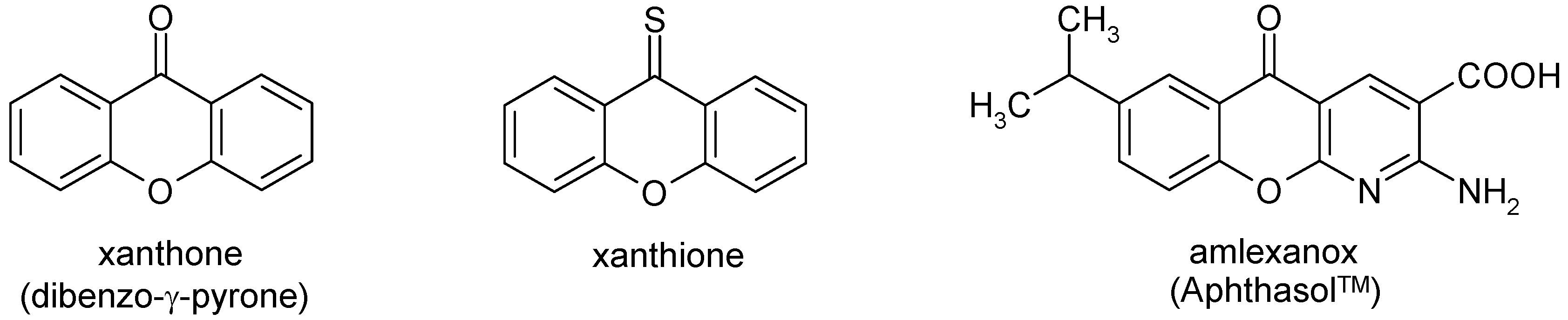
Heterocyclic Analogues of Xanthone and Xanthione. 1H-Pyrano[2,3-c:6,5-c]dipyrazol-4(7H)-ones and Thiones: Synthesis and NMR Data

Abstract
:1. Introduction


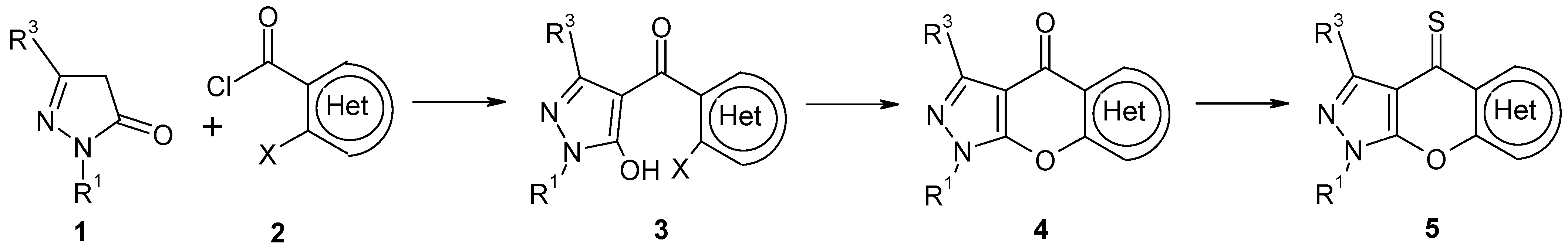
2. Results and Discussion
2.1. Chemistry


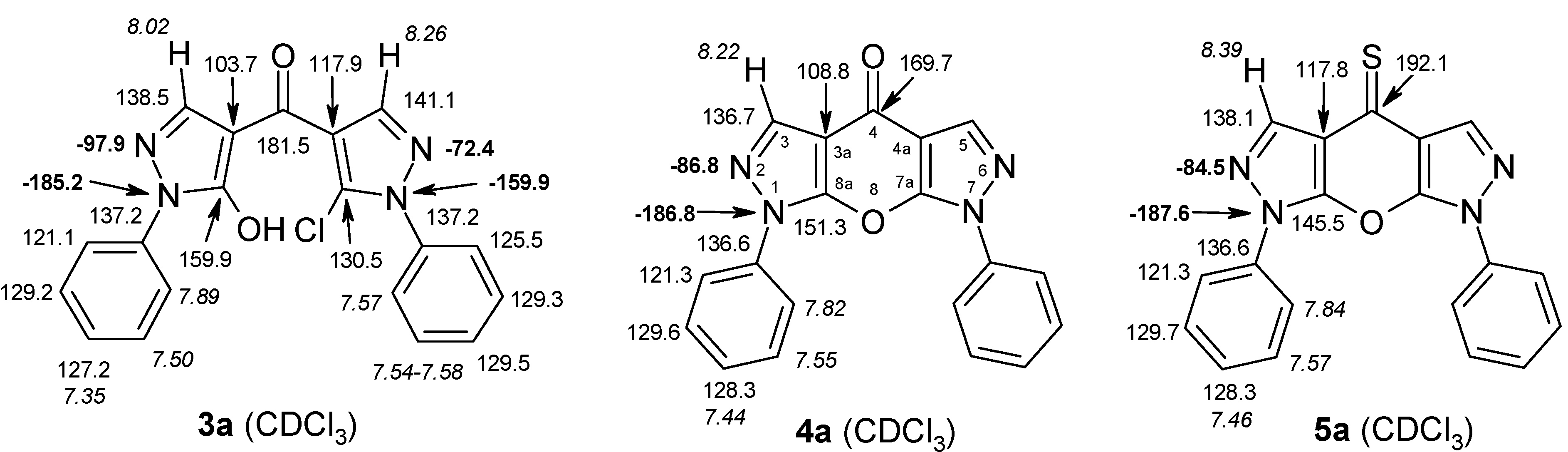
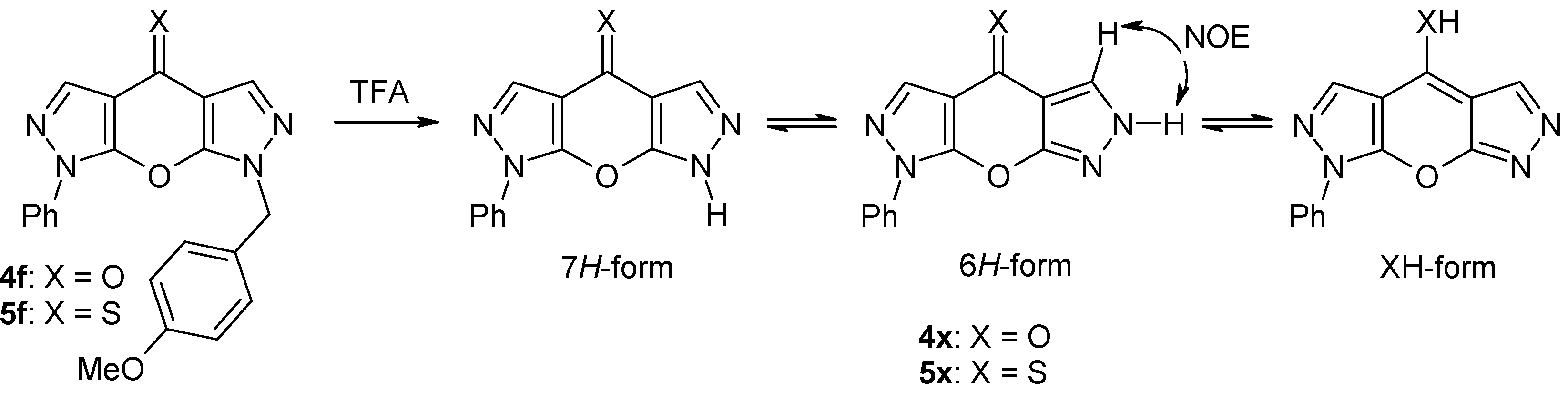
2.2. NMR Spectroscopic Investigations

3. Experimental
3.1. General
3.2. Synthetic procedures
3.2.1. General procedure for the synthesis of the carbaldehydes 6a and 6b
3.2.2. Preparation of 5-chloro-1-phenyl-1H-pyrazole-4-carboxylic acid (7a)
3.2.3. Preparation of 5-chloro-3-methyl-1-phenyl-1H-pyrazole-4-carboxylic acid (7b)
3.2.4. General procedure for the synthesis of the acid chlorides 2a and 2b
3.2.5. Acylation of Pyrazolones: General procedure for the synthesis of 3a-g and 8a-b

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | Spectroscopic and analytical data |
|---|---|---|
| 3a |  | 1H-NMR (300 MHz, CDCl3): δ (ppm) 8.26 (s, 1H, H-3’), 8.02 (s, 1H, H-3), 7.89 (m, 2H, N1-Ph H-2,6), 7.54–7.58 (3H, N1’-Ph H-3,4,5), 7.57 (m, 2H, N1’-Ph H-2,6), 7.50 (m, 2H, N1-Ph H-3,5), 7.35 (m, 1H, N1-Ph H-4), 6.0–8.5 (very broad s, 1H, OH); 13C-NMR (75 MHz, CDCl3): δ (ppm) 181.5 (C=O), 159.9 (C-5, 3J(C-5,H-3) = 4.9 Hz), 141.1 (C-3’, 1J(C-3’,H-3’) = 190.7 Hz), 138.5 (C-3, 1J(C-3,H-3) = 189.3 Hz), 137.2 (N1-Ph C-1 and N-1’-Ph C-1), 130.5 (C-5’, 3J(C-5’,H-3’) = 5.9 Hz), 129.5 (N1’-Ph C-4), 129.3 (N1’-Ph C-3,5), 129.2 (N1-Ph C-3,5), 127.2 (N1-Ph C-4), 125.5 (N1’-Ph C-2,6), 121.1 (N1-Ph C-2,6), 117.9 (C-4’, 2J(C-4’,H-3’) = 10.3 Hz), 103.7 (C-4, 2J(C-4,H-3) = 11.1 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −185.2 (N-1), −159.9 (N-1’), −97.9 (N-2), −72.4 (N-2’); IR: 1656 (C=O) cm-1; MS m/z (%): 364/266 (M+, 23/8), 329 (38), 186 (100), 91 (18), 77 (52), 51 (22). Calcd. for C19H13ClN4O2 (364.79): C, 62.56; H, 3.59; N, 15.36. Found: C, 62.39; H, 3.50; N, 15.16. |
| 3b |  | 1H-NMR (500 MHz, CDCl3): δ (ppm) 10.18 (broad s, 1H, OH), 7.92 (s, 1H, H-3), 7.89 (m, 2H, N1-Ph H-2,6), 7.58 (m, 2H, N1’-Ph H-2,6), 7.53 (m, 2H, N1’-Ph H-3,5), 7.49 (m, 2H, N1-Ph H-3,5), 7.48 (1H, N1’-Ph H-4), 7.34 (m, 1H, N1-Ph H-4), 2.50 (s, 3H, 3’-Me); 13C-NMR (125 MHz, CDCl3): δ (ppm) 183.0 (C=O), 160.0 (C-5, 3J(C-5,H-3) = 4.7 Hz), 150.5 (C-3’, 2J(C-3’,3’-Me) = 6.9 Hz), 140.1 (C-3, 1J(C-3,H-3) = 191.1 Hz), 137.3 (N1’-Ph C-1), 137.2 (N1-Ph C-1), 129.2 (N1’-Ph C-3,5), 129.15 (N1-Ph C-3,5), 129.05 (N1’-Ph C-4), 127.6 (C-5’), 127.0 (N1-Ph C-4), 125.4 (N1’-Ph C-2,6), 120.9 (N1-Ph C-2,6), 117.4 (C-4’, 3J(C-4’,3’-Me) = 2.8 Hz), 104.2 (C-4, 2J(C-4,H-3) = 10.6 Hz), 13.8 (3’-Me, 1J = 129.1 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −186.0 (N-1), −167.8 (N-1’), −97.3 (N-2), −75.8 (N-2’); IR: 1559 (C=O) cm-1; MS m/z (%): 378/380 (M+, 12/4), 343 (45), 219 (11), 186 (100), 118 (12), 91 (15), 77 (43), 51 (16). Calcd. for C20H15ClN4O2 (378.81): C, 63.41; H, 3.99; N, 14.79. Found: C, 63.24; H, 3.81; N, 14.74. |
| 3c |  | 1H-NMR (500 MHz, CDCl3): δ (ppm) 8.01 (s, 1H, H-3’), 7.87 (m, 2H, N1-Ph H-2,6), 7.60 (m, 2H, N1’-Ph H-2,6), 7.55 (m, 2H, N1’-Ph H-3,5), 7.52 (m, 1H, N1’-Ph H-4), 7.48 (m, 2H, N1-Ph H-3,5), 7.32 (m, N1-Ph H-4), 2.39 (s, 3H, 3-Me), OH not found; 13C-NMR (125 MHz, CDCl3): δ (ppm) 182.9 (C=O), 161.0 (C-5), 147.3 (C-3, 2J(C-3,3-Me) = 6.8 Hz), 140.8 (C-3’, 1J(C-3’,H-3’) = 192.0 Hz), 137.3 (N1’-Ph C-1), 137.1 (N1-Ph C-1), 129.3 (N1’-Ph C-3,4,5), 129.1 (N1-Ph C-3,5), 129.0 (C-5’, 3J(C-5’,H-3’) = 5.6 Hz), 126.9 (N1-Ph C-4), 125.4 (N1’-Ph C-2,6), 120.9 (N1-Ph C-2,6), 118.4 (C-4’, 2J(C-4’,H-3’) = 10.1 Hz), 104.4 (C-4, 3J(C-4,3-Me) = 2.7 Hz), 15.6 (3-Me, 1J = 128.8 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −190.3 (N-1), −161.7 (N-1’), −100.8 (N-2), −74.2 (N-2’); IR: 1619 (C=O) cm-1; MS m/z (%): 378/380 (M+, 8/3), 342 (48), 200 (100), 91 (37), 77 (58), 51 (25). Calcd. for C20H15ClN4O2 (378.81): C, 63.41; H, 3.99; N, 14.79. Found: C, 63.71; H, 3.91; N, 14.69. |
| 3d |  | 1H-NMR (500 MHz, CDCl3): δ (ppm) 9.20 (broad s, 1H, OH), 7.87 (m, 2H, N1-Ph H-2,6), 7.56 (m, 2H, N1’-Ph H-2,6), 7.52 (m, 2H, N1’-Ph H-3,5), 7.47 (m, 2H, N1-Ph H-3,5), 7.47 (m, 1H, N1’-Ph H-4), 7.31 (m, 1H, N1-Ph H-4), 2.41 (s, 3H, 3’-Me), 2.23 (s, 3H, 3-Me); 13C-NMR (125 MHz, CDCl3): δ (ppm) 183.8 (C=O), 160.8 (C-5), 148.8 (C-3’, 2J(C-3’,3’-Me) = 6.8 Hz), 148.1 (C-3, 2J(C-3,3-Me) = 6.7 Hz), 137.4 (N1’-Ph C-1), 137.1 (N1-Ph C-1), 129.2 (N1’-Ph C-3,5), 129.1 (N1-Ph C-3,5), 128.9 (N1’-Ph C-4), 126.8 (N1-Ph C-4), 126.7 (C-5’), 125.2 (N1’-Ph C-2,6), 120.7 (N1-Ph C-2,6), 117.8 (C-4’, 3J(C-4’,3’-Me) = 3.0 Hz), 105.5 (C-4, 3J(C-4,3-Me) = 2.8 Hz), 13.9 (3-Me, 1J = 129.0 Hz), 13.0 (3’-Me, 1J = 128.9 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −190.3 (N-1), −168.8 (N-1’), −100.0 (N-2), −76.6 (N-2’); MS m/z (%): 392/394 (M+, 5/2), 356 (39), 219 (10), 200 (100), 132 (13), 91 (31), 77 (40), 67 (12), 51 (15). Calcd. for C21H17ClN4O2 (392.84): C, 64.21; H, 4.36; N, 14.26. Found: C, 64.04; H, 4.18; N, 14.21. |
| 3e |  | 1H-NMR (300 MHz, CDCl3): δ (ppm) 10.35 (broad s, 1H, OH), 7.94 (s, 1H, H-3’), 7.44–7.59 (m, 5H, N1’-Ph), 3.62 (s, 3H, N1-Me), 2.27 (s, 3H, 3-Me); 13C-NMR (75 MHz, CDCl3): δ (ppm) 183.4 (C=O), 160.2 (C-5, 3J(C-5,N1-Me) = 2.3 Hz), 146.2 (C-3, 2J(C-3,3-Me) = 6.9 Hz), 140.6 (C-3’, 1J(C-3’,H-3’) = 191.7 Hz), 137.4 (N1’-Ph C-1), 129.2 (N1’-Ph C-3,4,5), 128.6 (C-5’, 3J(C-5’,H-3’) = 5.7 Hz), 125.3 (N1’-Ph C-2,6), 119.0 (C-4’, 2J(C-4’,H-3’) = 10.1 Hz), 103.2 (C-4, 3J(C-4,3-Me) = 2.7 Hz), 32.5 (N1-Me, 1J = 140.9 Hz), 15.2 (3-Me, 1J = 128.6 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −207.9 (N-1), −162.2 (N-1’), −100.5 (N-2), −74.7 (N-2’); IR: 1636 (C=O) cm-1;MS m/z (%): 316/318 (M+, 11/4), 281 (26), 138 (100), 77 (21), 51 (17). Calcd. for C15H13ClN4O2 (316.74): C, 56.88; H, 4.14; N, 17.69. Found: C, 57.12; H, 3.97; N, 17.61. |
| 3f |  | 1H-NMR (300 MHz, CDCl3): δ (ppm) 8.18 (s, 1H, H-3’), 7.30–8.00 (very broad s, 1H, OH), 7.82 (broad, s, 1H, H-3), 7.55 (m, 2H, N1’-Ph H-2,6), 7.52 (m, 3H, N1’-Ph H-3,4,5), 7.31 (m, 2H, CH2-Ph H-2,6), 6.88 (CH2-Ph H-3,-5), 5.13 (broad s, 2H, CH2), 3.79 (s, 3H, OMe); 13C-NMR (75 MHz, CDCl3): δ (ppm) 181.7 (C=O), 159.5 (CH2-Ph C-4), 159.0 (C-5), 141.1 (C-3’, 1J(C-3’,H-3’) = 190.7 Hz), 137.9 (C-3, 1J(C-3,H-3) = 188.8 Hz), 137.3 (N1’-Ph C-1), 130.3 (C-5’), 129.6 (CH2-Ph C-2,6), 129.4 (N1’-Ph C-4), 129.2 (N1’-Ph C-3,5), 127.5 (CH2-Ph C-1), 125.5 (N1’-Ph C-2,-6), 118.2 (C-4’, 2J(C-4’,H-3’) = 10.0 Hz), 114.2 (CH2-Ph C3,5), 103.1 (C-4), 55.3 (OMe, 1J = 143.9 Hz), 49.8 (CH2, 1J = 140.6 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −189.2 (N-1), −160.3 (N-1’), −98.3 (N-2), −73.1 (N-2’); IR: 1621 (C=O) cm-1;MS m/z (%): 408/410 (M+, 7/2), 373 (25), 121 (100), 77 (23). Calcd. for C21H17ClN4O3 (408.84): C, 61.69; H, 4.19; N, 13.70. Found: C, 61.47; H, 4.13, N, 13.55. |
| 3g |  | 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.70–8.20 (broad s, 1H, OH), 7.96 (s, 1H, H-3’), 7.58 (m, 2H, N1’-Ph H-2,6), 7.53 (m, 2H, N1’-Ph H-3,5), 7.48 (m, 1H, N1’-Ph H-4), 7.28–7.38 (m, 5H, CH2-Ph), 5.12 (s, 2H, CH2), 2.28 (s, 3H, 3-Me); 13C-NMR (75 MHz, CDCl3): δ (ppm) 183.3 (C=O), 160.3 (C-5, 3J(C-5,CH2) = 2.4 Hz), 146.6 (C-3, 2J(C-3,3-Me) = 7.0 Hz), 140.7 (C-3’, 1J(C-3’,H-3’) = 192.0 Hz), 137.4 (N1’-Ph C-1), 135.5 (CH2-Ph C-1), 129.2 (N1’-Ph C-3,4,5), 128.7 (CH2-Ph C-3,5 and C-5’, 3J(C-5’,H-3’) = 5.7 Hz), 128.0 (CH2-Ph C-2,4,6), 125.3 (N1’-Ph C-2,6), 118.9 (C-4’, 2J(C-4’,H-3’) = 10.1 Hz), 103.4 (C-4, 3J(C-4,3-Me) = 2.6 Hz), 49.8 (CH2, 1J = 140.2 Hz), 15.4 (3-Me, 1J = 128.6 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −196.4 (N-1), −162.1 (N-1’), −101.1 (N-2), −74.7 (N-2’); IR: 1633 (C=O) cm-1; MS m/z (%): 392/394 (M+, 8/3), 356 (91), 91 (100), 77 (25), 51 (18). Calcd. for C21H17ClN4O2 (392.84): C, 64.21; H, 4.36; N, 14.26. Found: C, 64.13; H, 4.27; N, 14.11. |
| 8a |  | 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.52 (m, 5H, Ph-H), 6.04 (s, 1H, H-4), 3.74 (s, 3H, N-Me), 2.59 (s, 3H, 3’-Me), 2.25 (s, 3H, 3-Me); 13C-NMR (75Hz, CDCl3): δ (ppm) 157.3 (C=O), 153.1 (C-3’, 2J(C-3’,3’-Me) = 6.9 Hz), 147.2 (C-3, 2J(C-3,3-Me) = 6.7 Hz, 2J(C-3,H-4) = 4.3 Hz), 144.6 (C-5, 3J(C-5,N-CH3) = 2.2 Hz, 2J(C 5,H-4) = 4.4 Hz), 137.2 (Ph-C-1), 132.2 (C-5’), 129.3 (Ph-C-4), 129.2 (Ph-C-3,5), 125.5 (Ph-C-2,-6), 108.3 (C-4’, 3J(C-4’,3’-Me) = 2.7 Hz), 93.9 (C-4, 1J(C-4,H-4) = 181.2 Hz, 3J(C-4,3-Me) = 3.5 Hz), 14.8 (3’-Me, 1J = 129.5Hz), 14.2 (3-Me, 1J = 127.5 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −202.2 (N-1), −165.8 (N-1’), −98.9 (N-2), −75.7 (N-2’); IR: 1745 (C=O) cm-1; MS m/z (%): 330 (M+, 0.1), 219 (100), 77 (42), 51 (19). Calcd. for C16H15ClN4O2 (330.77): C, 58.10; H, 4.57; N, 16.94. Found: C, 58.14; H, 4.37; N, 16.90. |
| 8b |  | 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.53 (d, 1H, H-3, 3J(H3,H4) = 2.1 Hz), 7.52 (m, 5H, N1’-Ph), 7.12 (m, 2H, CH2-Ph H-2,6), 6.82 (m, 2H, CH2-Ph H-3,5), 6.30 (d, 1H, H-4, 3J(H4,H3) = 2.1 Hz), 5.28 (s, 2H, CH2), 3.75 (s, 3H, O-Me), 2.52 (s, 3H, 3’-Me); 13C-NMR (75 MHz, CDCl3): δ (ppm) 159.2 (CH2-Ph C4), 157.2 (C=O), 153.2 (C-3’, 2J(C-3’,3’-Me) = 7.0 Hz), 144.3 (C-5), 138.7 (C-3, 1J(C-3,H-3) = 187.6 Hz, 2J(C-3,H-4) = 4.8 Hz), 137.2 (N1’-Ph C-1), 132.2 (C-5’), 129.4 (N1’-Ph C-4), 129.2 (N1’-Ph C-3,5), 128.4 (CH2-Ph C-2,6), 128.3 (CH2-Ph C-1), 125.5 (N1’-Ph C-2,6), 114.1 (CH2-Ph C-3,5), 108.2 (C-4’, 3J(C-4’,3’-Me) = 2.7 Hz), 94.9 (C-4, 1J(C-4,H-4) = 183.4 Hz, 2J(C-4,H-5) = 10.5 Hz), 55.2 (O-Me, 1J = 143.8 Hz), 51.3 (CH2, 1J = 140.0 Hz, 3J(CH2,Ph-H-2,6) = 4.3 Hz), 14.8 (3’-Me, 1J = 129.6 Hz); 15N-NMR (50 MHz, CDCl3): δ (ppm) −185.1 (N-1), −165.7 (N-1’), −95.3 (N-2), −75.7 (N-2’); IR: 1745 (C=O) cm-1; MS m/z (%): 422 (M+, 0.1), 219 (100), 121 (25), 77 (20). Calcd. for C22H19ClN4O3 (422.86): C, 62.49; H, 4.53; N, 13.25. Found: C, 62.45; H, 4.40; N, 13.15. |
3.2.6. Cyclization of 4-Aroylpyrazolones 3a-g: General procedure for the synthesis of 4a-b, 4d-g
3.2.7. General procedure for the synthesis of 5a-b and 5d-g
3.2.8. General procedure for the synthesis of 4x and 5x
| Comp | Solvent | H of R1 | H of R3 | H of R5 | H of R7 |
|---|---|---|---|---|---|
| 4a | CDCl3 | Ph: 7.82 (2,6), 7.55 (3,5), 7.44 (4) | 8.22 (H-3) | 8.22 (H-5) | Ph: 7.82 (2,6), 7.55 (3,5), 7.44 (4) |
| 4b | CDCl3 | Ph: 7.78 (2,6), 7.52 (3,5), 7.40 (4) | 2.66 (Me) | 8.17 (H-5) | Ph: 7.80 (2,6), 7.53 (3,5), 7.43 (4) |
| 4d | CDCl3 | Ph: 7.78 (2,6), 7.51 (3,5), 7.39 (4) | 2.65 (Me) | 2.65 (Me) | Ph: 7.78 (2,6), 7.51 (3,5), 7.39 (4) |
| 4e | CDCl3 | 3.87 (Me) | 2.55 (Me) | 8.13 (H-5) | Ph: 7.79 (2,6), 7.56 (3,5), 7.43 (4) |
| 4f | CDCl3 | Ph: 7.28 (2,6), 6.89 (3,5); 5.37 (CH2), 3.79 (OMe) | 8.05 (H-3) | 8.17 (H-5) | Ph: 7.66 (2,6), 7.56 (3,5), 7.46 (4) |
| 4g | CDCl3 | Ph: 7.37 (4), 7.36 (3,5), 7.31 (2,6); 5.35 (CH2) | 2.60 (Me) | 8.12 (H-5) | Ph: 7.60 (2,6), 7.52 (3,5), 7.42 (4) |
| 5a | CDCl3 | Ph: 7.84 (2,6), 7.57 (3,5), 7.46 (4) | 8.39 (H-3) | 8.39 (H-5) | Ph: 7.84 (2,6), 7.57 (3,5), 7.46 (4) |
| 5b | CDCl3 | Ph: 7.81 (2,6), 7.54 (3,5), 7.42 (4) | 2.78 (Me) | 8.32 (H-5) | Ph: 7.82 (2,6), 7.55 (3,5), 7.44 (4) |
| 5d | CDCl3 | Ph: 7.80 (2,6), 7.53 (3,5), 7.41 (4) | 2.80 (Me) | 2.80 (Me) | Ph: 7.80 (2,6), 7.53 (3,5), 7.41 (4) |
| 5e | CDCl3 | 3.89 (Me) | 2.65 (Me) | 8.27 (H-5) | Ph: 7.81 (2,6), 7.57 (3,5), 7.45 (4) |
| 5f | CDCl3 | Ph: 7.28 (2,6), 6.90 (3,5); 5.38 (CH2), 3.79 (OMe) | 8.20 (H-3) | 8.32 (H-5) | Ph: 7.66 (2,6), 7.56 (3,5), 7.47 (4) |
| 5g | CDCl3 | Ph: 7.38 (3,4,5), 7.33 (2,6); 5.37 (CH2) | 2.73 (Me) | 8.29 (H-5) | Ph: 7.61 (2,6), 7.53 (3,5), 7.43 (4) |
| 4x | DMSO- d6 | Ph: 7.85 (2,6), 7.62 (3,5), 7.48 (4) | 8.24 (H-3) | 8.56 (H-5) | 13.78 (NH) |
| 5x | DMSO- d6 | Ph: 7.87 (2,6), 7.64 (3,5), 7.50 (4) | 8.34 (H-3) | 8.66 (H-5) | 14.00 (NH) |
| Comp | C-3 | C-3a | C-4 | C-4a | C-5 | C-7a | C-8a | C of R1 | C of R3 | C of R5 | C of R7 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 136.7 | 108.8 | 169.7 | 108.8 | 136.7 | 151.3 | 151.3 | Ph: 136.6 (1), 129.6 (3,5), 128.3 (4), 121.3 (2,6) | - | - | Ph: 136.6 (1), 129.6 (3,5), 128.3 (4), 121.3 (2,6) |
| 4b | 148.1 | 106.5 | 170.6 | 108.9 | 136.6 | 151.3 | 151.4 | Ph: 136.6 (1), 129.48 (3,5), 127.8 (4), 121.1 (2,6) | 14.0 (Me) | - | Ph: 136.7 (1), 129.53 (3,5), 128.1 (4), 121.2 (2,6) |
| 4d | 148.0 | 106.6 | 171.8 | 106.6 | 148.0 | 151.5 | 151.5 | Ph: 136.7 (1), 129.5 (3,5), 127.7 (4), 121.0 (2,6) | 14.0 (Me) | 14.0 (Me) | Ph: 136.7 (1), 129.5 (3,5), 127.7 (4), 121.0 (2,6) |
| 4e | 146.8 | 105.0 | 170.7 | 108.7 | 136.6 | 151.3 | 152.6 | 34.1 (Me) | 13.9 (Me) | - | Ph: 136.7 (1), 129.6 (3,5), 128.1 (4), 121.5 (2,6) |
| 4f | 135.5 | 107.8 | 169.8 | 108.6 | 136.7 | 151.4 | 152.0 | Ph: 159.9 (4), 129.4 (2,6), 126.2 (1), 114.4 (3,5); 55.3 (OMe), 52.4 (CH2) | - | - | Ph: 136.6 (1), 129.6 (3,5), 128.2 (4), 121.6 (2,6) |
| 4g | 146.9 | 105.5 | 170.7 | 108.8 | 136.6 | 151.3 | 152.4 | Ph: 134.6 (1), 129.0 (3,5), 128.6 (4), 127.7 (2,6); 52.3 (CH2) | 14.0 (Me) | - | Ph: 136.7 (1), 129.5 (3,5), 128.0 (4), 121.4 (2,6) |
| 5a | 138.1 | 117.8 | 192.1 | 117.8 | 138.1 | 145.5 | 145.5 | Ph: 136.6 (1), 129.7 (3,5), 128.3 (4), 121.3 (2,6) | - | - | Ph: 136.6 (1), 129.7 (3,5), 128.3 (4), 121.3 (2,6) |
| 5b | 150.1 | 114.8 | 193.5 | 118.1 | 138.2 | 145.1 | 146.0 | Ph: 136.4 (1), 129.56 (3,5), 128.0 (4), 121.2 (2,6) | 15.6 (Me) | - | Ph: 136.6 (1), 129.6 (3,5), 128.2 (4), 121.1 (2,6) |
| 5d | 150.2 | 115.1 | 195.5 | 115.1 | 150.2 | 145.8 | 145.8 | Ph: 136.5 (1), 129.6 (3,5), 127.9 (4), 121.2 (2,6) | 15.9 (Me) | 15.9 (Me) | Ph: 136.5 (1), 129.6 (3,5), 127.9 (4), 121.2 (2,6) |
| 5e | 148.8 | 113.5 | 193.5 | 117.7 | 138.1 | 145.3 | 147.3 | 34.2 (Me) | 15.2 (Me) | - | Ph: 136.7 (1), 129.6 (3,5), 128.2 (4), 121.4 (2,6) |
| 5f | 136.9 | 117.0 | 192.2 | 117.5 | 138.1 | 145.5 | 146.2 | Ph: 159.9 (4), 129.4 (2,6), 126.0 (1), 114.4 (3,5); 55.3 (OMe), 52.5 (CH2) | - | - | Ph: 136.5 (1), 129.6 (3,5), 128.3 (4), 121.5 (2,6) |
| 5g | 148.9 | 114.1 | 193.6 | 117.9 | 138.2 | 145.2 | 147.0 | Ph: 134.4 (1), 129.1 (3,5), 128.7 (4), 127.8 (2,6); 52.4 (CH2) | 15.4 (Me) | - | Ph: 136.6 (1), 129.6 (3,5), 128.1 (4), 121.3 (2,6) |
| 4x | 136.3 | 107.4 | 170.5 | 106.7 | 128.6 | 160.6 | 153.0 | Ph: 136.5 (1), 129.6 (3,5), 128.0 (4), 121.8 (2,6) | - | - | - |
| 5x | 137.7 | 116.9 | 193.5 | 115.8 | 130.2 | 155.0 | 147.6 | Ph: 136.3 (1), 129.6 (3,5), 128.2 (4), 121.8 (2,6) | - | - | - |
| Comp | J of C-3 | J of C-3a | J of C-4a | J of C-5 | J of C-7a | J of C-8a | other couplings |
|---|---|---|---|---|---|---|---|
| 4a | 1J = 194.7 | 2J(H-3) = 9.9 | 2J(H-5) = 9.9 | 1J = 194.7 | 3J(H-5) = 5.2 | 3J(H-3) = 5.2 | |
| 4b | 2J(3-Me) = 7.2 | 3J(3-Me) = 2.9 | 2J(H-5) = 10.0 | 1J = 194.4 | 3J(H-5) = 5.1 | 1J(3-Me) = 129.4 | |
| 4d | 2J(3-Me) = 7.1 | 3J(3-Me) = 2.7 | 3J(5-Me) = 2.7 | 2J(5-Me) = 7.1 | 1J(3-Me) = 129.3, 1J(5-Me) = 129.3 | ||
| 4e | 2J(3-Me) = 7.1 | 3J(3-Me) = 2.6 | 2J(H-5) = 9.9 | 1J = 194.1 | 3J(H-5) = 5.2 | 3J(N-Me) = 2.1 | 1J(N-Me) = 141.7, 1J(3-Me) = 129.1 |
| 4f | 1J = 194.1 | 2J(H-3) = 10.0 | 2J(H-5) = 9.9 | 1J = 194.6 | 3J(H-5) = 5.2 | 3J(H-3) ~ 5.2, 3J(N-CH2) = 2.8 | 1J(OMe) = 144.1, 1J(N-CH2) = 141.5, 3J(NCH2,Ph H-2,6) = 4.9, 2J(Ph C-1,NCH2) = 4.7, 3J(Ph C-2/6,NCH2) = 4.4 |
| 4g | 2J(3-Me) = 7.1 | 3J(3-Me) = 2.8 | 2J(H-5) = 10.0 | 1J = 194.2 | 3J(H-5) = 5.2 | 3J(N-CH2) = 2.7 | 1J(N-CH2) = 141.0, 1J(3-Me) = 129.2, 3J(NCH2,Ph H-2,6) = 4.7 |
| 5a | 1J = 195.8 | 2J(H-3) = 9.3 | 2J(H-5) = 9.3 | 1J = 195.8 | 3J(H-5) = 5.1 | 3J(H-3) = 5.1 | |
| 5b | 2J(3-Me) = 7.1 | 3J(3-Me) = 2.6 | 2J(H-5) = 9.1 | 1J = 195.5 | 3J(H-5) = 5.0 | 1J(3-Me) = 129.7 | |
| 5d | 2J(3-Me) = 7.2 | 3J(3-Me) = 2.5 | 3J(5-Me) = 2.5 | 2J(5-Me) = 7.2 | 1J(3-Me) = 129.6, 1J(5-Me) = 129.6 | ||
| 5e | 2J(3-Me) = 7.1 | 3J(3-Me) = 2.7 | 2J(H-5) = 9.1 | 1J = 195.3 | 3J(H-5) = 5.1 | 3J(N-Me) = 2.4 | 1J(N-Me) = 141.9, 1J(3-Me) = 129.4 |
| 5f | 1J = 195.0 | 2J(H-3) = 9.4 | 2J(H-5) = 9.3 | 1J = 195.5 | 3J(H-5) = 5.1 | 3J(H-3) = 5.1, 3J(N-CH2) = 2.6 | 1J(OMe) = 144.1, 1J(N-CH2) = 141.4, 3J(NCH2,Ph H-2,6) = 4.6 |
| 5g | 2J(3-Me) = 7.1 | 3J(3-Me) = 2.7 | 2J(H-5) = 9.2 | 1J = 195.3 | 3J(H-5) = 5.1 | 3J(N-CH2) = 2.8 | 1J(N-CH2) = 141.2, 1J(3-Me) = 129.5, 3J(NCH2, Ph H-2,6) = 4.4 |
| 4x | 1J = 193.8 | 2J(H-3) = 10.2 | 2J(H-5) ~ 8.5 | 1J ~ 195.0 | 3J(H-5) =not resolved | 3J(H-3) = 5.2 | |
| 5x | 1J = 194.7 | 2J(H-3) = 9.5 | 2J(H-5) = 7.8 | 1J = 195.9 | 3J(H-5) = 8.5 | 3J(H-3) = 4.9 |
| Comp | N-1 | N-2 | N-6 | N-7 |
|---|---|---|---|---|
| 4a | −186.8 | −86.8 | −86.8 | −186.8 |
| 4b | −193.1 | −93.6 | −87.7 | −187.1 |
| 4d | −193.4 | −94.2 | −94.2 | −193.4 |
| 4e | −211.8 | −91.7 | −88.0 | −187.5 |
| 4f | −191.8 | −85.2 | −87.5 | −187.3 |
| 4g | −200.0 | −91.3 | −88.2 | −187.4 |
| 5a | −187.6 | −84.5 | −84.5 | −187.6 |
| 5b | −195.3 | −92.1 | −85.5 | −188.0 |
| 5d | −196.2 | −92.9 | −92.9 | −196.2 |
| 5e | −213.7 | −89.7 | −85.6 | −188.2 |
| 5f | −192.4 | −82.2 | −84.9 | −187.9 |
| 5g | −201.9 | −89.5 | −85.8 | −188.2 |
| 4x | −186.4 | −87.7 | −179.2* | −179.2* |
| 5x | −186.7 | −84.3 | −175.5* | −175.5* |
3.2.9. General procedure for the synthesis of 9a and 9b
4. Conclusions
Acknowledgements
References and Notes
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone Derivatives: New insights in biological activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef]
- El-Seedi, H.R.; El-Barbary, M.A.; El-Ghorab, D.M.H.; Bohlin, L.; Borg-Karlson, A.-K.; Goeransson, U.; Verpoorte, R. Recent insights into the biosynthesis and biological activities of natural xanthones. Curr. Med. Chem. 2010, 17, 854–901. [Google Scholar] [CrossRef]
- Na, Y. Recent cancer drug development with xanthone structures. J. Pharm. Pharmacol. 2009, 61, 707–712. [Google Scholar] [CrossRef]
- Suphavanich, K.; Maitarad, P.; Hannongbua, S.; Sudta, P.; Suksamrarn, S.; Tantirungrotechai, Y.; Limtrakul, J. CoMFA and CoMSIA studies on a new series of xanthone derivatives against the oral human epidermoid carcinoma (KB) cancer cell line. Monatsh. Chem. 2009, 140, 273–280. [Google Scholar] [CrossRef]
- Woo, S.; Jung, J.; Lee, C.; Kwon, Y.; Na, Y. Synthesis of new xanthone analogues and their biological activity test – Cytotoxicity, topoisomerase II inhibition, and DNA cross-linking study. Bioorg. Med. Chem. Lett. 2007, 17, 1163–1166. [Google Scholar] [CrossRef]
- Demirkiran, O. Xanthones in Hypericum: synthesis and biological activities. Topics Heterocycl. Chem. 2007, 9, 139–178. [Google Scholar] [CrossRef]
- Fotie, J.; Bohle, D.S. Pharmacological and biological activities of xanthones. Anti-infective Agents Med. Chem. 2006, 5, 15–31. [Google Scholar] [CrossRef]
- Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Syntheses, Patents, Applications, 4th ed; Thieme: Stuttgart, Germany, 2001; p. 99. [Google Scholar]
- Eller, G.A.; Wimmer, V.; Haring, A.W.; Holzer, W. An Efficient Approach to Heterocyclic Analogues of Xanthone: A Short Synthesis of all possible Pyrido[5,6]pyrano[2,3-c]pyrazol-4(1H)-ones. Synthesis 2006, 4219–4229. [Google Scholar]
- Eller, G.A.; Haring, A.W.; Datterl, B.; Zwettler, M.; Holzer, W. Tri- and Tetracyclic Heteroaromatic Systems: Synthesis of Novel Benzo-, Benzothieno- and Thieno-Fused pyrano[2,3-c]pyrazol-4(1H)-ones. Heterocycles 2007, 71, 87–104. [Google Scholar] [CrossRef]
- Eller, G.A.; Holzer, W. A Convenient Approach to Heterocyclic Building Blocks: Synthesis of Novel Ring Systems Containing a [5,6]pyrano[2,3-c]pyrazol-4(1H)-one Moiety. Molecules 2007, 12, 60–73. [Google Scholar] [CrossRef]
- Eller, G.A.; Datterl, B.; Holzer, W. Pyrazolo[4’,3’:5,6]pyrano[2,3-b]quinoxalin-4(1H)-one: Synthesis and Characterization of a Novel Tetracyclic Ring System. J. Heterocycl. Chem. 2007, 44, 1139–1143. [Google Scholar] [CrossRef]
- Eller, G.A.; Wimmer, V.; Holzer, W. Synthesis of Novel Polycyclic Ring Systems Containing two Pyrano[2,3-c]pyrazol-4(1H)-one Moieties. Khim. Geterotsikl. Soedin. 2007, 1251–1255, (Chem. Heterocycl. Comp. 2007, 43, 1060-1064). [Google Scholar]
- Eller, G.A.; Habicht, D.; Holzer, W. Synthesis of a Novel Pentacycle: 8-Methyl-10-phenylpyrazolo[4’,3’:5,6]pyrano[3,2-c][1,10]phenanthrolin-7(10H)-one. Khim. Geterotsikl. Soedin. 2008, 884–890, (Chem. Heterocycl. Comp. 2008, 44, 709-714). [Google Scholar]
- Eller, G.A.; Zhang, Q.; Habicht, D.; Datterl, B.; Holzer, W. Synthesis and NMR Data of Pyrazolo[4’,3’:5,6]pyrano[2,3-b]pyrazin-4(1H)-ones: Derivatives of a Novel Tricyclic Ring System. Acta Chim. Slov. 2009, 56, 521–526. [Google Scholar]
- Batezila, G.; Holzer, W. (2-Chlorophenyl)-3-methylchromeno[2,3-c]pyrazol-4(1H)-one. Molbank 2010, M 661. [Google Scholar]
- Jensen, B.S. The Synthesis of 1-Phenyl-3-methyl-4-acyl-pyrazolones-5. Acta Chem. Scand. 1959, 13, 1668–1670, (Chem. Abstr. 1962, 56, 66890). [Google Scholar] [CrossRef]
- Williams, A.C.; Camp, N. Product Class 4: Benzopyranones and Benzopyranthiones. Sci. Synth. 2003, 14, 347–638. [Google Scholar]
- Huemer, V.; Eller, G.A.; Holzer, W. Heterocyclic analogs of xanthiones: 5,6-fused 3-methyl-1-phenylpyrano[2,3-c]pyrazol-4(1H)thiones – synthesis and NMR (1H, 13C, 15N) data. Magn. Reson. Chem. 2010, 48, 476–482. [Google Scholar]
- Huemer, V.; Holzer, W. 1-Phenylpyrazolo[4’,3’:5,6]pyrano[2,3-c]pyridine-4(1H)thione. Molbank 2010, M 678. [Google Scholar]
- Eller, G.A.; Holzer, W. The 4-methoxybenzyl (PMB) function as a versatile protecting group in the synthesis of N-unsubstituted pyrazolones. Heterocycles 2004, 63, 2537–2555. [Google Scholar] [CrossRef]
- Jones, G.; Stanforth, S.P. The Vilsmeier reaction of fully conjugated carbocycles and heterocycles. Org. React. 1997, 49, 1–330. [Google Scholar]
- Zhang, X.-Li; Lu, X.-H.; Jiang, W.-Q. Synthesis of 5-chloro-N-[[(fluorophenyl)amino]thioxomethyl]-3-methyl-1-phenyl-1H-pyrazole-4-carboxamide derivatives and determination of their activity as agrochemical fungicides. Yingyong Huaxue 2008, 25, 459–463. [Google Scholar]
- Baraldi, P.G.; Tabtizi, M.A.; Preti, D.; Bovero, A.; Fruttarolo, F.; Romagnoli, R.; Zaid, N.A.; Moorman, A.R.; Varani, K.; Borea, P.A. New 2-Arylpyrazolo[4,3-c]quinoline Derivatives as Potent and Selective Human A3 Adenosine Receptor Antagonists. J. Med. Chem. 2005, 48, 5001–5008. [Google Scholar]
- Haider, N.; Heinisch, G. Pyridazines. III. A Novel Approach to Pyrido[2,3-d]pyridazines by Annelation of the Pyridine Ring to the 1,2-Diazine System. Synthesis 1986, 862–864. [Google Scholar] [CrossRef]
- Cherkasov, R.A.; Kutyrev, G.A.; Pudovik, A.N. Organothiophosphorus reagents in organic synthesis. Tetrahedron 1985, 41, 2567–2624. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Scheibye, S.; Nilsson, N.H.; Lawesson, S.-O. Studies on organophosphorus compounds. XX. Syntheses of thioketones. Bull. Soc. Chim. Belg. 1978, 87, 223–228. [Google Scholar]
- Jesberger, M.; Davis, T.P.; Barner, L. Applications of Lawesson’s reagent in organic and organometallic syntheses. Synthesis 2003, 13, 1929–1958. [Google Scholar]
- Sarenko, A.S.; Kvitko, I.Ya.; Efros, L.S. Heterocyclic analoges of xanthones. II. C-Acylation of 5-pyrazolinones and synthesis of chromono[3,2-d]pyrazoles. Khim. Geterotsikl. Soedin. 1972, 6, 799–804, (Chem. Heterocycl. Comp. 1972, 8, 722-727). [Google Scholar]
- Braun, S.; Kalinowski, H.-O.; Berger, S. 150 and More Basic NMR Experiments, 2nd ed; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar]
- Bax, A. Structure determination and spectral assignment by pulsed polarization transfer via long-range proton-carbon-13 couplings. J. Magn. Reson. 1984, 57, 314–318. [Google Scholar] [CrossRef]
- Jippo, T.; Kamo, O.; Nagayama, N. Determination of long-range proton-carbon 13 coupling constants with selective two-dimensional INEPT. J. Magn. Reson. 1986, 66, 344–348. [Google Scholar] [CrossRef]
- Kalchhauser, H.; Robien, W. CSEARCH: A Computer Program for Identification of Organic Compounds and Fully Automated Assignment of Carbon-13 Nuclear Magnetic Resonance Spectra. J. Chem. Inf. Comput. Sci. 1985, 25, 103–108. [Google Scholar] [CrossRef]
- NMR Predict, version 4.7; Modgraph Consultants, Ltd.: Herts, UK, 2010. Available online: www.modgraph.co.uk/product_nmr.htm (accessed on 14 June 2010).
- ACD/C+H Predictors and DB, version 12.0; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2008. Available online: www.acdlabs.com (accessed on 14 June 2010).
- Huemer, V.; Eller, G.A.; Holzer, W. Heterocyclic analogs of xanthiones: 5,6-fused 3-methyl-1-phenylpyrano[2,3-c]pyrazol-4(1H)thiones – synthesis and NMR (1H, 13C, 15N) data. Magn. Reson. Chem. 2010, 48, 476–482. [Google Scholar]
- Holzer, W.; Kautsch, C.; Laggner, C.; Claramunt, R.M.; Perez-Torralba, M.; Alkorta, I.; Elguero, J. On the tautomerism of pyrazolones: the geminal 2J[pyrazole C-4,H-3(5)] spin coupling constant as a diagnostic tool. Tetrahedron 2004, 60, 6791–6805. [Google Scholar] [CrossRef]
- Begtrup, M.; Boyer, G.; Cabildo, P.; Cativiela, C.; Claramunt, R.M.; Elguero, J.; Ignacio Garcia, J.; Toiron, C.; Vesdo, P. Carbon-13 NMR of pyrazoles. Magn. Reson. Chem. 1993, 31, 107–168. [Google Scholar]
- Sucrow, W.; Slopianka, M. Enehydrazines, 19. Pyrazolium Betaines from 1,1-Dialkylhydrazines and Acetylenecarboxylic Esters. Chem. Ber. 1978, 111, 780–790. [Google Scholar] [CrossRef]
- ACD Name, version 12.0; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2010. Available online: www.acdlabs.com/ (accessed on 14 June 2010).
- Eller, G.A. Improving the quality of published chemical names with nomenclature software. Molecules 2006, 11, 915–928. [Google Scholar] [CrossRef]
- Koshelev, Yu.N.; Kvitko, I.Ya.; Efros, L.S. Transfer of substituent effects in thieno[3,2-d]pyrazole. Zh. Org. Khim. 1972, 8, 1750–1754, (J. Org. Chem. 1972, 8, 1789-1793).. [Google Scholar]
- Brack, A. Condensed pyrazolopyridines. Liebigs Ann. Chem. 1965, 681, 105–110. [Google Scholar] [CrossRef]
- Dickopp, H. Sydnones. II. 5-Halopyrazolecarboxylic acids. Chem. Ber. 1974, 107, 3036–3042. [Google Scholar] [CrossRef]
- Duquenois, P.; Amal, H. The action of PCl5 and SOCl2 on antipyrine-4-carboxylic acid. Bull. Soc. Chim. Fr. 1942, 9, 721–725. [Google Scholar]
- Beck, J.R.; Gajewski, R.P.; Lynch, M.P.; Wright, F.L. Nonaqueous Diazotiation of 5-Amino-1-aryl-1H-pyrazole-4- carboxylate Esters. J. Het. Chem. 1987, 24, 267–270. [Google Scholar] [CrossRef]
- Rojahn, C.A.; Fahr, K. Synthesis of pyrazole aldehydes. I. Liebigs Ann. Chem. 1923, 434, 252–264. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Datterl, B.; Tröstner, N.; Kucharski, D.; Holzer, W. Heterocyclic Analogues of Xanthone and Xanthione. 1H-Pyrano[2,3-c:6,5-c]dipyrazol-4(7H)-ones and Thiones: Synthesis and NMR Data. Molecules 2010, 15, 6106-6126. https://doi.org/10.3390/molecules15096106
Datterl B, Tröstner N, Kucharski D, Holzer W. Heterocyclic Analogues of Xanthone and Xanthione. 1H-Pyrano[2,3-c:6,5-c]dipyrazol-4(7H)-ones and Thiones: Synthesis and NMR Data. Molecules. 2010; 15(9):6106-6126. https://doi.org/10.3390/molecules15096106
Chicago/Turabian StyleDatterl, Barbara, Nicole Tröstner, Dorota Kucharski, and Wolfgang Holzer. 2010. "Heterocyclic Analogues of Xanthone and Xanthione. 1H-Pyrano[2,3-c:6,5-c]dipyrazol-4(7H)-ones and Thiones: Synthesis and NMR Data" Molecules 15, no. 9: 6106-6126. https://doi.org/10.3390/molecules15096106
APA StyleDatterl, B., Tröstner, N., Kucharski, D., & Holzer, W. (2010). Heterocyclic Analogues of Xanthone and Xanthione. 1H-Pyrano[2,3-c:6,5-c]dipyrazol-4(7H)-ones and Thiones: Synthesis and NMR Data. Molecules, 15(9), 6106-6126. https://doi.org/10.3390/molecules15096106