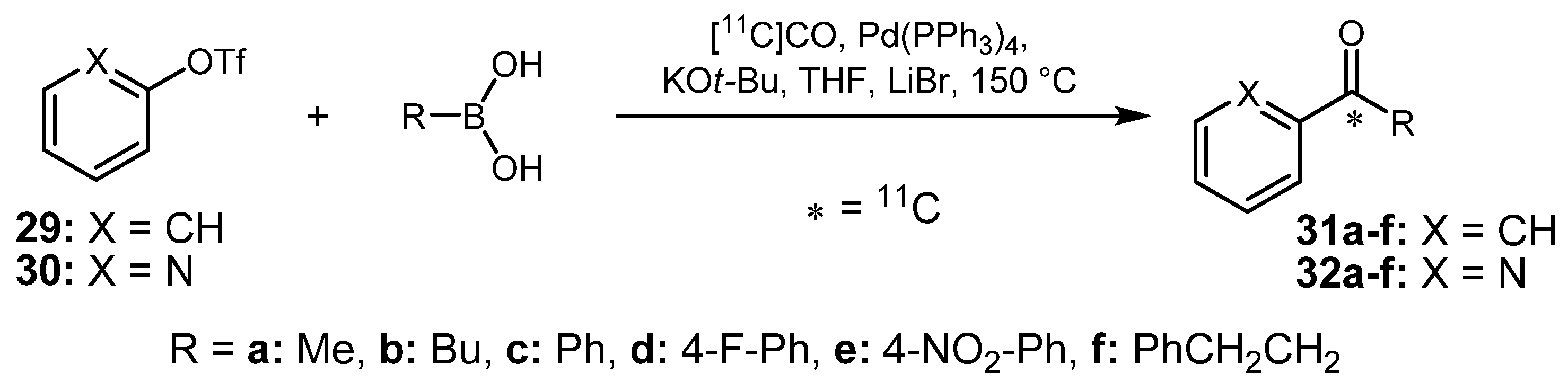{kind=link}
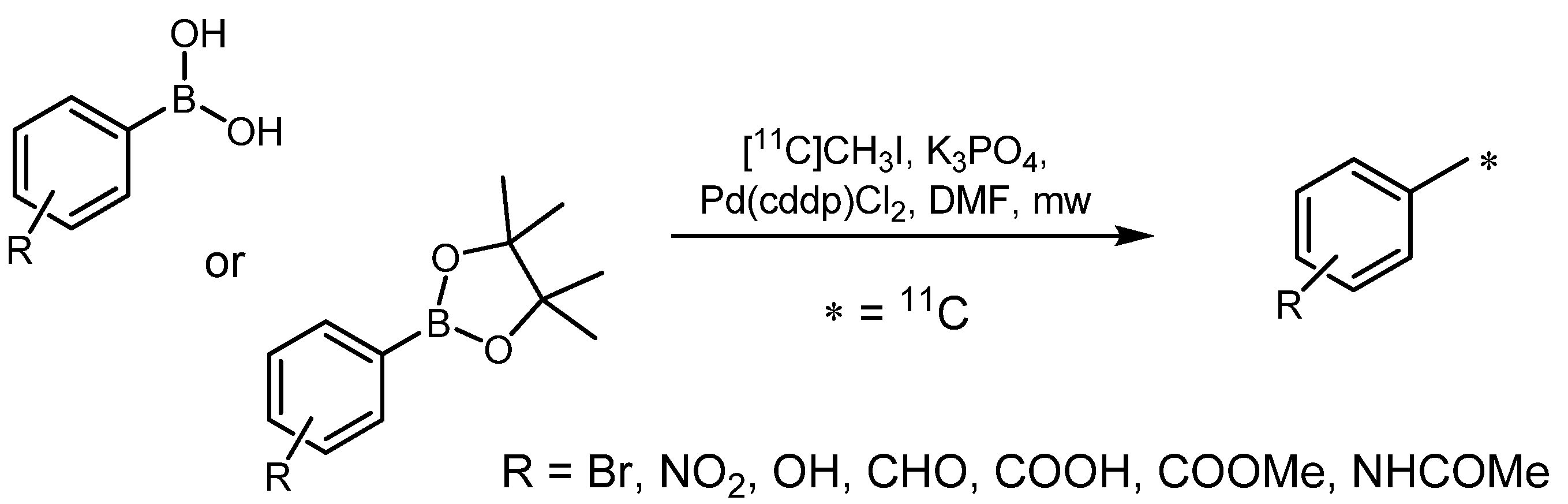{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
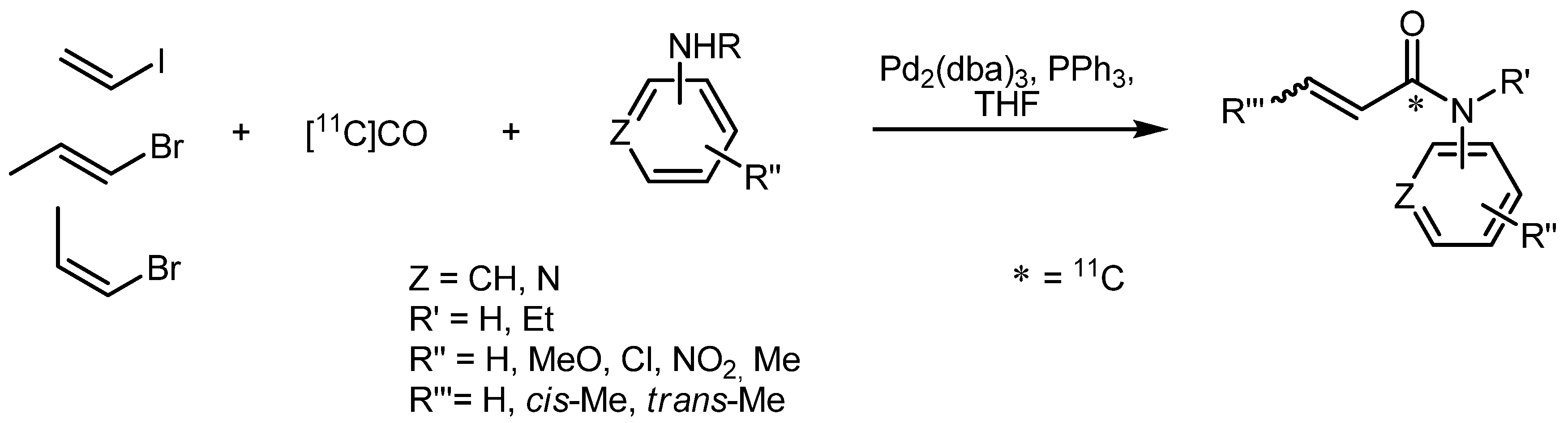{kind=link}
{kind=link}
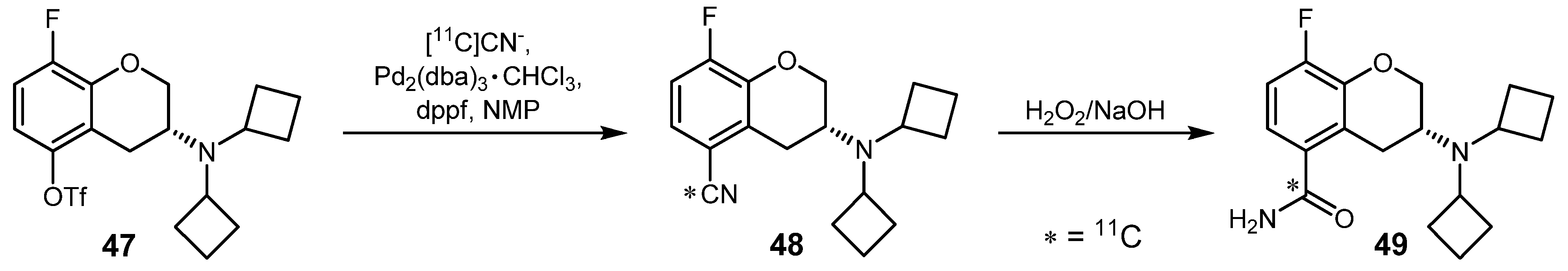{kind=link}
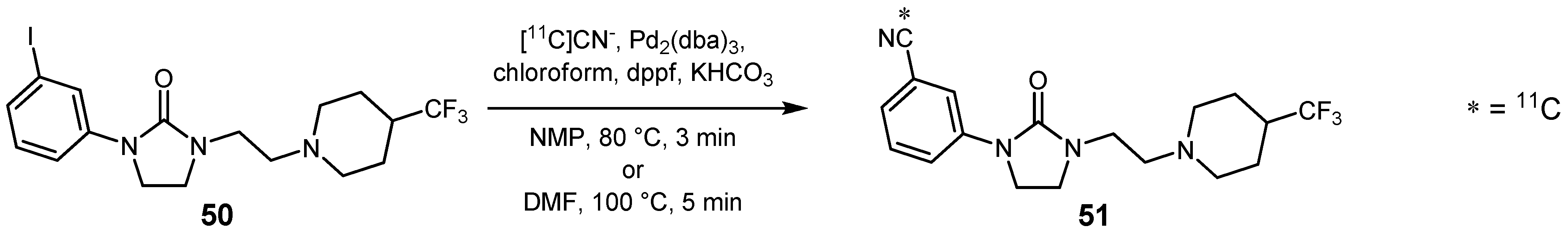{kind=link}
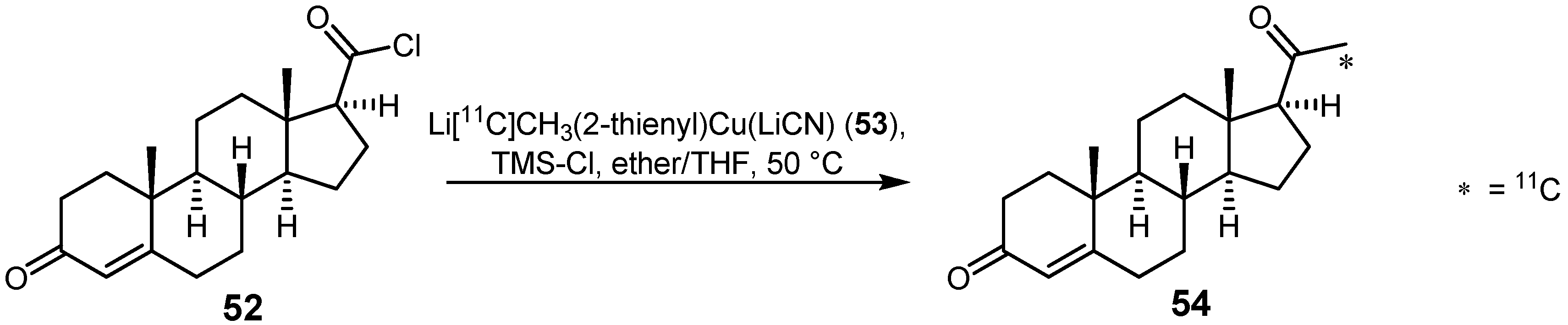{kind=link}
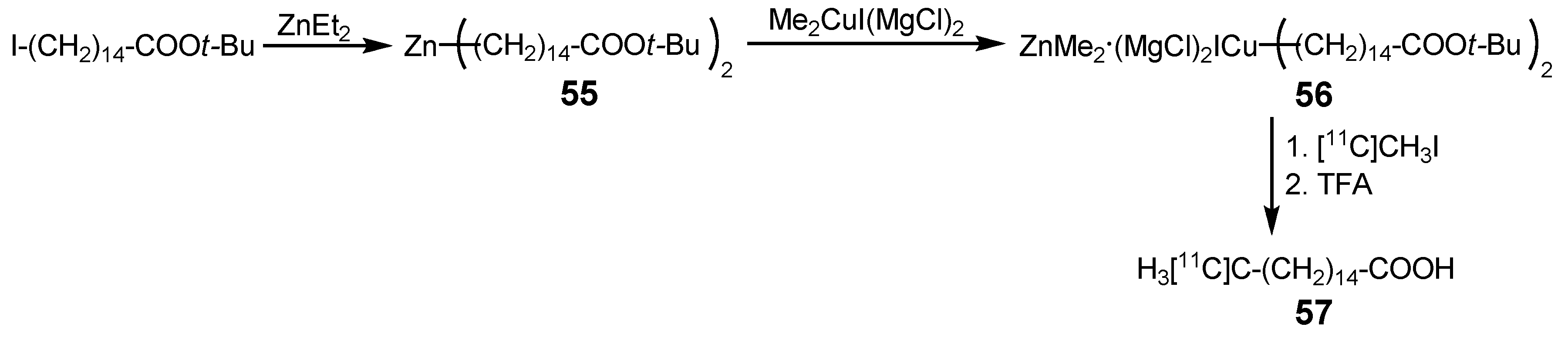{kind=link}
{kind=link}
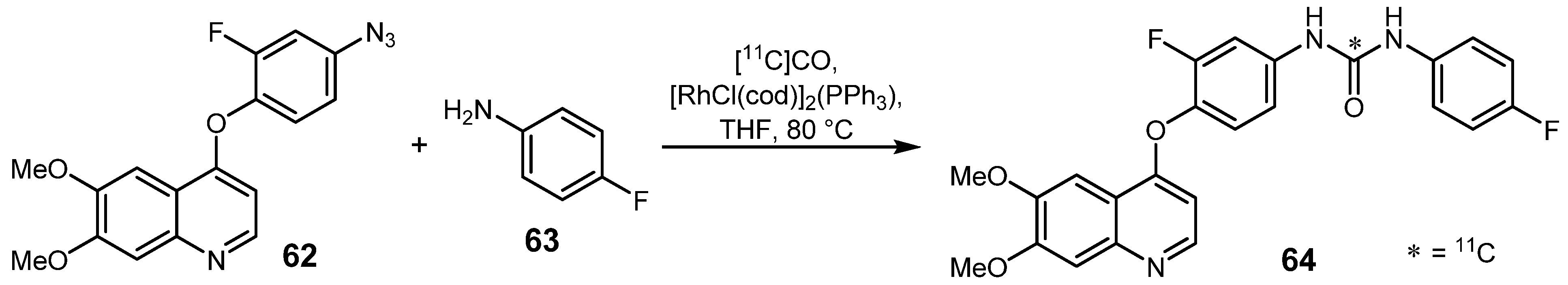{kind=link}
{kind=link}
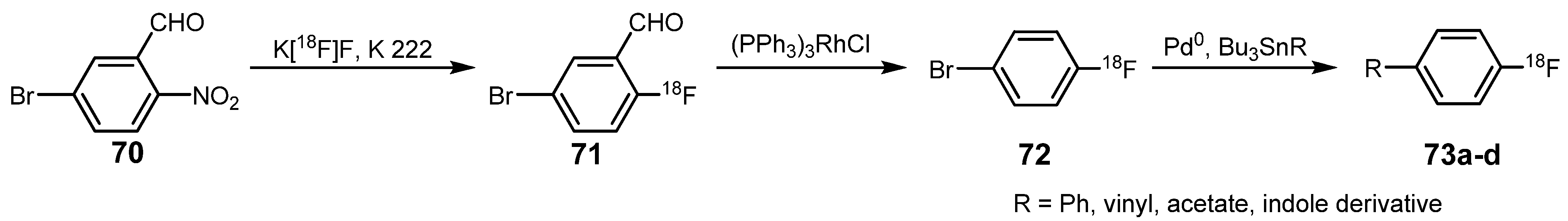{kind=link}
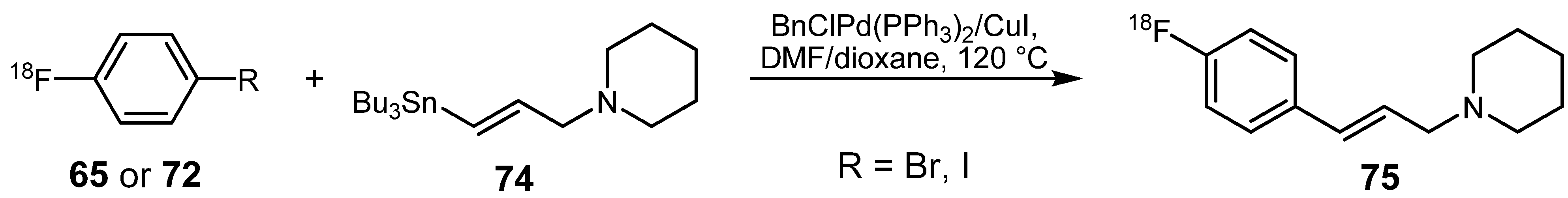{kind=link}
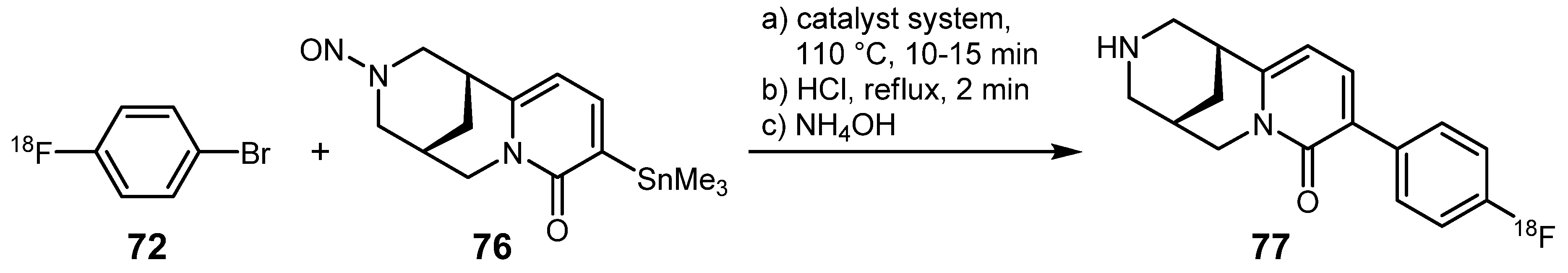{kind=link}
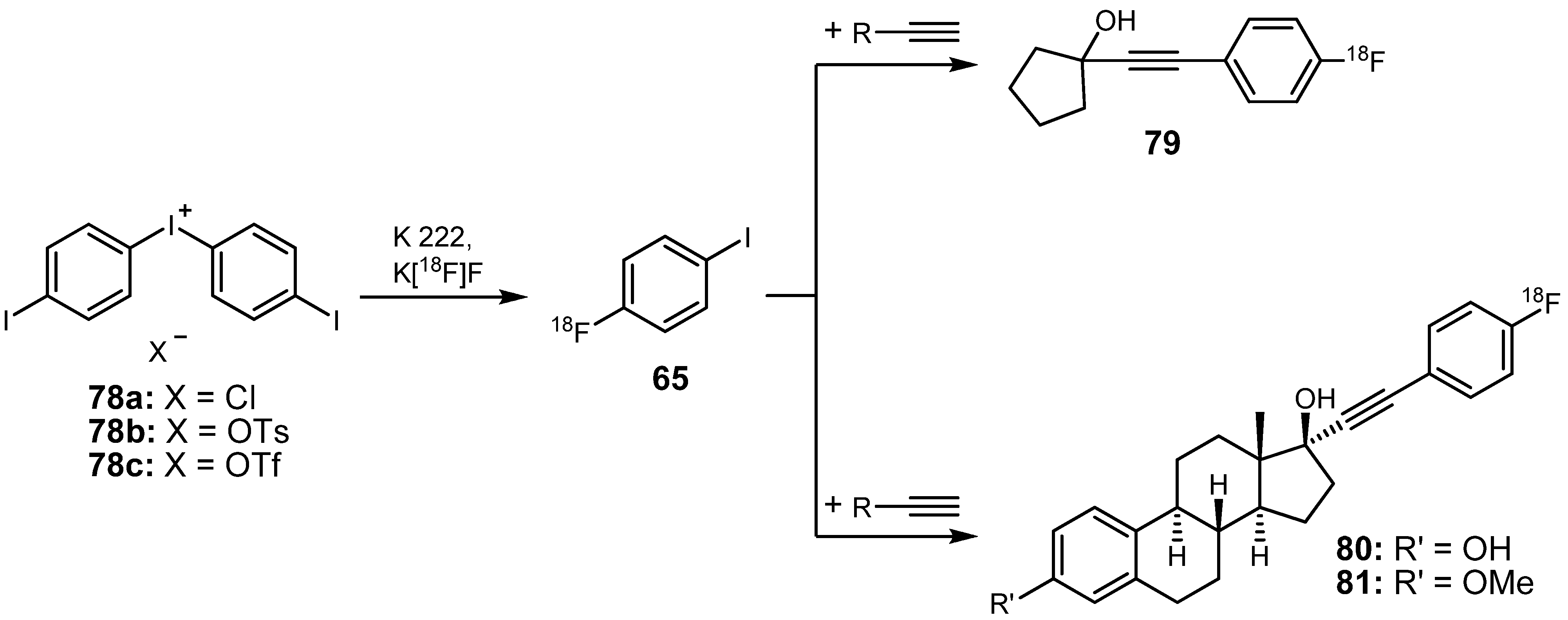{kind=link}
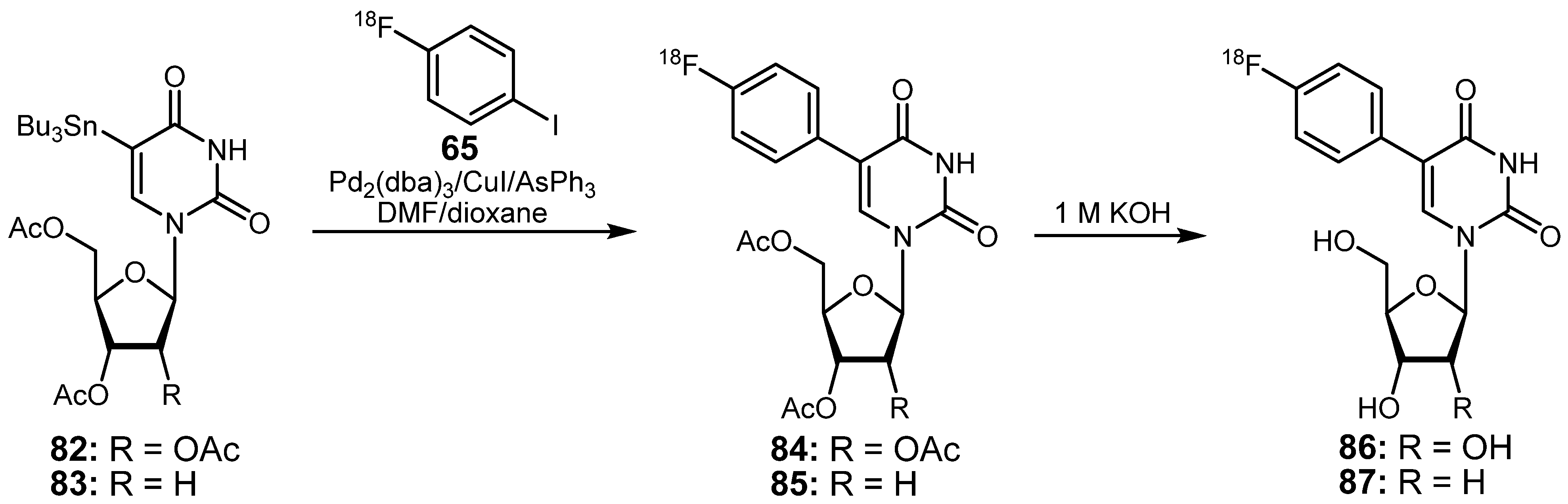{kind=link}
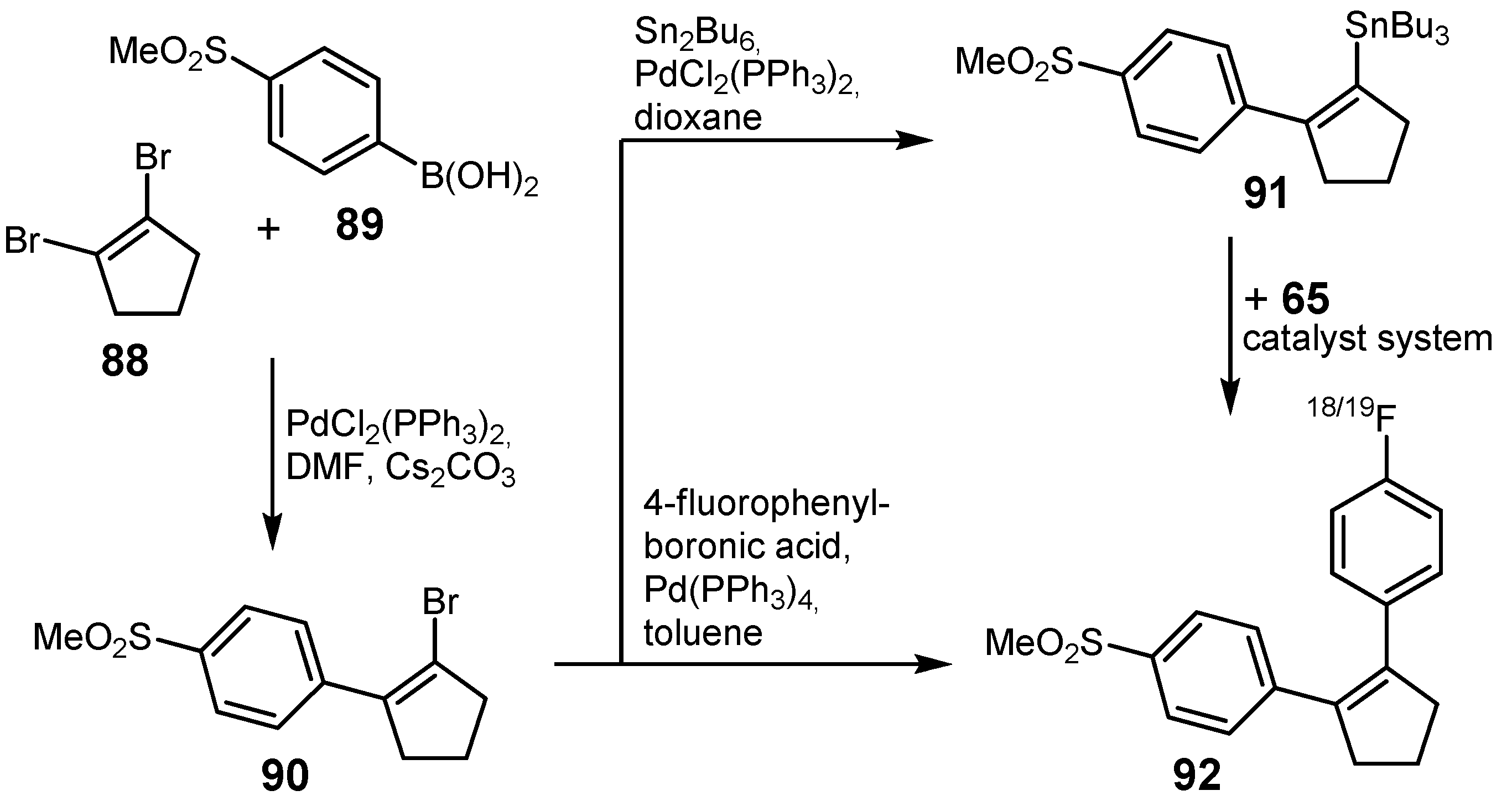{kind=link}
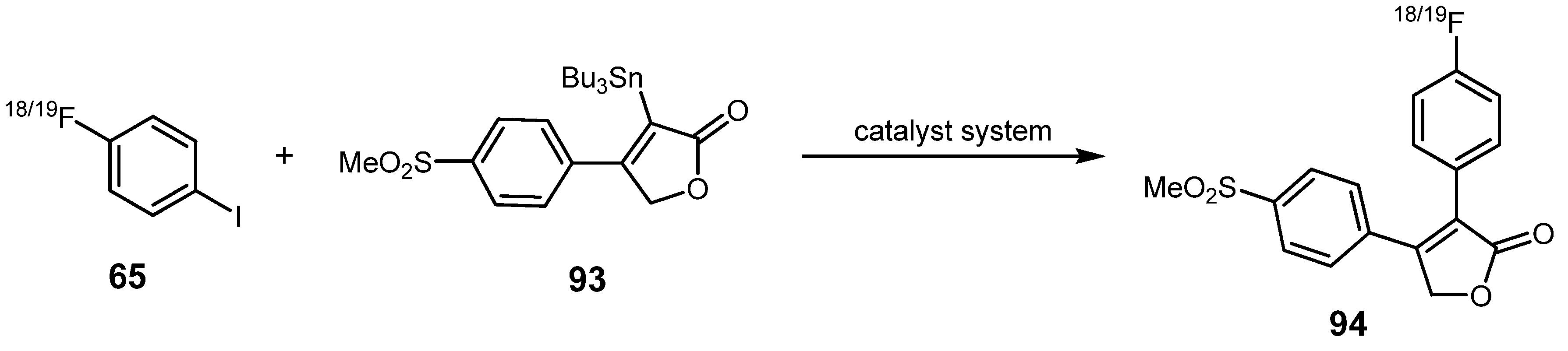{kind=link}
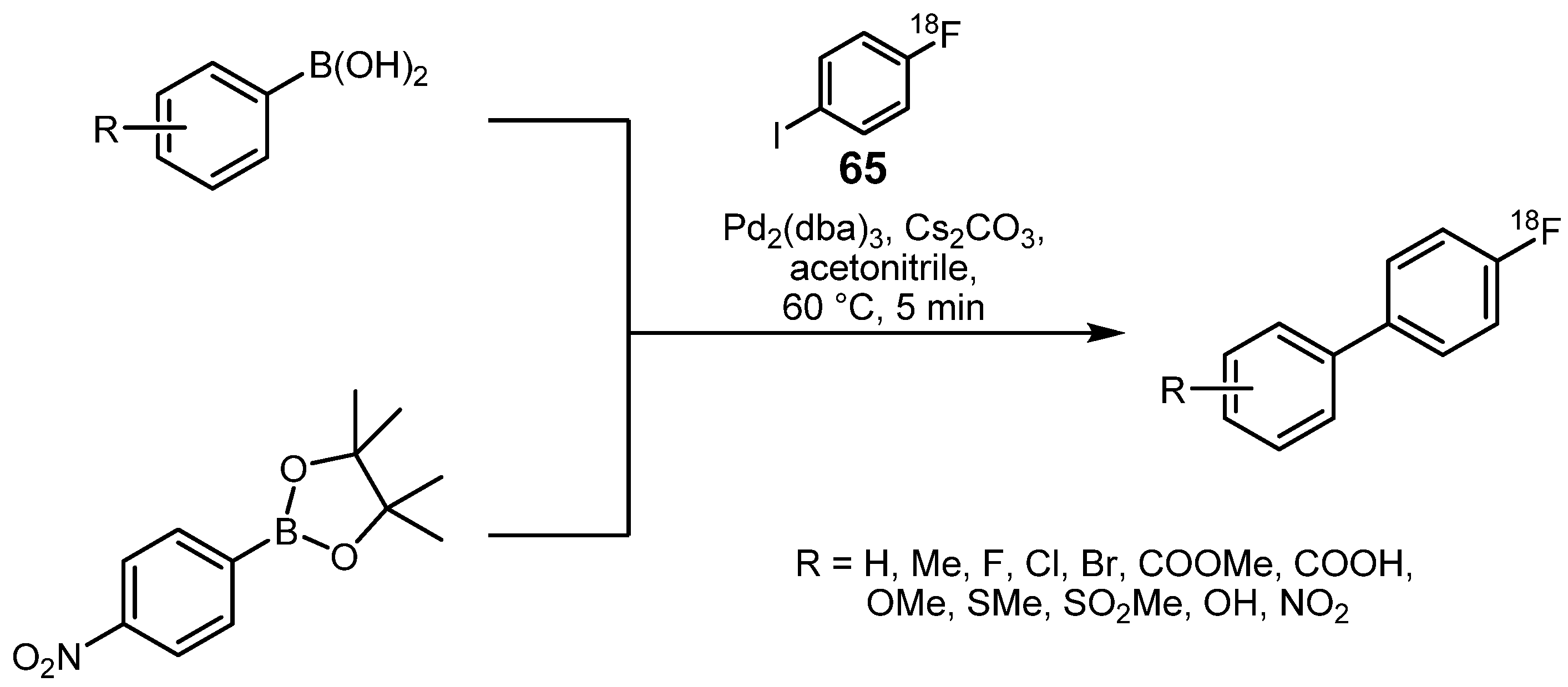{kind=link}
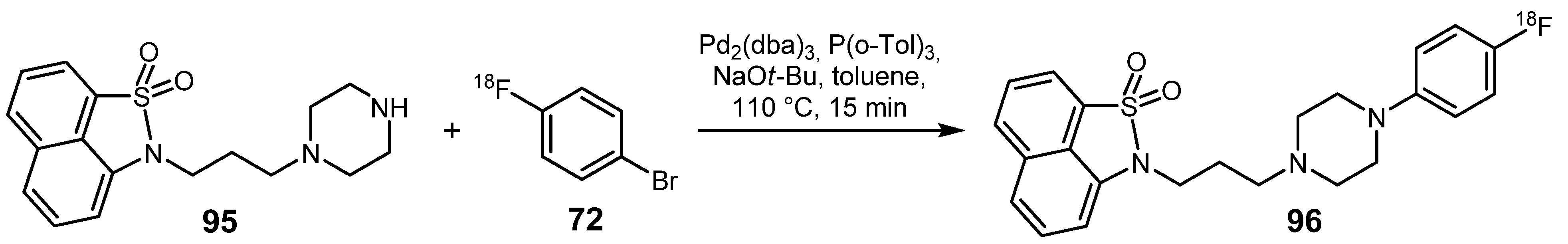{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | T / °C | t / min | Product | RCY / % |
|---|---|---|---|---|
| Bu3SnPh | 115 | 15 | 4-[18F]fluorobiphenyl 73a | >90 |
| Bu3SnCH=CH2 | 115 | 30 | 4-[18F]fluorostyrole 73b | >80 |
| Bu3SnOOCCH3 | 120 | 5 | 4-[18F]fluorophenyl acetate 73c | 78 |
| 1-isopropyl-2-methyl-3-(tributylstannyl)indole | 115 | 15 | 1-isopropyl-2-methyl-3-(4-[18F]fluorophenyl)indole 73d | 15 |







3.2. 18F-labeled compounds prepared via carbon-heteroatom cross-coupling reactions




4. Conclusions
Acknowledgements
References and Notes
- Diederich, F.; Stang, P.J. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Wu, X.-F.; Anbarasan, P.; Neumann, H. Beller, M. Vom Edelmetall zum Nobelpreis: Palladiumkatalysierte Kupplungen als Schlüsselmethode in der organischen Chemie. Angew. Chem. 2010, 122, 9231–9234. [Google Scholar]
- Mamat, C.; Ramenda, T.; Wuest, F.R. Recent Applications of Click Chemistry for the Synthesis of Radiotracers for Molecular Imaging. Mini-Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Glaser, M.; Robins, E.G. Click labelling’ in PET radiochemistry. J. Labelled Compd. Radiopharm. 2009, 52, 407–414. [Google Scholar] [CrossRef]
- Pfennig, G.; Klewe-Nebenius, H.; Seelmann-Eggebert, W. Karlsruher Nuklidkarte; Haberbeck: Lage, Germany, 2009. [Google Scholar]
- Antoni, G.; Kihlberg, T.; Långström, B. Handbook of Radiopharmaceuticals, Radiochemistry and Applications; Welch, M.J., Redvanly, C.S., Eds.; John Wiley & Sons: Chichester, UK, 2003; pp. 141–195, Chapter 5. [Google Scholar]
- Scott, P.J.H. Methoden für den Einbau von Kohlenstoff-11 zur Erzeugung von Radiopharmaka für die Positronenemissionstomographie. Angew. Chem. 2009, 121, 6115–6118. [Google Scholar] [CrossRef]
- Allard, M.; Fouquet, E.; James, D.; Szlosek-Pinaud, M. State of the Art in 11C Labelled Radiotracers Synthesis. Curr. Med. Chem. 2008, 15, 235–277. [Google Scholar] [CrossRef]
- Oberdorfer, F.; Hanisch, M.; Helus, F.; Maier-Borst, W. A New Procedure for the Preparation of 11C-Labelled Methyl Iodide. Int. J. Appl. Radiat. Isot. 1985, 36, 435–438. [Google Scholar] [CrossRef]
- Holschbach, M.; Schüller, M. A New and Simple On-line Method for the Preparation of n.c.a. [11C]Methyl Iodide. Appl. Radiat. Isot. 1993, 44, 779–780. [Google Scholar] [CrossRef]
- Larsen, P.; Ulin, J.; Dahlstrom, K.; Jensen, M. Synthesis of [11C]Iodomethane by of [11C]Methane Iodination. Appl. Radiat. Isot. 1997, 48, 153–157. [Google Scholar] [CrossRef]
- Buckley, K.R.; Jivan, S.; Ruth, T.J. Improved yields for the in situ production of [11C]CH4 using a niobium target chamber. Nucl. Med. Biol. 2004, 31, 825–827. [Google Scholar] [CrossRef]
- Zhang, M.-R.; Suzuki, K. Sources of carbon which decrease the specific activity of [11C]CH3I synthesized by the single pass I2 method. Appl. Radiat. Isot. 2005, 62, 447–450. [Google Scholar] [CrossRef]
- Matarrese, M.; Soloviev, D.; Todde, S.; Neutro, F.; Petta, P.; Carpinelli, A.; Brüssermann, M.; Kienle, M.G.; Fazio, F. Preparation of [11C] radioligands with high specific radioactivity on a commercial PET tracer synthesizer. Nucl. Med. Biol. 2003, 30, 79–83. [Google Scholar] [CrossRef]
- Stille, J.K. Palladium-katalysierte Kupplungsreaktionen organischer Elektrophile mit Organo-zinn-Verbindungen. Angew. Chem. 1986, 98, 504–519. [Google Scholar] [CrossRef]
- Mitchell, T.N. Palladium-Catalysed Reactions of Organotin Compounds. Synthesis 1992, 803–815. [Google Scholar] [CrossRef]
- Andersson, Yvonne; Cheng, Aiping; Långström, Bengt. Palladium-promoted Coupling Reactions of [11C]Methyl Iodide with Organotin and Organoboron Compounds. Acta. Chem. Scand. 1995, 49, 683–688. [Google Scholar] [CrossRef]
- Suzuki, M.; Doi, H.; Björkman, M.; Andersson, Y.; Långström, B.; Watanabe, Y.; Noyori, R. Rapid Coupling of Methyl Iodide with Aryltributylstannanes Mediated by Palladium(0) Complexes: A General Protocol for the Synthesis of 11CH3-Labeled PET Tracers. Chem. Eur. J. 1997, 3, 2039–2042. [Google Scholar] [CrossRef]
- Björkman, M.; Doi, H.; Resul, B.; Suzuki, M.; Noyori, R.; Watanabe, Y.; Långström, B. F2α Analogue Using An Improved Method For Stille Reactions With [11C]Methyl Iodide. J. Labelled Compd. Radiopharm. 2000, 43, 1327–1334. [Google Scholar] [CrossRef]
- Sandell, J.; Halldin, C.; Sovago, J.; Chou, Y.-H.; Gulyás, B.; Yu, M.; Emond, P.; Någren, K.; Guilloteau, D.; Farde, L. PET examination of [11C]5-methyl-6-nitroquipazine, a radioligand for visualization of the serotonin transporter. Nucl. Med. Biol. 2002, 29, 651–656. [Google Scholar] [CrossRef]
- Langer, O.; Forngren, T.; Sandell, J.; Dollé, F.; Långström, B.; Någren, K.; Halldin, C. Preparation of 4-[11C]methylmetaraminol, a potential PET tracer for assessment of myocardial sympathetic innervation. J. Labelled Compd. Radiopharm. 2003, 46, 55–65. [Google Scholar] [CrossRef]
- Forngren, T.; Samuelsson, L.; Långström, B. A 11C-Methyl stannane (5-[11C]methyl-1-aza-5-stannabicyclo[3.3.3]undecane) for use in palladium-mediated [11C]C–C bond forming reactions with organohalides. J. Labelled Compd. Radiopharm. 2004, 47, 71–78. [Google Scholar] [CrossRef]
- Reiffers, S.; Vaalburg, W.; Wiegman, T.; Wynberg, H.; Woldring, M.G. Carbon-11 labelled methyllithium as methyl donating agent: The addition to 17-Keto steroids. Int. J. Appl. Radiat. Isot. 1980, 31, 535–539. [Google Scholar] [CrossRef]
- Karimi, F.; Barletta, J.; Långström, B. Palladium-Mediated 11C-Carbonylative Cross-Coupling of Alkyl/Aryl Iodides with Organostannanes: An Efficient Synthesis of Unsymmetrical Alkyl/Aryl [11C-carbonyl]Ketones. Eur. J. Org. Chem. 2005, 2374–2378. [Google Scholar]
- Arai, T.; Kato, K.; Zhang, M.R. Synthesis of [carbonyl-11C]acetophenone via the Stille cross-coupling reaction of [1-11C]acetyl chloride with tributylphenylstannane mediated by Pd2(dba)3/P(MeNCH2CH2)3N·HCl. Tetrahedron Lett. 2009, 50, 4788–4791. [Google Scholar] [CrossRef]
- Liu, X.; Verkade, J.G. Free and Polymer-Bound Tricyclic Azaphosphatranes HP(RNCH2CH2)3N+: Procatalysts in Dehydrohalogenations and Debrominations with NaH. J. Org. Chem. 1999, 64, 4840–4843. [Google Scholar] [CrossRef]
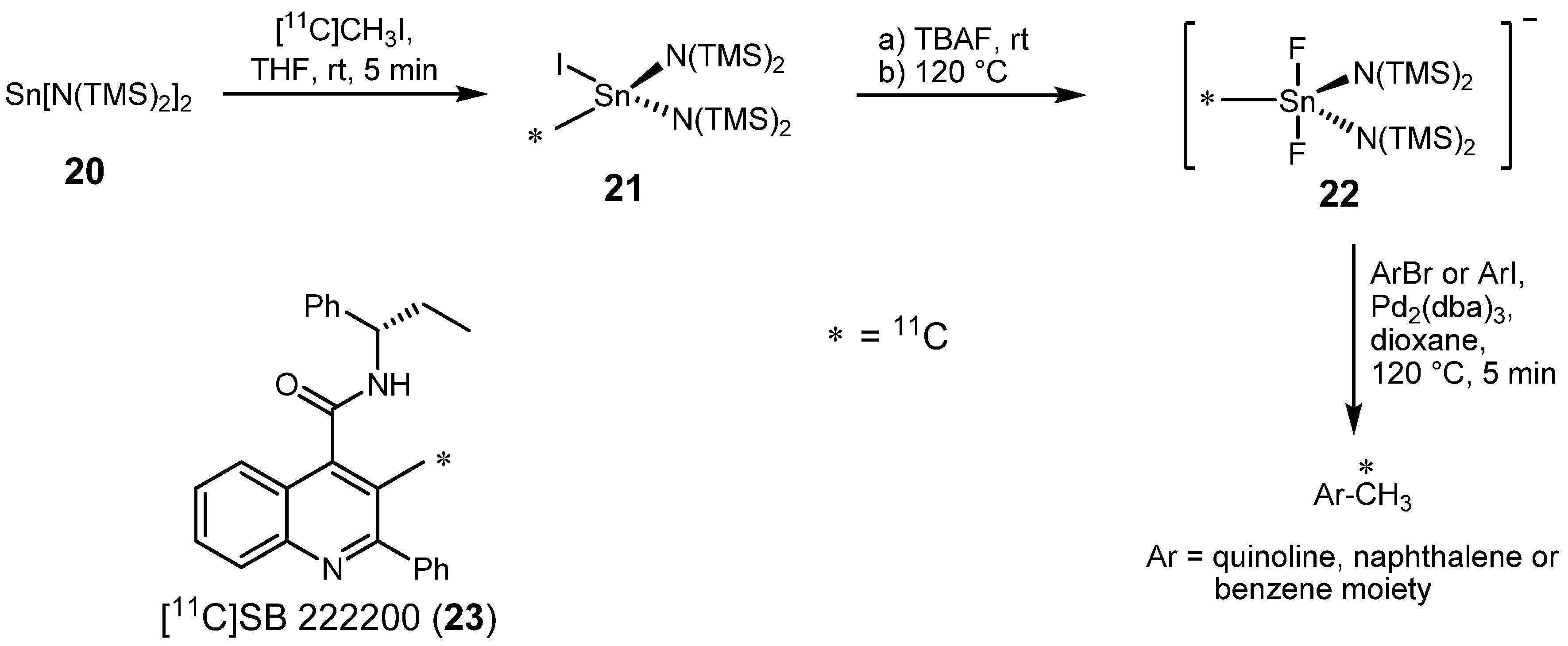
- Huiban, M.; Huet, A.; Barré, L.; Sobrio, F.; Fouquet, E.; Perrio, C. Methyl transfer reaction from monomethyltin reagent under palladium(0) catalysis: a versatile method for labelling with carbon-11. Chem. Commun. 2006, 97–99. [Google Scholar]
- Harris, D.H.; Lappert, M.F.J. Monomeric, volatile bivalent amides of group IVB elements, M(NR12)2 and M(NR1R2)2(M=Ge, Sn, or Pb; R1=Me3Si, R2=Me3C. J. Chem. Soc. Chem. Commun. 1974, 895–896. [Google Scholar]
- Giardina, G.A.M.; Sarau, H.M.; Farina, C.; Medhurst, A.D.; Grugni, M.; Foley, J.J.; Raveglia, L.F.; Schmidt, D.B.; Rigolio, R.; Vassallo, M.; Vecchietti, V.; Hay, D.W.P. 2-Phenyl-4-quinolinecarboxamides: A Novel Class of Potent and Selective Non-Peptide Competitive Antagonists for the Human Neurokinin-3 Receptor. J. Med. Chem. 1996, 39, 2281–2284. [Google Scholar]
- Bourdier, T.; Huiban, M.; Huet, A.; Sobrio, F.; Fouquet, E.; Perrio, C.; Barré, L. Tetra- and Monoorganotin Reagents in Palladium-Mediated Cross-Coupling Reactions for the Labeling with Carbon-11 of PET Tracers. Synthesis 2008, 978–984. [Google Scholar]
- Langer, O.; Forngren, T.; Sandell, J.; Dollé, F.; Långström, B.; Någren, K.; Halldin, C. Preparation of 4-[11C]methylmetaraminol, a potential PET tracer for assessment of myocardial sympathetic innervation. J. Labelled Compd. Radiopharm. 2003, 46, 55–65. [Google Scholar] [CrossRef]
- Samuelsson, L.; Långström, B. Synthesis of 1-(2'-deoxy-2'-fluoro-ß-d-arabinofuranosyl)-[Methyl-11C]thymine ([11C]FMAU) via a Stille cross-coupling reaction with [11C]methyl iodide. J. Labelled Compd. Radiopharm. 2003, 46, 263–272. [Google Scholar] [CrossRef]
- Toyohara, J.; Okada, M.; Toramatsu, C.; Suzuki, K.; Irie, T. Feasibility studies of 4'-[methyl-11C]thiothymidine as a tumor proliferation imaging agent in mice. Nucl. Med. Biol. 2008, 35, 67–74. [Google Scholar] [CrossRef]
- Kawamura, K.; Shi, K.; Tsukada, H.; Nishiyama, S.; Mori, H.; Ishiwata, K. Synthesis and evaluation of vesamicol analog (–)-o-[11C]methylvesamicol as a PET ligand for vesicular acetylcholine transporter. Ann. Nucl. Med. 2006, 20, 417–424. [Google Scholar] [CrossRef]
- Toyohara, J.; Sakata, M.; Wu, J.; Ishikawa, M.; Oda, K.; Ishii, K.; Iyo, M.; Hashimoto, K.; Ishiwata, K. Preclinical and the first clinical studies on [11C]CHIBA-1001 for mapping α7 nicotinic receptors by positron emission tomography. Ann. Nucl. Med. 2009, 23, 301–309. [Google Scholar] [CrossRef]
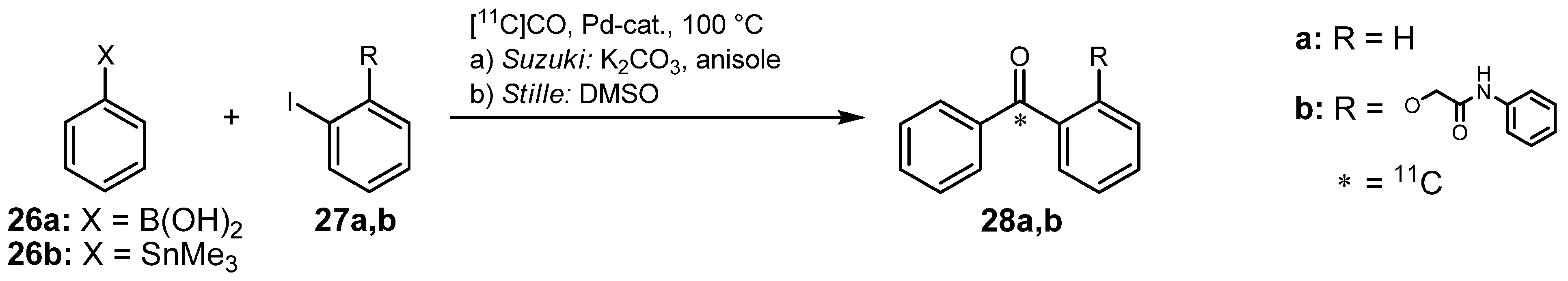
- Lu, S.; Hong, J.; Itoh, T.; Fujita, M.; Inoue, O.; Innis, R.B.; Pike, V.W. [carbonyl-11C]Benzyl acetate: Automated radiosynthesis via Pd-mediated [11C]carbon monoxide chemistry and PET measurement of brain uptake in monkey. J. Labelled Compd. Radiopharm. 2010, 53, 548–551. [Google Scholar] [CrossRef]
- Hosoya, T.; Sumi, K.; Doi, H.; Wakao, M.; Suzuki, M. Rapid methylation on carbon frameworks useful for the synthesis of 11CH3-incorporated PET tracers: Pd(0)-mediated rapid coupling of methyl iodide with an alkenyltributylstannane leading to a 1-methylalkene. Org. Biomol. Chem. 2006, 4, 410–415. [Google Scholar] [CrossRef]
- Kawamura, K.; Naganawa, M.; Konno, F.; Yui, J.; Wakizaka, H.; Yamasaki, T.; Yanamoto, K.; Hatori, A.; Takei, M.; Yoshida, Y.; Sakaguchi, K.; Fukumura, T.; Kimura, Y.; Zhang, M.-R. Imaging of I2-imidazoline receptors by small-animal PET using 2-(3-fluoro-[4-11C]tolyl)-4,5-dihydro-1H-imidazole ([11C]FTIMD). Nucl. Med. Biol. 2010, 37, 625–635. [Google Scholar]
- Bennacef, I.; Perrio, C.; Lasne, M.-C.; Barré, L. Functionalization through Lithiation of (S)-N-(1-Phenylpropyl)-2-phenylquinoline-4-carboxamide. Application to the Labeling with Carbon-11 of NK-3 Receptor Antagonist SB 222200. J. Org. Chem. 2007, 72, 2161–2165. [Google Scholar]
- Madsen, J.; Merachtsaki, P.; Davoodpour, P.; Bergström, M.; Långström, B.; Andersen, K.; Thomsen, C.; Martiny, L.; Knudsen, G. M. Synthesis and Biological Evaluation of Novel Carbon-11-Labelled Analogues of Citalopram as Potential Radioligands for the Serotonin Transporter. Bioorg. Med. Chem. 2003, 11, 3447–3456. [Google Scholar]
- Tarkiainen, J.; Vercouillie, J.; Emond, P.; Sandell, J.; Hiltunen, J.; Frangin, Y.; Guilloteau, D.; Halldin, C. Carbon-11 labelling of MADAM in two different positions: a highly selective PET radioligand for the serotonin transporter. J. Labelled Compd. Radiopharm. 2001, 44, 1013–1023. [Google Scholar] [CrossRef]
- Yu, M.; Tueckmantel, W.; Wang, X.; Zhu, A.; Kozikowski, A.P.; Brownell, A.-L. Methoxyphenylethynyl, methoxypyridylethynyl and phenylethynyl derivatives of pyridine: synthesis, radiolabeling and evaluation of new PET ligands for metabotropic glutamate subtype 5 receptors. Nucl. Med. Biol. 2005, 32, 631–640. [Google Scholar] [CrossRef]
- Hamill, T.G.; Krause, S.; Ryan, C.; Bonnefous, C.; Govek, S.; Seiders, T.J.; Cosford, N.D.P.; Roppe, J.; Kamenecka, T.; Patel, S.; Gibson, R.E.; Sanabria, S.; Riffel, K.; Eng, W.; King, C.; Yang, X.; Green, M.D.; O’Malley, S.S.; Hargreaves, R.; Burns, H.D. Synthesis, Characterization, and First Successful Monkey Imaging Studies of Metabotropic Glutamate Receptor Subtype 5 (mGluR5) PET Radiotracers. Synapse 2005, 56, 205–216. [Google Scholar] [CrossRef]
- Huang, Y.; Narendran, R.; Bischoff, F.; Guo, N.; Zhu, Z.; Bae, S.; Lesage, A.S.; Laruelle, M. A Positron Emission Tomography Radioligand for the in Vivo Labeling of Metabotropic Glutamate 1 Receptor: (3-Ethyl-2-[11C]methyl-6-quinolinyl)(cis-4-methoxycyclohexyl)methanone. J. Med. Chem. 2005, 48, 5096–5099. [Google Scholar]
- Björkman, M.; Andersson, Y.; Doi, H.; Kato, K.; Suzuki, M.; Noyori, R.; Watanabe, Y.; Långström, B. Synthesis of 11C/13C-Labelled Prostacyclins. Acta Chem. Scand. 1998, 52, 635–640. [Google Scholar] [CrossRef]
- Suzuki, M.; Doi, H.; Kato, K.; Björkman, M.; Långström, B.; Watanabe, Y.; Noyori, R. Rapid Methylation for the Synthesis of a 11C-Labeled Tolylisocarbacyclin Imaging the IP2 Receptor in a Living Human Brain. Tetrahedron 2000, 56, 8263–8273. [Google Scholar] [Green Version]
- Suzuki, M.; Doi, H.; Hosoya, T.; Långström, B.; Watanabe, Y. Rapid methylation on carbon frameworks leading to the synthesis of a PET tracer capable of imaging a novel CNS-type prostacyclin receptor in living human brain. Trends Anal. Chem. 2004, 23, 595–607. [Google Scholar] [Green Version]
- Prabhakaran, J.; Majo, V.J.; Simpson, N.R.; Van Heertum, R.L.; Mann, J.J.; Kumar, J.S.D. Synthesis of [11C]celecoxib: A potential PET probe for imaging COX-2 expression. J. Labelled Compd. Radiopharm. 2005, 48, 887–895. [Google Scholar] [CrossRef]
- Karimi, F.; Långström, B. Synthesis of 3-[(2S)-azetidin-2-ylmethoxy]-5-[11C]-methylpyridine, an analogue of A-85380, via a Stille coupling. J. Labelled Compd. Radiopharm. 2002, 45, 423–434. [Google Scholar] [CrossRef]
- Iida, Y.; Ogawa, M.; Ueda, M.; Tominaga, A.; Kawashima, H.; Magata, Y.; Nishiyama, S.; Tsukada, H.; Mukai, T.; Saji, H. Evaluation of 5-11C-Methyl-A-85380 as an Imaging Agent for PET Investigations of Brain Nicotinic Acetylcholine Receptors. J. Nucl. Med. 2004, 45, 878–884. [Google Scholar]
- Suzuki, A. Synthetic studies via the cross-coupling reaction of organoboron derivatives with organic halides. Pure and Appl. Chem. 1991, 63, 419–422. [Google Scholar] [CrossRef]
- Ishiyama, T.; Kizaki, H.; Miyaura, N.; Suzuki, A. Synthesis of unsymmetrical biaryl ketones via palladiumcatalyzed cross-coupling reaction of aryboronic acids with iodoarenes. Tetrahedron Lett. 1993, 34, 7595–7598. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Zeisler, S.K.; Nader, M.; Theobald, A.; Oberdorfer, F. Conversion of No-carrier-added [11C]carbon dioxide to [11C]carbon monoxide on molybdenum for the synthesis of 11C-labelled aromatic ketones. Appl. Radiat. Isot. 1997, 48, 1091–1095. [Google Scholar] [CrossRef]
- Nader, M.W.; Oberdorfer, F. Syntheses of [carbonyl-11C]2-(2-benzoylphenoxy)-N-phenylacet-amide from [11C]carbon monoxide by the Suzuki and the Stille reactions. Appl. Radiat. Isot. 2002, 57, 681–685. [Google Scholar] [CrossRef]
- Rahman, O.; Llop, J.; Långström, B. Organic Bases as Additives to Improve the Radiochemical Yields of [11C]Ketones Prepared by the Suzuki Coupling Reaction. Eur. J. Org. Chem. 2004, 2674–2678. [Google Scholar]
- Colquhoun, H.M.; Thompson, D.J.; Twigg, M.V. Carbonylation; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Rahman, O.; Kihlberg, T.; Långström, B. Synthesis of 11C-/13C-Ketones by Suzuki Coupling. Eur. J. Org. Chem. 2004, 474–478. [Google Scholar]
- Hostetler, E.D.; Terry, G.E.; Burns, H.D. An improved synthesis of substituted [11C]toluenes via Suzuki coupling with [11C]methyl iodide. J. Labelled Compd. Radiopharm. 2005, 48, 629–634. [Google Scholar] [CrossRef]
- De Meijere, A.; Meyer, F.E. Kleider machen Leute: Heck-Reaktion im neuen Gewand. Angew. Chem. 1994, 106, 2473–2506. [Google Scholar] [CrossRef]
- Björkman, M.; Långström, B. Functionalisation of 11C-labelled olefins via a Heck coupling reac-tion. J. Chem. Soc. Perkin Trans. 1 2000, 3031–3034. [Google Scholar]
- Chinchilla, R.; Nájera, C. The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar]
- Wuest, F.; Zessin, J.; Johannsen, B. A new approach for 11C–C bond formation: synthesis of 17α-(3'-[11C]prop-1-yn-1-yl)-3-methoxy-3,17ß-estradiol. J. Labelled Compd. Radiopharm. 2003, 46, 333–342. [Google Scholar] [CrossRef]
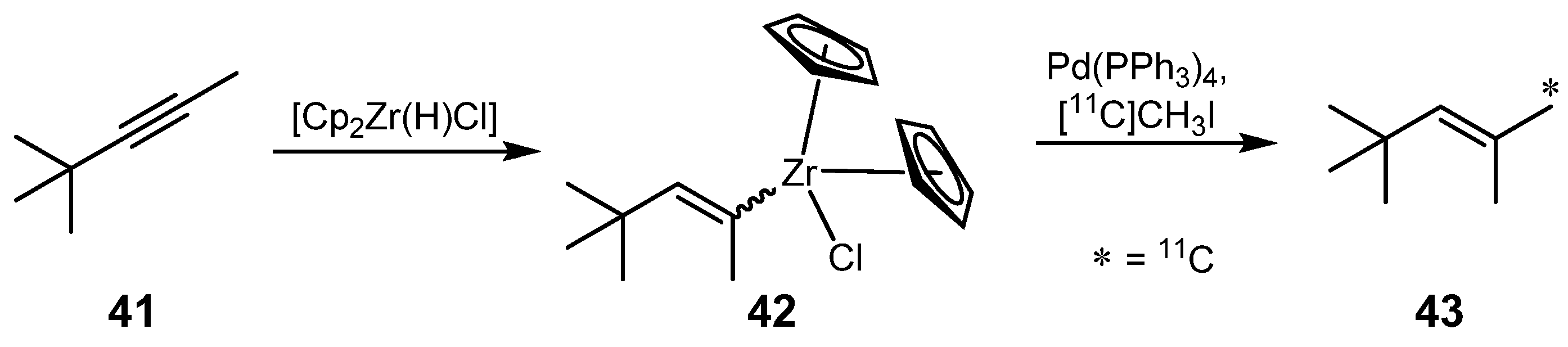
- Wuest, F.; Berndt, M. 11C–C bond formation by palladium-mediated cross-coupling of alkenylzirconocenes with [11C]methyl iodide. J. Labelled Compd. Radiopharm. 2006, 49, 91–100. [Google Scholar] [CrossRef]
- Hart, D.W.; Schwartz, J. Hydrozirconation. Organic Synthesis via organozirconium intermediates. Synthesis and rearrangement of alkylzirconium(IV) complexes and their reaction with electrophiles. J. Am. Chem. Soc. 1974, 96, 8115–8116. [Google Scholar] [CrossRef]
- Tarkiainen, J.; Vercouillie, J.; Emond, P.; Sandell, J.; Hiltunen, J.; Frangin, Y.; Guilloteau, D.; Halldin, C. Carbon-11 labelling of MADAM in two different positions: a highly selective PET radioligand for the serotonin transporter. J. Label. Compd. Radiopharm. 2001, 44, 1013–1023. [Google Scholar] [CrossRef]
- Lasne, M.C.; Pike, V.W.; Turton, D,R. The radiosynthesis of [-methyl-11C]-sertraline. Int. J. Radiat. Appl. Instrum., Part A 1989, 40, 147–151. [Google Scholar] [CrossRef]
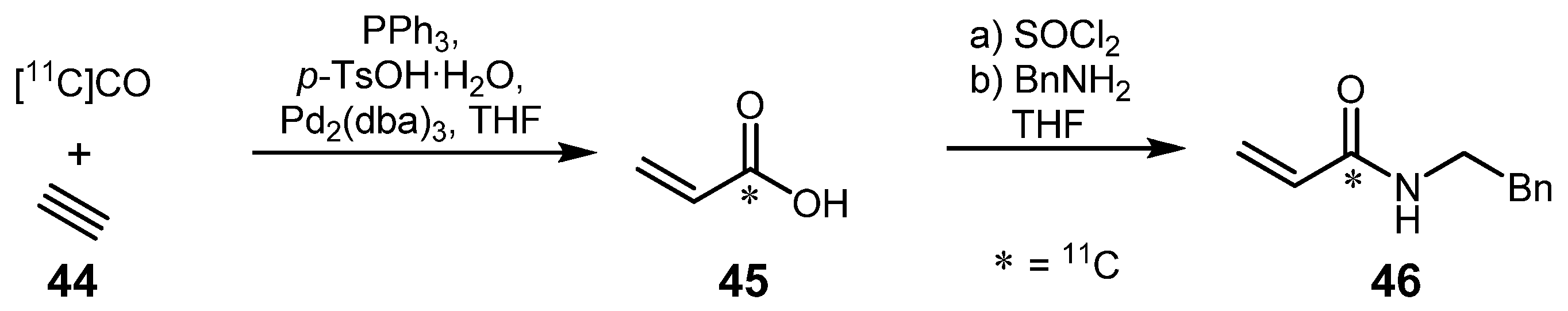
- Eriksson, J.; Åberg, O.; Långström, B. Synthesis of [11C]/[13C]Acrylamides by Palladium-Media-ted Carbonylation. Eur. J. Org. Chem. 2007, 455–461. [Google Scholar]
- Amatore, C.; Jutand, A. Role of dba in the reactivity of palladium(0) complexes generated in situ from mixtures of Pd(dba)2 and phosphines. Coord. Chem. Rev. 1998, 178-180, 511–528. [Google Scholar]
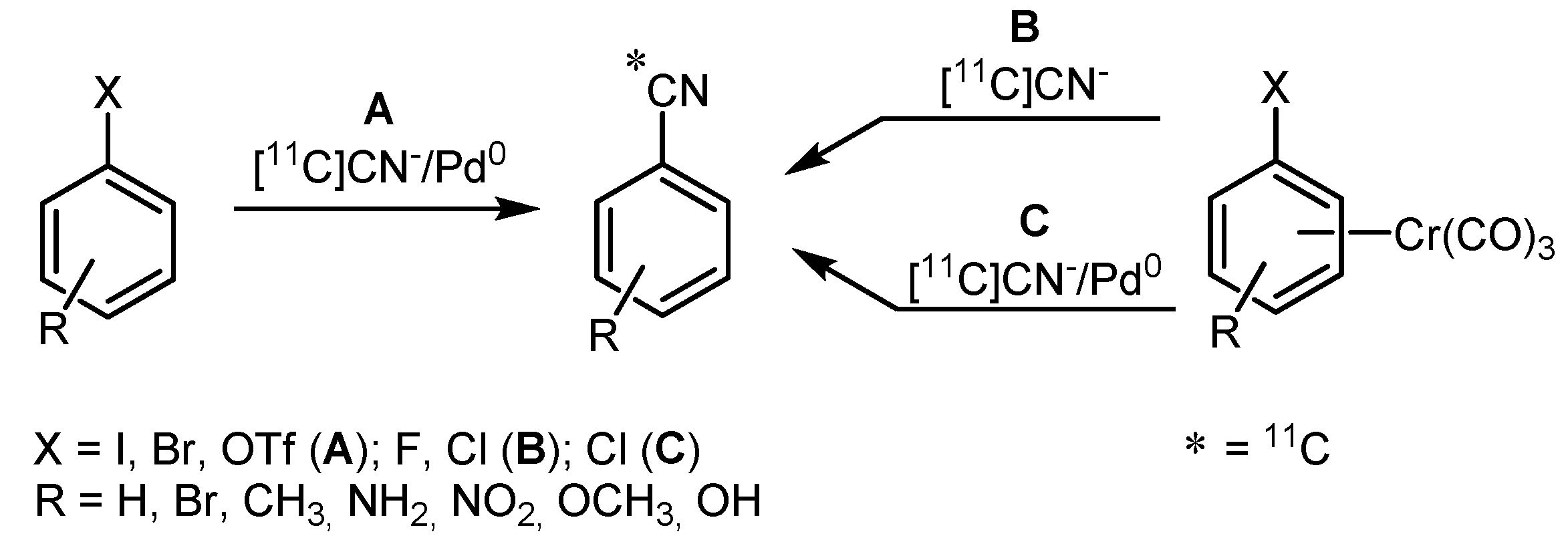
- Andersson, Y.; Långström, B. Transition Metal-mediated Reactions using [11C]Cyanide in Synthesis of 11C-labelled Aromatic Compounds. J. Chem. Soc. Perkin Trans. 1 1994, 1395–1400. [Google Scholar] [CrossRef]
- Sandell, J.; Halldin, C.; Hall, H.; Thorberg, S.-O.; Werner, T.; Sohn, D.; Sedvall, G.; Farde, L. Radiosynthesis and Autoradiographic Evaluation of [11C]NAD-299, a Radioligand for Visualization of the 5-HT1A Receptor. Nucl. Med. Biol. 1999, 26, 159–164. [Google Scholar] [CrossRef]
- Bennacef, I.; Salinas, C.A.; Bonasera, T.A.; Gunn, R.N.; Audrain, H.; Jakobsen, S.; Nabulsi, N.; Weinzimmer, D.; Carson, R.E.; Huang, Y.; Holmes, I.; Micheli, F.; Heidbreder, C.; Gentile, G.; Rossi, T.; Laruelle, M. Dopamine D3 receptor antagonists: The quest for a potentially selective PET ligand. Part 3: Radiosynthesis and in vivo studies. Bioorg. Med. Chem. Lett. 2009, 19, 5056–5059. [Google Scholar]
- Lidström, P.; Neu, H.; Långström, B. Syntheses of [21-11C] and (21-13C) progesterone. J. Labelled Compd. Radiopharm. 1997, 40, 695–704, J. Labelled Compd. Radiopharm. 1997, 40, 783-784.. [Google Scholar]
- Madsen, J.; Merachtsaki, P.; Davoodpour, P.; Bergström, M.; Långström, B.; Andersen, K.; Thomsen, C.; Martiny, L.; Knudsen, G.M. Synthesis and Biological Evaluation of Novel Carbon-11-Labelled Analogues of Citalopram as Potential Radioligands for the Serotonin Transporter. Bioorg. Med. Chem. 2003, 11, 3447–3456. [Google Scholar] [CrossRef]
- Tucker, C.E.; Knochel, P. Cross-Coupling between Functionalized Alkylcopper Reagents and Functionalized Alkyl Halides. J. Org. Chem. 1993, 58, 4781–4782. [Google Scholar] [CrossRef]
- Wuest, F.; Dence, C.S.; McCarthy, T.J.; Welch, M.J. A New Approach for the Synthesis of [11C]-Labeled Fatty Acids. J. Labelled Compd. Radiopharm. 2000, 43, 1289–1300. [Google Scholar] [CrossRef]
- Cenini, S.; Pizzotti, M.; Porta, F.; La Monica, G. Isocyanate, urea and amide complexes from the reactions of organic azides with low oxidation state complexes of transition metals. J. Organomet. Chem. 1975, 88, 237–248. [Google Scholar]
- Doi, H.; Barletta, J.; Suzuki, M.; Noyori, R.; Watanabe, Y.; Långström, B. Synthesis of 11C-labelled N,N'-diphenylurea and ethyl phenylcarbamate by a rhodium-promoted carbonylation via [11C]isocyanatobenzene using phenyl azide and [11C]carbon monoxide. Org. Biomol. Chem. 2004, 2, 3063–3066. [Google Scholar] [CrossRef]
- Ilovich, O.; Åberg, O.; Långström, B.; Mishania, E. Rhodium-mediated [11C]Carbonylation: a li-brary of N-phenyl-N'-{4-(4-quinolyloxy)-phenyl}-[11C]-urea derivatives as potential PET angio-genic probes. J. Labelled Compd. Radiopharm. 2008, 52, 151–157. [Google Scholar]
- La Monica, G.; Ardizzoia, G. Five-membered Heterocycles by the Rh(I)-Catalysed Carbonylation of Aromatic ortho-Substituted Azides. J. Mol. Catal. 1986, 38, 327–330. [Google Scholar] [CrossRef]
- Vallabhajosula, S. Molecular Imaging; Springer: Berlin, Heidelberg, Germany, 2008; pp. 151–164. [Google Scholar]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthese von 11C-, 18F-, 15O- und 13N-Radiotracern für die Positronenemissionstomographie. Angew. Chem. 2008, 120, 9136–9172. [Google Scholar]
- Kima, D.W.; Choeb, Y.S.; Chi, D.Y. A new nucleophilic fluorine-18 labeling method for aliphatic mesylates: reaction in ionic liquids shows tolerance for water. Nucl. Med. Biol. 2003, 30, 345–350. [Google Scholar] [CrossRef]
- Le Bars, D. Fluorine-18 and medical imaging: Radiopharmaceuticals for positron emission tomography. J. Fluorine Chem. 2006, 127, 1488–1493. [Google Scholar] [CrossRef]
- Wuest, F.; Berndt, M.; Kniess, T. Recent Advances of Bioconjugation Chemistry in Molecular Imaging; Chen, X., Ed.; Research Signpost: Kerala, India, 2008; pp. 155–173. [Google Scholar]
- Piarraud, A.; Lasne, M.C.; Barré, L.; Vaugeois, J.M.; Lancelot, J.C. Synthesis of no carrier added [18f] Gbr 12936 via a wittig reaction for use in a the dopamine reuptake site study. J. Labelled Compd. Radiopharm. 1993, 32, 253–254. [Google Scholar]
- Hantraye, P.; Brownell, A.-L.; Elmaleh, D.; Spealman, R.D.; Wüllner, U.; Brownell, G.L.; Madras, B.K.; Isacson, O. Dopamine fiber detection by [11C]-CFT and PET in a primate model of parkinsonism. Neuroreport 1992, 3, 265–268. [Google Scholar] [CrossRef]
- Allain-Barbier, L.; Lasne, M.C.; Huard, C.; Barré, L. Palladium Mediated Synthesis of [18F]Fluorophenyl Alkenes or Arenes. J. Labelled Compd. Radiopharm. 1995, 37, 572–574. [Google Scholar]
- Allain-Barbier, L.; Lasne, M.C.; Perrio-Huard, C.; Moreau, B.; Barré, L. Synthesis of [18F]Fluorophenyl Alkenes or Arenes via Palladium-Catalyzed Coupling of 4-[18F]Fluoroiodo-benzene with Vinyl and Aryl Tin Reagents. Acta Chem. Scand. 1998, 52, 480–489. [Google Scholar] [CrossRef]
- Gail, R.; Coenen, H.H. A one step preparation of the n.c.a. fluorine-18 labelled synthons: 4-Fluorobromobenzene and 4-fluoroiodobenzene. Appl. Radiat. Isot. 1994, 45, 105–111. [Google Scholar] [CrossRef]
- Forngren, T.; Andersson, Y.; Lamm, B.; Långström, B. Palladium-Mediated Coupling Reactions of 18F-Labelled Arylhalides with Organotin Compounds. J. Labelled Compd. Radiopharm. 1995, 37, 595–596. [Google Scholar]
- Plenevaux, A.; Lemaire, C.; Palmer, A.J.; Damhaut, P.; Comar, D. Synthesis of non-activated 18F-fluorinated aromatic compounds through nucleophilic substitution and decarboxylation reactions. Int. J. Radiat. Appl. Instrument. Part A; Appl. Radiat. Isot. 1992, 43, 1035–1040. [Google Scholar] [CrossRef]
- Forngren, T.; Andersson, Y.; Lamm, B.; Långström, B. Synthesis of [4-18F]-1-Bromo-4-fluorobenzene and its Use in Palladium-Promoted Cross-Coupling Reactions with Organostannanes. Acta Chem. Scand. 1998, 52, 475–479. [Google Scholar] [CrossRef]
- Marrière, E.; Rouden, J.; Tadino, V.; Lasne, M.C. Synthesis of Analogues of (−)-Cytisine for in Vivo Studies of Nicotinic Receptors Using Positron Emission Tomography. Org. Lett. 2000, 2, 1121–1124. [Google Scholar] [CrossRef]
- Holladay, M.W.; Dart, M.J.; Lynch, J.K. Neuronal Nicotinic Acetylcholine Receptors as Targets for Drug Discovery. J. Med. Chem. 1997, 40, 4169–4194. [Google Scholar] [CrossRef]
- Wüst, F.R.; Kniess, T. Synthesis of 4-[18F]fluoroiodobenzene and its application in sonogashira cross-coupling reactions. J. Labelled Compd. Radiopharm. 2003, 46, 699–713. [Google Scholar] [CrossRef]
- Mangner, T.J.; Klecker, R.W.; Anderson, L.; Shields, A.F. Synthesis of 2′-deoxy-2′-[18F]fluoro-β-D-arabinofuranosyl nucleosides, [18F]FAU, [18F]FMAU, [18F]FBAU and [18F]FIAU, as potential PET agents for imaging cellular proliferation: Synthesis of [18F]labelled FAU, FMAU, FBAU. Nucl. Med. Biol. 2003, 30, 215–224. [Google Scholar] [CrossRef]
- Toyohara, J.; Fujibayashi, Y. Trends in nucleoside tracers for PET imaging of cell proliferation. Nucl. Med. Biol. 2003, 30, 681–685. [Google Scholar] [CrossRef]
- Brust, P.; Haubner, R.; Friedrich, A.; Scheunemann, M.; Anton, M.; Koufaki, O.-N.; Hauses, M.; Noll, S.; Noll, B.; Haberkorn, U.; Schackert, G.; Schackert, H.K.; Avril, N.; Johannsen, B. Comparison of [18F]FHPG and [124/125I]FIAU for imaging herpes simplex virus type 1 thymidine kinase gene expression. Eur. J. Nucl. Med. Mol. Imaging 2001, 28, 721–729. [Google Scholar] [CrossRef]
- Wüst, F.R.; Kniess, T. No-carrier added synthesis of 18F-labelled nucleosides using Stille cross-coupling reactions with 4-[18F]fluoroiodobenzene. J. Labelled Compd. Radiopharm. 2004, 47, 457–468. [Google Scholar] [CrossRef]
- Marnett, L.J. Cyclooxygenase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 545–552. [Google Scholar] [CrossRef]
- Wüst, F.R.; Höhne, A.; Metz, P. Synthesis of 18F-labelled cyclooxygenase-2 (COX-2) inhibitors via Stille reaction with 4-[18F]fluoroiodobenzene as radiotracers for positron emission tomography (PET). Org. Biomol. Chem. 2005, 3, 503–507. [Google Scholar] [CrossRef]
- Li, J.J.; Anderson, G.D.; Burton, E.G.; Cogburn, J.N.; Collins, J.T.; Garland, J.D.; Gregory, S.A.; Huang, H.; Isakson, P.C.; Koboldt, C.M.; Logusch, E.W.; Norton, M.B.; Perkins, W.E.; Reinhard, E.J.; Seibert, K.; Veenhuizen, A.W.; Zhang, Y.; Reitz, D.B. 1,2-Diarylcyclopentenes as Selective Cyclooxygenase-2 Inhibitors and Orally Active Anti-inflammatory Agents. J. Med. Chem. 1995, 38, 4570–4578. [Google Scholar]
- Steininger, B.; Wuest, F.R. Synthesis of 18F-labelled biphenyls via SUZUKI cross-coupling with 4-[18F]fluoroiodobenzene. J. Labelled Compd. Radiopharm. 2006, 49, 817–827. [Google Scholar] [CrossRef]
- Marrière, E.; Chazalviel, L.; Dhilly, M.; Toutain, J.; Perrio, C.; Dauphin, F.; Lasne, M.C. Synthesis of [18F]RP 62203, a potent and selective Serotonin 5-HT2A Receptor Antagonist and Biological Evaluation with ex vivo Autoradiography. J. Labelled Compd. Radiopharm. 1999, 42, S69–S71. [Google Scholar]
- Malleron, J.L.; Comte, M.T.; Gueremy, C.; Peyronel, J.F.; Truchon, A.; Blanchard, J.C.; Doble, A.; Piot, O.; Zundel, J.L. Naphthosultam Derivatives: A New Class of Potent and Selective 5-HT2 Antagonists. J. Med. Chem. 1991, 34, 2477–2483. [Google Scholar] [CrossRef]
- Das, M.K.; Mukherjee, J. [18F]RP 62203: A High Affinity and Selective Radioligand as a Potential PET Tracer for Serotonin 5HT2 Receptor. J. Labelled Compd. Radiopharm. 1994, 35, 497–499. [Google Scholar]
- Collins, M.; Lasne, M.-C.; Barré, L. Rapid synthesis of N,N′-disubstituted piperazines. Application to the preparation of No carrier added 1-(4-[18F]fluorophenyl)piperazine and of an [18F]-selective ligand of serotoninergic receptors (5HT2 antagonist). J. Chem. Soc. Perkin Trans. 1 1992, 23, 3185–3188. [Google Scholar]
- Lasne, M.C.; Le Secq, B.; Barre, L.; Collins, M.; Huard, C.; Baron, J.C. Synthesis of [18F]RP 62203, a Selective Antagonist of 5HT2 Receptors for Positron Emission Tomography Studies. J. Labelled Compd. Radiopharm. 1994, 35, 441–443. [Google Scholar]
- Stabler, S. R.; Jahangir. Preparation of N-Arylated Heterocycles by Nucleophilic Aromatic Substitution. Synth. Commun. 1994, 24, 123–129. [Google Scholar] [CrossRef]
- Mor, M.; Plazzi, P.V.; Rivara, S. 2-[N-Acylamino(C1−C3)alkyl]indoles as MT1 Melatonin Receptor Partial Agonists, Antagonists, and Putative Inverse Agonists. J. Med. Chem. 1998, 41, 3624–3634. [Google Scholar] [CrossRef]
- Andersen, K.; Liljefors, T.; Hyttel, J.; Perregaard, J. Serotonin 5-HT2 Receptor, Dopamine D2 Receptor, and α1 Adrenoceptor Antagonist. Conformationally Flexible Analogues of the Atypical Antipsychotic Sertindole. J. Med. Chem. 1996, 39, 3723–3738. [Google Scholar] [CrossRef]
- Perregaard, J.; Moltzen, E.K.; Meier, E.; Sanchez, C. σ Ligands with Subnanomolar Affinity and Preference for the σ2 Binding Site. 1. 3-(ω-Aminoalkyl)-1H-indoles. J. Med. Chem. 1995, 38, 1998–2008. [Google Scholar] [CrossRef]
- Sánchez, C.; Arnt, J.; Costall, B.; Kelly, M.E.; Meier, E.; Naylor, R.J.; Perregaard, J. The Selective σ2-Ligand Lu 28-179 Has Potent Anxiolytic-Like Effects in Rodents. J. Pharmacol. Exp. Ther. 1997, 283, 1333–1341. [Google Scholar]
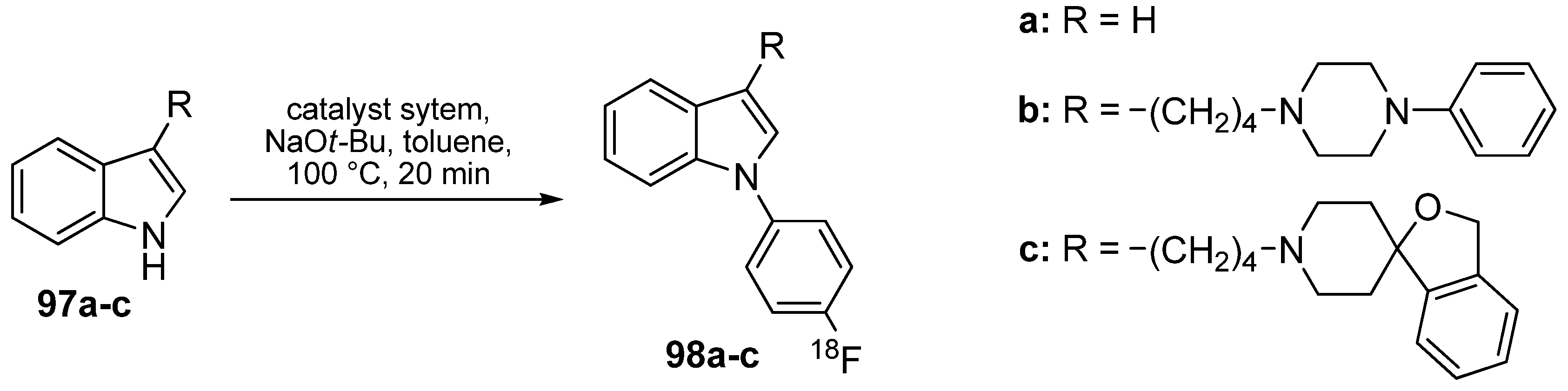
- Wüst, F.R.; Kniess, T. N-Arylation of indoles with 4-[18F]fluoroiodobenzene: synthesis of 18F-labelled σ2 receptor ligands for positron emission tomography (PET). J. Labelled Compd. Radiopharm. 2005, 48, 31–43. [Google Scholar] [CrossRef]
- Lee, B.-C.; Dence, C.S.; Zhou, H.; Parent, E.E.; Welch, M.J.; Katzenellenbogen, J.A. Fluorine-18 labeling and biodistribution studies on peroxisome proliferator-activated receptor-γ ligands: potential positron emission tomography imaging agents. Nucl. Med. Biol. 2009, 36, 147–153. [Google Scholar] [CrossRef]
- Henke, B.R.; Blanchard, S.G.; Brackeen, M.F.; Brown, K.K.; Cobb, J.E.; Collins, J.L.; Harrington, W.W., Jr.; Hashim, M.A.; Hull-Ryde, E.A.; Kaldor, I.; Kliewer, S.A.; Lake, D.H.; Leesnitzer, L.M.; Lehmann, J.M.; Lenhard, J.M.; Orband-Miller, L.A.; Miller, J.F.; Mook, R.A., Jr.; Noble, S.A.; Oliver, W., Jr.; Parks, D.J.; Plunket, K.D:; Szewczyk, J.R:; Willson, T.M. N-(2-Benzoylphenyl)-l-tyrosine PPARγ agonists: 1. Discovery of a Novel Series of Potent Antihyperglycemic and Antihyperlipidemic Agents. J. Med. Chem. 1998, 41, 5020–5036. [Google Scholar]
- Young, P.W.; Buckle, D.R.; Cantello, B.C.C.; Chapman, H.; Clapham, J.C.; Coyle, P.J.; Haigh, D.; Hindley, R.M.; Holder, J.C.; Kallender, H.; Latter, A.J.; Lawrie, K.W.M.; Mossakowska, D.; Murphy, G.J.; Cox, L.R.; Smith, S.A. Identification of High-Affinity Binding Sites for the Insulin Sensitizer Rosiglitazone (BRL-49653) in Rodent and Human Adipocytes Using a Radioiodinated Ligand for Peroxisomal Proliferator-Activated Receptor γ. J. Pharmacol. Exp. Ther. 1998, 284, 751–759. [Google Scholar]
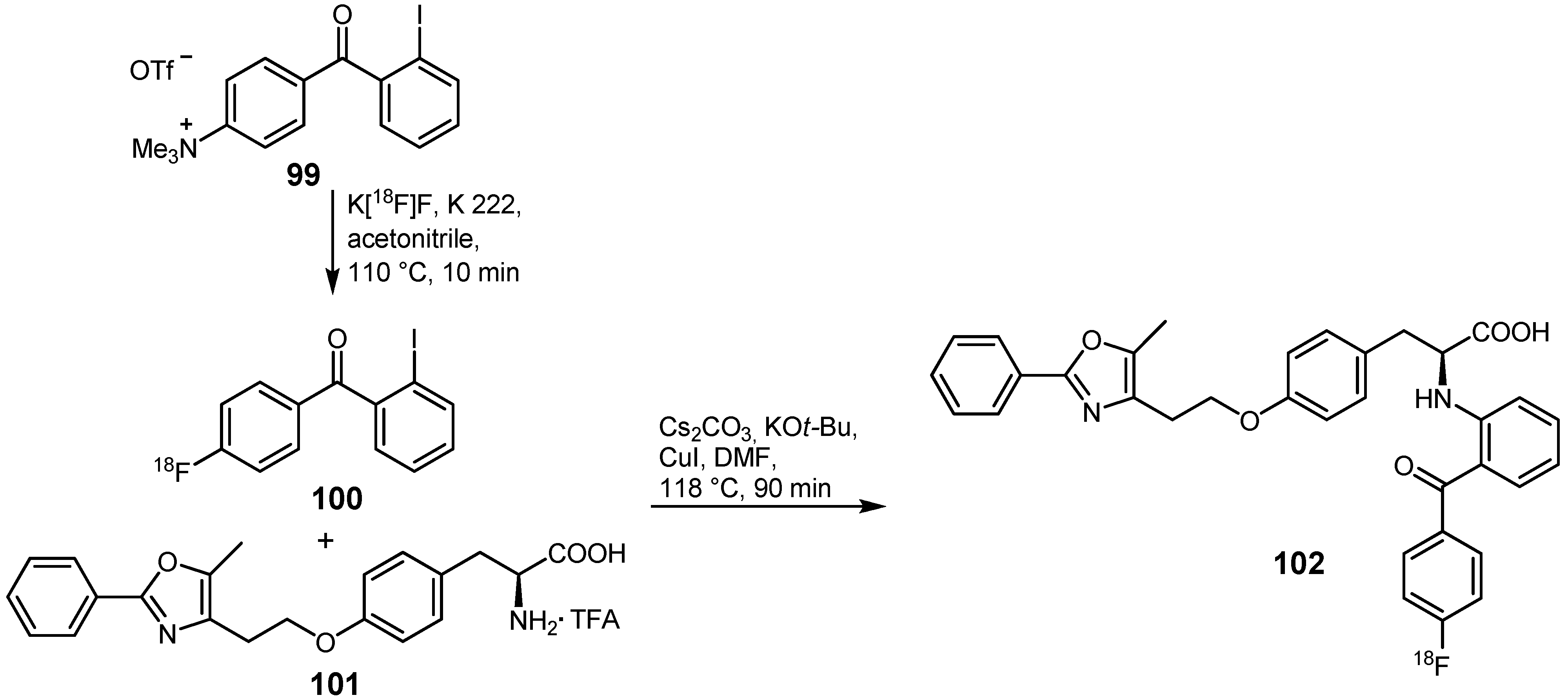
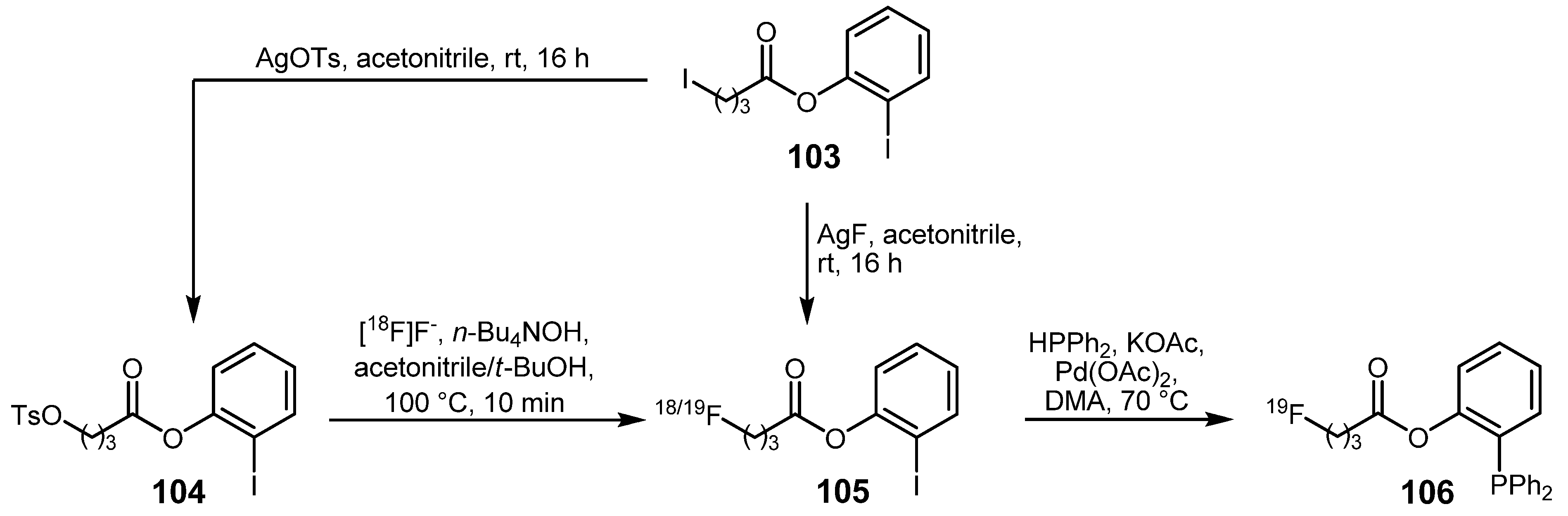
- Pretze, M.; Flemming, A.; Köckerling, M.; Mamat, C. Synthesis and Radiofluorination of Iodophenyl Esters as Tool for the Traceless Staudinger Ligation. Z. Naturforsch. 2010, 65b, 1128–1136. [Google Scholar]
- Pretze, M.; Wuest, F.; Peppel, T.; Köckerling, M.; Mamat, C. The traceless Staudinger ligation with fluorine-18: A novel and versatile labeling technique for the synthesis of PET-radiotracers. Tetrahedron Lett. 2010, 51, 6410–6414. [Google Scholar]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pretze, M.; Große-Gehling, P.; Mamat, C. Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers. Molecules 2011, 16, 1129-1165. https://doi.org/10.3390/molecules16021129
Pretze M, Große-Gehling P, Mamat C. Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers. Molecules. 2011; 16(2):1129-1165. https://doi.org/10.3390/molecules16021129
Chicago/Turabian StylePretze, Marc, Philipp Große-Gehling, and Constantin Mamat. 2011. "Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers" Molecules 16, no. 2: 1129-1165. https://doi.org/10.3390/molecules16021129
APA StylePretze, M., Große-Gehling, P., & Mamat, C. (2011). Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers. Molecules, 16(2), 1129-1165. https://doi.org/10.3390/molecules16021129