Antimony(V) and Bismuth(V) Complexes of Lapachol: Synthesis, Crystal Structure and Cytotoxic Activity

Abstract
:1. Introduction

2. Results and Discussion

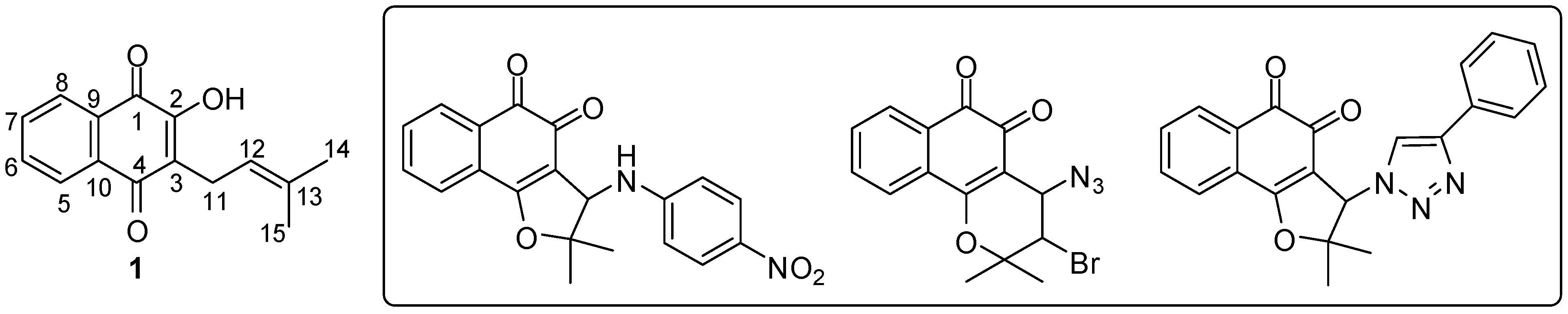{kind=link}
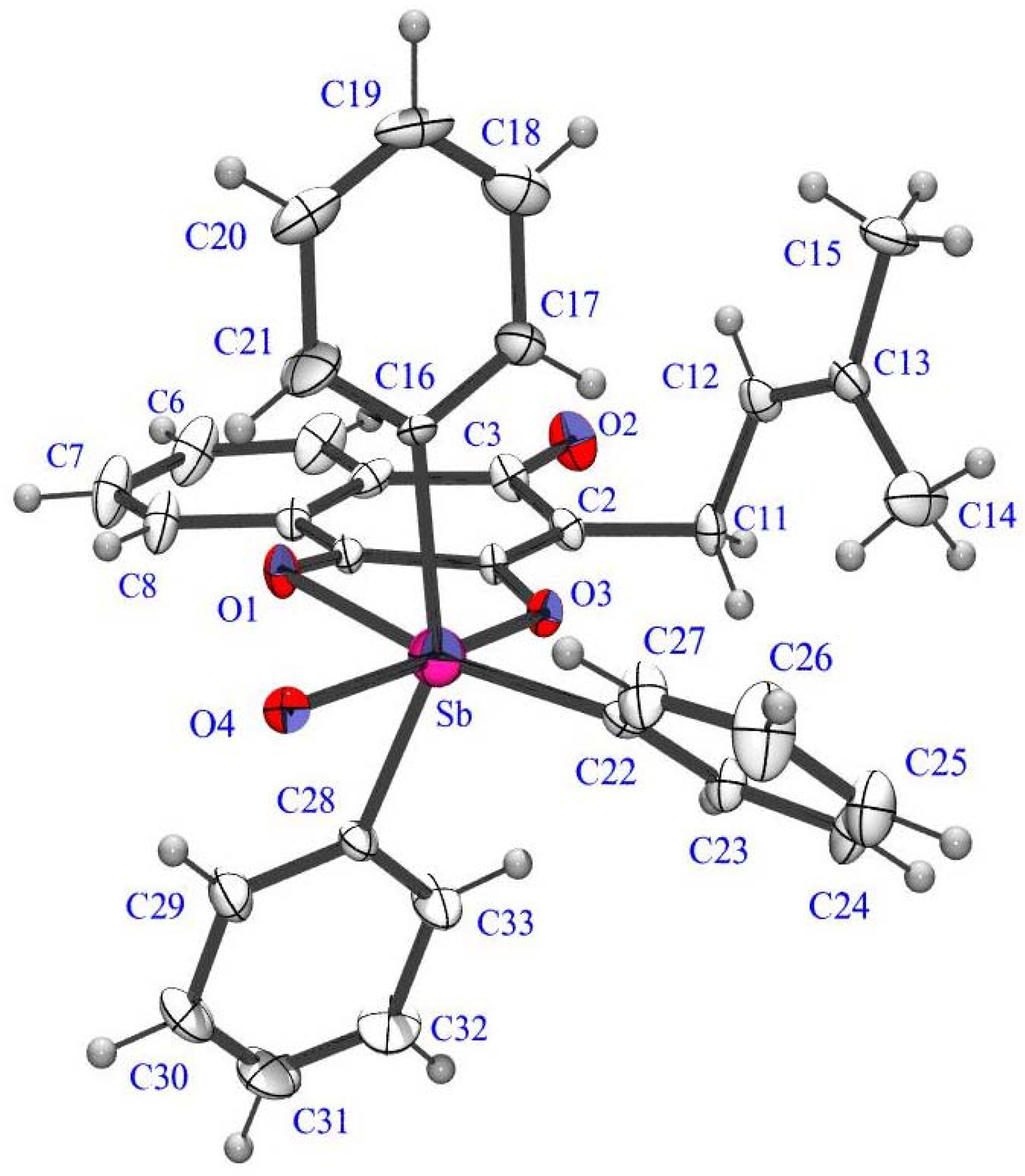{kind=link}
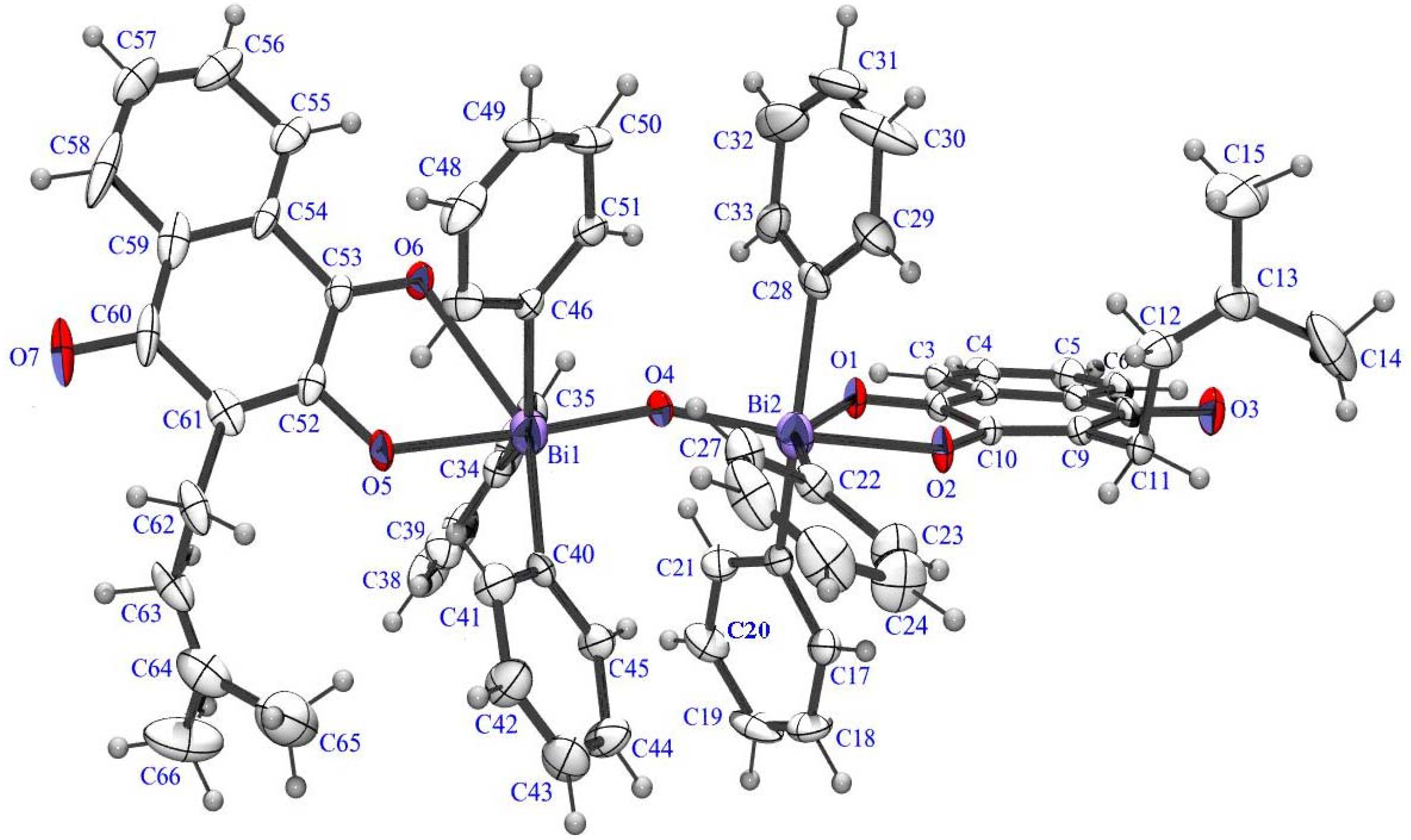{kind=link}
| Compound | Lp | (2) | (3) | Lp | (2) | (3) | |
|---|---|---|---|---|---|---|---|
| Solvent | CDCl3 | CDCl3 | CDCl3 | CDCl3 | CDCl3 | CDCl3 | |
| Number | δ 13C | δ 13C | δ 13C | δ 1H | δ 1H | δ 1H | |
| 1 | 184.5 | 182.6 | 183.3 | ||||
| 2 | 152.7 | 158.1 | 156.0 | ||||
| 3 | 123.5 | 126.0 | 122.1 | ||||
| 4 | 181.7 | 184.9 | 185.5 | ||||
| 5 | 126.0 | 126.1 | 125.7 | 8.1 (dd 6.4 and 1.3) | 8.0 (d, 7.6) | 8.2 (d, 7.6) | |
| 6 | 132.8 | 134.3 | 131.5 | 7.7 (td, 6.4 and 1.3) | 7.6 (t, 7.5) | 7.7 (t, 7.8) | |
| 7 | 134.8 | 133.4 | 134.0 | 7.7 (td, 6.4 and 1.3) | 7.5 (t, 7.6) | 8.0 (t, 7.6) | |
| 8 | 126.7 | 126.3 | 125.9 | 8.1 (dd, 6.4 and 1.3) | 8.1 (d,7.6) | 8.5 (d, 7.6) | |
| 9 | 129.4 | 132.0 | 130.2 | ||||
| 10 | 132.9 | 133.4 | 133.6 | ||||
| 11 | 22.6 | 23.2 | 23.2 | 3.3 (d, 6.7) | 3.3 (d, 7.2) | 3.4 (d, 6.8) | |
| 12 | 119.6 | 121.3 | 121.9 | 5.2 (m, 6.7) | 5.1 (m, 6.8) | 5.1 (m, 6.8) | |
| 13 | 133.8 | 132.8 | 133.2 | ||||
| 14 or 15 | 25.7 | 25.6 | 25.7 | 1.7 s | 1.6 s | 1.6 s | |
| 15 or 14 | 17.8 | 17.7 | 17.8 | 1.6 s | 1.5 s | 1.7 s | |


| Empirical formula | C66H58Bi2O7 | C33H28SbO4 | ||||
| Formula weight | 1381.08 | 610.30 | ||||
| Temperature | 295 (2) K | 295 (2) K | ||||
| Wavelength | 0.71073 Å | 0.71073 Ă | ||||
| Crystal system, space group | Monoclinic, C2/c | Monoclinic, P21/c | ||||
| Unit cell dimensions | a = 27.8310(5) Å | a = 14.9030(2) Å | ||||
| b = 15.6580(3) Å | β = 100.58(1)° | b = 8.9130(3) Å | β = 99.09(1)° | |||
| c = 27.1660(5) Å | c = 21.8200(3) Å | |||||
| Volume | 11637.2(4) Å3 | 2861.9(6) Å3 | ||||
| Z, Calculated density | 4, 1.58 Mg/m3 | 4, 1.42 Mg/m3 | ||||
| Absorption coefficient | 2.406 mm−1 | 0.99 mm−1 | ||||
| F(000) | 5408 | 1236 | ||||
| Crystal size | 0.42 × 0.31 × 0.29 mm | 0.32 × 0.27 × 0.22 mm | ||||
| Theta range for data collection | 2.6 to 27.53° | 2.6 to 27.4° | ||||
| Limiting indices | −36 ≤ h ≤ 36, −20 ≤ k ≤ 20, −35 ≤ l ≤ 35 | −19 ≤ h ≤ 19, −11 ≤ k ≤ 11, −27 ≤ l ≤28 | ||||
| Reflections collected/unique | 68287/13271 [Rint = 0.052] | 42238/6275 [Rint = 0.102] | ||||
| Completeness to theta = 27.52 | 98.8% | 98.8% | ||||
| Refinement method | Full-matrix least-squares | Full-matrix least-squares | ||||
| Data/restraints/parameters | 10165/0/676 | 4902/0/344 | ||||
| Goodness-of-fit on F2 | 1.242 | 1.027 | ||||
| Final R indices [I > 2σ(I) ] | R1 = 0.087, wR2 = 0.122 | R1 = 0.080, wR2 = 0.098 | ||||
| R índices (all data) | R1 = 0.232, wR2 = 0.289 | R1 = 0.245, wR2 = 0.266 | ||||
| Largest diff. peak and hole | 1.401 and −2.035 e.Å-3 | 1.935 and −0.965 e.Å−3 | ||||
| Lp | Ph3BiCl2 | Ph3SbCl2 | (Lp)2(Ph3Bi)2O | (Lp)(Ph3Sb)OH | |
|---|---|---|---|---|---|
| a IC50 (μM) | 9.2 ± 0.9 | 30.1 ± 0.1 | 17.6 ± 1.6 | 1.8 ± 0.3 | 36.4 ± 1.8 |
3. Experimental
3.1. General
3.2. Cell Line, Culture and Cytotoxicity Assays
3.3. Synthesis of the Complex (Lp)(Ph3Sb)OH (2) and (Lp)2(Ph3Bi)2O (3)
3.3.1. Preparation of (Lp)(Ph3Sb)OH (2)
3.3.2. Preparation of (Lp)2(Ph3Bi)2O (3)
3.4. X-Ray Analysis
4. Conclusions
Acknowledgements
References and Notes
- Rao, K.V.; McBride, T.J.; Oleson, J.J. Recognition and evaluation of lapachol as an antitumor agent. Cancer Res. 1968, 28, 1952–1954. [Google Scholar]
- Nagata, K.; Hirai, K.-I.; Koyama, J.; Wada, Y.; Tamura, T. Antimicrobial activity of novel furanonaphthoquinone analogs. Antimicrob. Agents Chemother. 1998, 42, 700–702. [Google Scholar]
- de Andrade-Neto, V.F.; Goulart, M.O.F.; da Silva Filho, J.F.; da Silva, M.J.; Pinto, M.C.F.R.; Pinto, A.V.; Zalis, M.G.; Carvalho, L.H.; Krettli, A.U. Antimalarial activity of phenazines from lapachol, β-lapachone and its derivatives against Plasmodium falciparum in vitro and Plasmodium berghei in vivo. Bioorg. Med. Chem. Lett. 2004, 14, 1145–1149. [Google Scholar]
- de Moura, K.C.G.; Emery, F.S.; Pinto, C.N.; Pinto, M.C.F.R.; Dantas, A.P.; Salomão, K.; de Castro, S.L.; Pinto, A.V. Trypanocidal activity of isolated naphthoquinones from Tabebuia and some heterocyclic derivatives: A review from an interdisciplinary study. J. Braz. Chem. Soc. 2001, 12, 325–338. [Google Scholar]
- Lima, N.M.F.; Correia, C.S.; Leon, L.L.; Machado, G.M.C.; Madeira, M.F.; Santana, A.E.G.; Goulart, M.O.F. Antileishmanial activity of lapachol analogues. Mem. Inst. Oswaldo Cruz 2004, 99, 757–761. [Google Scholar] [CrossRef]
- da Silva Júnior, E.N.; Guimarães, T.T.; Menna-Barreto, R.F.S.; Pinto, M.C.F.R.; de Simone, C.A.; Pessoa, C.; Cavalcanti, B.C.; Sabino, J.R.; Andrade, C.K.Z.; Goulart, M.O.F.; et al. The evaluation of quinonoid compounds against Trypanosoma cruzi: Synthesis of imidazolic anthraquinones, nor-β-lapachone derivatives and β-lapachone-based 1,2,3-triazoles. Bioorg. Med. Chem. 2010, 18, 3224–3230. [Google Scholar]
- da Silva Júnior, E.N.; de Deus, C.F.; Cavalcanti, B.C.; Pessoa, C.; Costa-Lotufo, L.V.; Montenegro, R.C.; de Moraes, M.O.; Pinto, M.C.F.R.; de Simone, C.A.; Ferreira, V.F.; et al. 3-Arylamino and 3-alkoxy-nor-β-lapachone derivatives: synthesis and cytotoxicity against cancer cell lines. J. Med. Chem. 2010, 53, 504–508. [Google Scholar]
- Tonholo, J.; Freitas, L.R.; de Abreu, F.C.; Azevedo, D.C.; Zani, C.L.; de Oliveira, A.B.; Goulart, M.O.F. Electrochemical properties of biologically active heterocyclic naphthoquinones. J. Braz. Chem. Soc. 1998, 9, 163–169. [Google Scholar] [CrossRef]
- Hillard, E.A.; Abreu, F.C.; Ferreira, D.C.M.; Jaouen, G.; Goulart, M.O.F.; Amatore, C. Electrochemical parameters and techniques in drug development, with an emphasis on quinones and related compounds. Chem. Commun. 2008, 2612–2628. [Google Scholar]
- Ferraz, P.A.L.; de Abreu, F.C.; Pinto, A.V.; Glezer, V.; Tonholo, J.; Goulart, M.O.F. Electrochemical aspects of the reduction of biologically active 2-hydroxy-1,4-naphthoquinones. J. Electroanal. Chem. 2001, 507, 275–286. [Google Scholar] [CrossRef]
- Bodini, M.E.; Arancibia, V. Manganese complexes with 2-hydroxy-3(3-methyl-2-butenyl)-1,4-naphthoquinone (Lapachol). Redox chemistry and spectroscopy in dimethylsulphoxide. Polyhedron 1989, 8, 1407–1412. [Google Scholar] [CrossRef]
- Sawhney, S.S.; Vohra, N.; Chandel, S.K. Formation constants and thermodynamic functions of Cd(II), Zn(II), Pb(II), VO2+ and Ce(IV) with Lapachol. Thermochim. Acta 1982, 52, 349–350. [Google Scholar] [CrossRef]
- Sawhney, S.S.; Bains, S.S. Thermal and pH-metric studies on complexes of Hg(II) and Er(III) with 2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinone (lapachol). Thermochim. Acta 1983, 71, 381–386. [Google Scholar] [CrossRef]
- Hernández-Molina, R.; Kalinina, I.; Esparza, P.; Sokolov, M.; Gonzalez-Platas, J.; Estévez-Braun, A.; Pérez-Sacau, E. Complexes of Co(II), Ni(II) and Cu(II) with lapachol. Polyhedron 2007, 26, 4860–4864. [Google Scholar] [CrossRef]
- Caruso, F.; Martýnez, M.; Rossi, M.; Goldberg, A.; ChaconVillalba, M.E.; Aymonin, P.J. Crystal and molecular structure of manganese(II) lapacholate, a novel polymeric species undergoing temperature-reversible metal to ligand electron transfer. Inorg. Chem. 2009, 48, 3529–3534. [Google Scholar]
- Ge, R.; Sun, H. Bioinorganic chemistry of bismuth and antimony: Target sites of metallodrugs. Acc. Chem. Res. 2007, 40, 267–274. [Google Scholar] [CrossRef]
- Frézard, F.; Demicheli, C.; Ribeiro, R.R. Pentavalent antimonials: New perspectives for old drugs. Molecules 2009, 14, 2317–2336. [Google Scholar] [CrossRef]
- Tiekink, E.R. Antimony and bismuth compounds in oncology. Crit. Rev. Oncol. Hematol. 2002, 42, 217–224. [Google Scholar] [CrossRef]
- Sharma, P.; Perez, D.; Cabrera, A.; Rosas, N.; Arias, J.L. Perspectives of antimony compounds in oncology. Acta Pharmacol. Sin. 2008, 29, 881–890. [Google Scholar] [CrossRef]
- Silvestru, C.; Socaciu, C.; Bara, A.; Haiduc, I. The first organoantimony(III) compounds possessing antitumor properties: Diphenylantimony(III) derivatives of dithiophosphorus ligands. Anticancer Res. 1990, 10, 803–804. [Google Scholar]
- Bara, A.; Socaciu, C.; Silvestru, C.; Haiduc, I. Antitumour organometallics. I. Activity of some diphenyltin(IV) and diphenylantimony(III) derivatives on in vitro and in vivo Ehrlich ascites tumor. Anticancer Res. 1991, 11, 1651–1655. [Google Scholar]
- Carraher, C.E., Jr.; Nass, M.D.; Giron, D.J.; Cerutis, D.R. Structural and biological characterization of antimony(V) polyamines. J. Macromol. Sci. Chem. 1983, 19, 1101–1120. [Google Scholar] [CrossRef]
- Wang, G.C.; Xiao, J.; Lu, Y.; Li, J.S.; Cui, J.R.; Wang, R.Q.; Ran, F.X. Synthesis, crystal structures and in vitro antitumor activities of some arylantimony derivatives of analogues of demethylcantharimide. J. Organomet. Chem. 2004, 689, 1631–1638. [Google Scholar] [CrossRef]
- Li, J.S.; Ma, Y.Q.; Cui, J.R.; Wang, R.Q. Synthesis and in vitro antitumor activity of some tetraphenylantimony derivatives of exo-7-oxa-bicyclo[2,2,1] heptane (ene)-3-arylamide-2-acid. Appl. Organomet. Chem. 2001, 15, 639–645. [Google Scholar] [CrossRef]
- Desoize, B. Metals and metal compounds in cancer treatment. Anticancer Res. 2004, 24, 1529–1544. [Google Scholar]
- Murafuji, T.; Miyoshi, Y.; Ishibashi, M.; Rahman, A.F.M.M.; Sugihara, Y.; Miyakawa, I.; Uno, H. Antifungal activity of organobismuth compounds against the yeast Saccharomyces cerevisiae: Structure-activity relationship. J. Inorg. Biochem. 2004, 98, 547–552. [Google Scholar] [CrossRef]
- Agrawal, R.; Sharma, J.; Nandani, D.; Batra, A.; Singh, Y. Triphenylarsenic(V) and -antimony(V) derivatives of multidentate Schiff bases: Synthesis, characterization, and antimicrobial activitie. J. Coord. Chem. 2011, 64, 554–563. [Google Scholar] [CrossRef]
- Matano, Y.; Nomura, H.; Hisanaga, T.; Nakano, H.; Shiro, M.; Imahori, H. Diverse structures and remarkable oxidizing ability of triarylbismuthane oxides. Comparative study on the structure and reactivity of a series of triarylpnictogen oxides. Organometallics 2004, 23, 5471–5480. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ohdoi, K.; Chen, X.; Kitano, M.; Akiba, K. Synthesis and structure of six-coordinated organobismuth compounds with bidentate ligands (12-Bi-6). Organometallics 1993, 12, 3297–3303. [Google Scholar] [CrossRef]
- Campi, E.M.; Deacon, G.B.; Jackson, W.R.; Skelton, B.W.; Smith, K.A.; White, A.H. Novel coordination complexes in the bismuth(III)/oxinate system. Z. Anorg. Allg. Chem. 2006, 632, 1483–1486. [Google Scholar] [CrossRef]
- Dittes, U.; Keppler, B.K.; Nuber, B. Synthesis and structure of seven-coordinate bismuth(V) complexes with benzenoid and non-benzenoid arene ligands: Tri(aryl)tropolonatobismuth(V) complexes. Angew. Chem. Int. Ed. 1996, 35, 67–68. [Google Scholar] [CrossRef]
- Enraf-Nonius. In Collect; Nonius BV: Delft, The Netherlands; pp. 1997–2000.
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology; Carter, C.W., Jr., Sweet, R.M., Eds.; Academic Press: New York, NY, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Sheldrick, G.M. SHELXS-97. Program for crystal structure resolution. University of Göttingen: Göttingen, Germany, 2008. [Google Scholar]
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sample Availability:Samples of the antimony(V) and bismuth(V) complexes of lapachol are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oliveira, L.G.d.; Silva, M.M.; Paula, F.C.S.d.; Pereira-Maia, E.C.; Donnici, C.L.; Simone, C.A.d.; Frézard, F.; Júnior, E.N.d.S.; Demicheli, C. Antimony(V) and Bismuth(V) Complexes of Lapachol: Synthesis, Crystal Structure and Cytotoxic Activity. Molecules 2011, 16, 10314-10323. https://doi.org/10.3390/molecules161210314
Oliveira LGd, Silva MM, Paula FCSd, Pereira-Maia EC, Donnici CL, Simone CAd, Frézard F, Júnior ENdS, Demicheli C. Antimony(V) and Bismuth(V) Complexes of Lapachol: Synthesis, Crystal Structure and Cytotoxic Activity. Molecules. 2011; 16(12):10314-10323. https://doi.org/10.3390/molecules161210314
Chicago/Turabian StyleOliveira, Ludmila G. de, Meiriane M. Silva, Flávia C. S. de Paula, Elene C. Pereira-Maia, Cláudio L. Donnici, Carlos A. de Simone, Frédéric Frézard, Eufrânio N. da Silva Júnior, and Cynthia Demicheli. 2011. "Antimony(V) and Bismuth(V) Complexes of Lapachol: Synthesis, Crystal Structure and Cytotoxic Activity" Molecules 16, no. 12: 10314-10323. https://doi.org/10.3390/molecules161210314