General
Melting points were determined on a Stuart SMP2 apparatus and are uncorrected. Ultraviolet spectra were recorded on methanol solutions with a Jasco V-530 UV-VIS spectrophotometer. Infrared spectra were recorded on Nujol mulls unless stated otherwise with a Perkin-Elmer Spectrum GX FT-IR spectrophotometer. 1H- and 13C-NMR magnetic resonance spectra were recorded on CDCl3 solutions, unless stated otherwise, at 300 MHz for 1H and 75 MHz for 13C with a Bruker AVANCE 300 spectrometer. Tetramethylsilane was used as the internal standard. Mass spectra were measured on a Hewlett Packard 5989B spectrometer. Elemental microanalyses were performed with a Perkin-Elmer 2400 elemental analyser.
2-Benzyloxy-6-bromo-3-methoxybenzaldehyde (
3a). Using standard conditions,
3a was obtained in 92.4% yield as a green solid, m.p.53-54°C (Lit. [
11] m.p. 49 °C);
1H-NMR: d 10.23 (1H, s, CHO), 7.45-7.32 (6H, m, Ar-H), 6.96 (1H, d,
J = 9.00 Hz, Ar-H), 5.10 (2H, s, PhCH
2), 3.89 (3H, s, OCH
3);
13C–NMR: d 190.4 (CH), 152.8 (C), 150.8 (C), 136.3 (C), 129.6 (CH), 129.1 (C), 128.7 (CH), 128.6 (CH), 128.5 (CH), 117.4 (CH), 112.4 (C), 76.5 (CH
2), 56.2 (OCH
3).
2-Benzyloxy-6-bromo-3-methoxybenzyl alcohol (3b). Standard sodium borohydride reduction of 3a gave 3b in 94.6% yield as colourless prisms from ethanol, m.p. 84-85°C; Anal.Calc. for C15H15BrO3: C, 55.8; H, 4.7. Found: C, 55.6; H, 4.9%; 1H-NMR: d 7.45-7.29 (6H, m, Ar-H), 6.79 (1H, d, J = 9.0 Hz, Ar-H), 5.06 (2H, s, PhCH2), 4.72 (2H, d, J = 5.40 Hz, CH2), 3.87 (3H, s, OCH3), 2.14 (1H, br t, J = 5.40 Hz, OH); 13C–NMR: d 152.3 (C), 147.3 (C), 137.0 (C), 134.2 (C), 128.6 (CH), 128.5 (CH), 128.4 (CH), 128.1 (CH), 115.0 (C), 113.3 (CH), 75.8 (CH2), 60.4 (CH2), 56.0 (OCH3).
2-Benzyloxy-6-bromo-3-methoxybenzyl chloride (3c). Treatment of 3b with thionyl chloride gave crude 3c as a white solid in 98.2% yield, m.p. 65-66°C. This was used in the next step without further purification. 1H-NMR: d 7.53-7.24 (6H, m, Ar-H), 6.82 (1H, d, J = 9.00 Hz, Ar-H), 5.11 (2H, s, PhCH2), 4.77 (2H, s, CH2), 3.87 (3H, s, OCH3); 13C–NMR: d 152.4 (C), 147.6 (C), 137.1 (C), 131.7 (C), 128.5 (CH), 128.4 (CH), 128.3 (CH), 128.2 (CH), 115.4 (C), 114.0 (CH), 75.6 (CH2), 56.1 (OCH3), 41.0 (CH2).
2-Benzyloxy-6-bromo-3-methoxybenzyl cyanide (3d). Treatment of 3c with sodium cyanide in acetone/water gave 3d as colourless needles in 94.9% yield, m.p. 54-55°C; EI-MS (70 eV) m/z 333 [(M+2)+, 13%], 331 (M+, 14), 91(100); Anal. calc. for C16H14BrNO2: C, 57.9; H, 4.3; N, 4.2. Found: C, 57.7; H, 4.5; N, 4.0%; 1H-NMR: d 7.48-7.28 (6H, m, Ar-H), 6.83 (1H, d, J = 8.70 Hz, Ar-H), 5.12 (2H, s, PhCH2), 3.88 (3H, s, OCH3), 3.75 (2H, s, CH2); 13C-NMR: d 152.3 (C), 146.9 (C), 136.7 (C), 128.7 (CH), 128.6 (CH), 128.5 (CH), 128.0 (CH), 125.1 (C), 117.1 (C), 114.7 (C), 113.8 (CH), 75.3 (CH2), 56.1 (OCH3), 19.0 (CH2).
2-Benzyloxy-6-bromo-3-methoxyphenylacetic acid (3e). Hydrolysis of 3d with potassium hydroxide in ethanol/water gave 3e as colourless needles in 88.7% yield, m.p. 99-100°C; EI-MS (70 eV) m/z 352 [(M+2)+, 14%], 350 (M+, 14), 270(84), 244(61), 91(100). Anal. calc. for C16H15BrO4: C, 54.7; H, 4.3. Found: C, 54.9; H, 4.1%; 1H-NMR: d 7.44-7.26 (6H, m, Ar-H), 6.79 (1H, d, J = 9.00 Hz, Ar-H), 5.02 (2H, s, PhCH2), 3.87 (3H, s, OCH3), 3.84 (2H, s, CH2); 13C–NMR: d 175.9 (C), 152.1 (C), 147.4 (C), 137.1 (C), 128.8 (C), 128.5 (CH), 128.3 (CH), 128.2 (CH), 127.6 (CH), 116.0 (C), 112.9 (CH), 75.1 (CH2), 56.0 (OCH3), 35.8 (CH2).
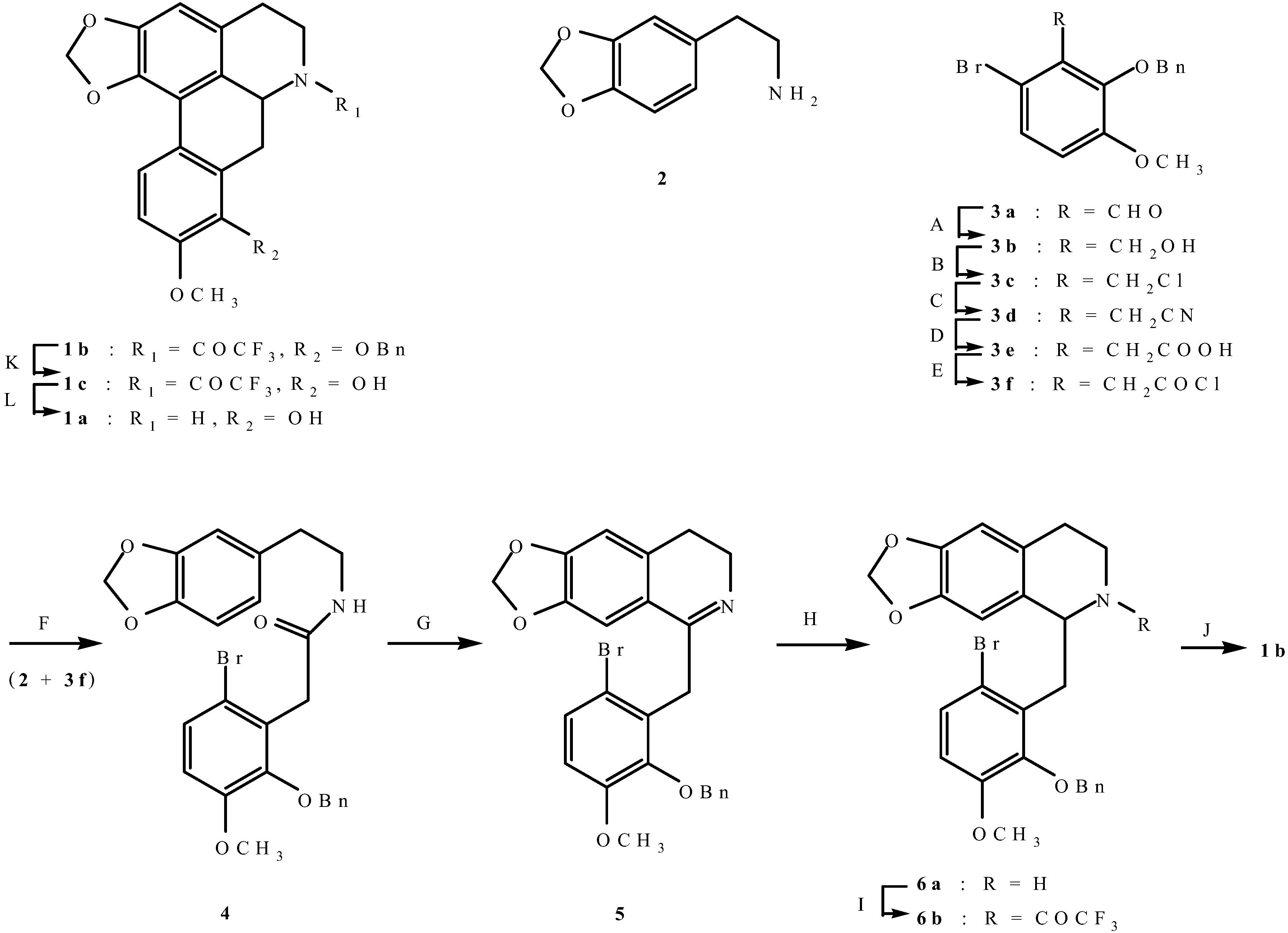
2-(2-Benzyloxy-6-bromo-3-methoxyphenyl)-N-(3,4-methylenedioxyphenethyl)acetamide (
4). A mixture of 2-benzyloxy-6-bromo-3-methoxyphenylacetic acid (
3e, 21.5 g) and thionyl chloride (20.3 g) in benzene (150 mL) was refluxed for 1 h, then the solvent and excess thionyl chloride were removed
in vacuo. The resulting crude 2-benzyloxy-6-bromo-3-methoxyphenylacetyl chloride (
3f) was dissolved in chloroform (200 mL) and added portionwise to a mixture of 2-(3,4-methylene-dioxyphenyl)ethylamine (
2, 11.2 g) [
9] in chloroform (100 mL), sodium hydrogen carbonate (25 g) and ice (200 g). The mixture was stirred at room temperature for 3 h. The chloroform layer was washed with 10% sodium carbonate (3 x100 mL), water (2 x100 mL), 5% HCl (3 x 100 mL), brine and then dried. Removal of the solvent under vacuum gave a pale yellow solid which was recrystallized from ethanol to give amide
4 as a pale yellow solid (21.6 g , 70.8%), m.p. 143-144°C; UV λ
max nm (MeOH) (log ε ) 231sh (4.09), 286 (3.77); IR ν
max (KBr): 3290 (NH), 3062, 3031, 3007, 2938, 2888, 2839, 1647 (C=O), 1550, 1504, 1489, 1471, 1441, 1406, 1366, 1344, 1274, 1249, 1129, 1073, 1040, 978, 927, 861, 805, 750, 696 cm
-1; EI-MS(70 eV)
m/z 499 [(M+2)
+, 4%], 497 (M
+, 4), 418 (10), 352(7), 270(9), 238(30), 223(14), 164(8), 148(82), 135(19), 91(100). Anal. calc. for C
25H
24BrNO
5: C, 60.3; H, 4.9; N, 2.8. Found: C, 60.5; H, 4.7; N, 2.7%;
1H-NMR: d 7.46-7.23 (6H, m, Ar-H), 6.78 (1H, d,
J = 8.70 Hz, Ar-H), 6.61 (1H, d,
J = 7.80 Hz, Ar-H), 6.54 (1H, d,
J = 1.50 Hz, Ar-H), 6.47 (1H, dd,
J = 7.80, 1.50 Hz, Ar-H), 5.49 (1H, br s, N-H), 4.98 (2H, s, PhCH
2), 3.88 (3H, s, OCH
3), 3.68 (2H, s, CH
2), 3.34 (2H, apparent q,
J = 6.60 Hz, CH
2N), 2.60 (2H, t,
J = 6.60 Hz, CH
2);
13C–NMR: d 169.3 (C), 152.3 (C) , 147.6 (C), 147.0 (C), 146.0 (C), 137.0 (C), 132.5 (C), 129.7 (C), 128.5 (C), 128.4 (CH), 128.3 (CH), 128.1 (CH), 121.6 (CH), 113.5 (CH), 112.7 (CH), 109.0 (CH), 108.2 (CH), 100.8 (CH
2), 75.1 (CH
2), 55.9 (OCH
3), 40.8 (CH
2), 38.4 (CH
2), 35.2 (CH
2).
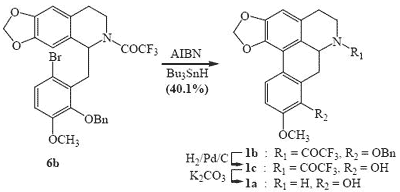
1-(2-Benzyloxy-6-bromo-3-methoxybenzyl)-6,7-methylenedioxy-3, 4-dihydroisoquinoline (5). A solution of amide 4 (6.03 g) and phosphorus oxychloride (19.4 g) in benzene (50 mL) was refluxed for 2 h. The excess reagent and solvent were removed under vacuum. The resulting brown residue was shaken with chloroform (80 mL) and dilute ammonium hydroxide (60 mL). The chloroform layer was washed with water (3 x 60 mL) and then dried. Removal of the solvent under vacuum followed by recrystallisation from ethanol gave dihydroisoquinoline 5 as pale yellow prisms (5.64 g, 97.1%), m.p. 81-82°C; UV λmax nm (log ε) 227sh (4.52), 280 (3.91), 313 (3.90); IR νmax(KBr): 1636, 1600, 1574, 1266, 1233, 1173, 1129, 1097, 1076, 1038, 1013, 980, 934, 865, 798, 751, 697 cm-1; Anal. calc. for C25H22BrNO4: C, 62.5; H, 4.6; N, 2.9. Found: C, 62.7; H, 4.4; N, 2.8%; 1H-NMR: d 7.40-7.26 (6H, m, Ar-H), 7.25 (1H, s, Ar-H), 6.75 (1H, d, J = 8.70 Hz, Ar-H), 6.63 (1H, s, Ar-H), 5.00 (2H, s, PhCH2), 4.17 (2H, s, CH2), 3.85 (3H, s, OCH3), 3.50 (2H, t, J = 7.20 Hz, CH2), 2.46 (2H, t, J = 7.20 Hz, CH2); 13C–NMR: d 163.8 (C), 152.2 (C), 148.8 (C), 147.4 (C), 146.3 (C), 137.7 (C), 133.3 (C), 132.6 (C), 128.3 (CH), 128.1 (CH), 127.8 (CH), 127.7 (CH), 123.5 (C), 116.3 (C), 112.0 (CH), 107.8 (CH), 105.6 (CH), 101.2 (CH2), 74.8 (CH2), 55.8 (OCH3), 46.8 (CH2), 37.0 (CH2), 26.3 (CH2).
1-(2-Benzyloxy-6-bromo-3-methoxybenzyl)-6,7-methylenedioxy-1,2,3,4-tetrahydroisoquinoline (6a). Sodium borohydride (2.0 g) was added portionwise to a stirred solution of dihydroisoquinoline 5 (18.2 g) in ethanol (310 mL) and the mixture was refluxed for 1 h. Chloroform (200 mL) was added and the mixture was washed with water (4 x 200 mL), brine and then dried. Removal of the solvent under vacuum gave tetrahydroisoquinoline 6a as a yellow-red viscous oil (14.8 g, 81.0%) which failed to crystallise and was used in the next step without further purification. 1H-NMR: d 7.47-7.26 (6H, m, Ar-H), 6.73 (1H, d, J = 8.70 Hz, Ar-H), 6.66 (1H, s, Ar-H), 6.50 (1H, s, Ar-H), 5.84 (2H, s, OCH2O), 5.05 and 4.92 (each 1H, d, J = 10.80 Hz, PhCH2), 4.23 (1H, dd, J = 10.50, 2.70 Hz, H-1), 3.85 (3H, s, OCH3), 3.30-2-43 (6H, m, CH2); 13C-NMR: d 152.1 (C), 147.6 (C), 145.8 (C), 145.6 (C), 137.3 (C), 133.5 (C), 131.9 (C), 128.5 (CH), 128.3 (CH), 128.2 (CH), 128.0 (CH), 127.6 (C), 115.9 (C), 112.0 (CH), 108.5 (CH), 106.9 (CH) 100.5 (CH2), 75.0 (CH2), 55.8 (OCH3), 55.5 (CH), 39.4 (CH2), 37.6 (CH2), 29.9 (CH2).
2-Trifluoroacetyl-1-(2-benzyloxy-6-bromo-3-methoxybenzyl)-6,7-methylenedioxy-1,2,3,4-tetrahydro-isoquinoline (6b). Trifluoroacetic anhydride (38.5 g) was added dropwise to a stirred mixture of tetra-hydroisoquinoline 6a (14.8 g) and triethylamine (26.6 g) in chloroform (230 mL) at 0-10°C. Stirring was continued at room temperature for 3 h. Chloroform (150 mL) was added and the chloroform layer was washed with 10% sodium hydrogen carbonate (4 x 200 mL), 10% HCl (3 x 250 mL), brine and then dried. Removal of the solvent gave a red-brown viscous oil which crystallized from ethanol to give trifluoroacetamide 6b as a pale yellow solid (7.0 g, 39.4%); m.p. 122-123°C; UV λmax nm (MeOH) (log ε ) 231sh (4.17), 289 (3.80); IR νmax (KBr): 1681, 1571, 1504, 1300, 1284, 1269, 1235, 1190, 1180, 1158, 1082, 1036, 984, 976, 961, 941, 912, 855, 795, 759, 699 cm-1; 1H-NMR (d6-DMSO): d 7.47-7.31 (5H, m, Ar-H), 7.23 (1H, d, J = 8.70 Hz, Ar-H), 6.82 (1H, d, J = 8.70 Hz, Ar-H), 6.54 (1H, s, Ar-H), 6.37 (1H, s, Ar-H), 5.91 (2H, s, OCH2O), 5.68 (1H, dd, J = 10.70, 3.20 Hz, H-6a), 5.16 and 4.95 (each 1H, d, J = 10.80 Hz, PhCH2), 3.88 (3H, s, OCH3), 3.85-3.65 (1H, m, CH), 3.30-2.54 (5H, m, CH2); 13C–NMR: (d6-DMSO): d 155.1 (C), 151.6 (C), 147.8 (C), 146.6 (C), 146.2 (C), 137.2 (C), 130.7 (C), 128.4 (CH), 128.3 (CH), 128.2 (CH), 128.1 (CH), 127.1 (C), 125.8 (C), 118.2 (C), 115.5 (C), 113.0 (CH), 108.0 (CH), 106.6 (CH), 100.8 (CH2), 74.7 (CH2), 55.9 (OCH3), 53.4 (CH), 39.8 (CH2), 36.6 (CH2), 29.0 (CH2).
8-Benzyloxy-9-methoxy-1,2-methylenedioxy-6-trifluoroacetylnoraporphine (1b). A solution of 2,2'-azobis(isobutyronitrile) (1.59 g) and tributyltin hydride (6.18 g) in toluene (70 mL) was added in 6 equal portions over 2.5 h to a refluxing solution of trifluoroacetamide 6b (6.23 g) in toluene (110 mL). The resulting mixture was then refluxed for 24 h. The solvent was removed under vacuum and the resulting yellow residue was dissolved in acetonitrile (500 mL) and washed with hexane (5 x 200 mL) and then dried. Removal of the solvent under vacuum gave a yellow solid which was triturated from ethanol to give noraporphine 1b as colourless needles (2.15 g, 40.1%); m.p. 218 -219°C; UV λmax nm (log ε ) 282 (4.33), 323sh (3.71); IR νmax (KBr): 1681 (C=O), 1601, 1573, 1273, 1245, 1236, 1215, 1202, 1187, 1173, 1153, 1136, 1080, 1061, 1037, 944, 919, 847, 816, 735, 694, 651 cm-1; EI-MS (70 eV) m/z 497(M+, 91%), 406(91), 374(45), 309(24), 91(100). Anal. calc. for C27H22F3NO5: C, 65.2; H, 4.5; N, 2.8. Found: C, 65.0; H, 4.7; N, 3.0%; 1H-NMR: d7.86 (1H, d, J = 8.70 Hz, H-11), 7.60-7.25 (5H, m, Ar-H), 6.91 (1H, d, J = 8.70 Hz, H-10), 6.54 (1H, s, H-3), 6.02 (2H, AB q, J = 1.20 Hz, OCH2O), 5.10 and 4.97 (each 1H, d, J = 10.80 Hz, PhCH2), 4.94-4.83 (1H, m, H-6a), 4.18 (1H, br d, J = 13.20 Hz, Ha-5), 3.91 (3H, s, OCH3), 3.63-2.35 (5H, m, CH2); 13C–NMR: d 155.8 (C), 152.6 (C), 147.1 (C), 145.2 (C), 142.8 (C), 137.6 (C), 129.6 (C), 128.6 (CH), 128.4 (CH), 128.0 (CH), 126.4 (C), 123.8 (C), 123.6 (CH), 118.3 (C), 117.5 (C), 114.5 (C), 110.6 (CH), 106.7 (CH), 101.0 (CH2), 75.1 (CH2), 55.8 (OCH3), 52.3 (CH), 41.4 (CH2), 30.4 (CH2), 26.8 (CH2).
8-Hydroxy-9-methoxy-1, 2-methylenedioxy-6-trifluoroacetylnoraporphine (1c). A solution of noraporphine 1b (2.0 g) in methanol (100 mL) and chloroform (250 mL) was hydrogenolysed in the presence of 10% Pd/C (2.8 g) at 45-50 psi for 4 h. The catalyst was filtered off and the solvent removed under vacuum. The resulting white residue was shaken with chloroform (150 mL) and 10% ammonium hydroxide (100 mL). Removal of the solvent under vacuum followed by recrystallisation form ethanol gave noraporphine 1c as pale brown needles (0.65 g , 40.0%); m.p. 287-289 °C; UV λmax nm (log ε ) 284 (4.24), 298 (4.12), 321sh (3.81); IR νmax (KBr): 3459 (OH), 1674 (C=O), 1613, 1583, 1275, 1240, 1196, 1186, 1175, 1161, 1145, 1078, 1061, 1023, 940, 916, 897, 845, 810, 764, 757 cm-1; EI-MS(70 eV) m/z 407(M+,74%), 376(13), 346(30), 327(20), 305(64), 281(100), 272(39), 256(39), 206(28), 167(30), 91(100); Anal. calc. for C20H16F3NO5: C, 59.0; H, 4.0; N, 3.4. Found: C, 59.2; H, 3.8; N, 3.2%; 1H-NMR: d 7.69 (1H, d, J = 8.70 Hz, H-11), 6.87 (1H, d, J = 8.70 Hz, H-10), 6.59 (1H, s, H-3), 6.06 (2H, AB q, J = 1.20 Hz, OCH2O), 5.13 (1H, dd, J = 13.50, 4.50 Hz, H-6a), 4.30-4.18 (1H, m, Ha-5), 3.96 (3H, s, OCH3), 3.66-2.48 (5H, m, CH2); 13C-NMR (d6-DMSO): d 154.6 (C), 146.9 (C), 146.0 (C), 142.6 (C), 141.4 (C), 127.8 (C), 126.8 (C), 124.1 (C), 122.4 (C), 118.1 (CH), 116.1(C), 109.0 (CH), 106.8 (CH), 100.1 (CH2), 55.6 (OCH3), 53.0 (CH), 43.0 (CH2), 29.1 (CH2), 29.0 (CH2).
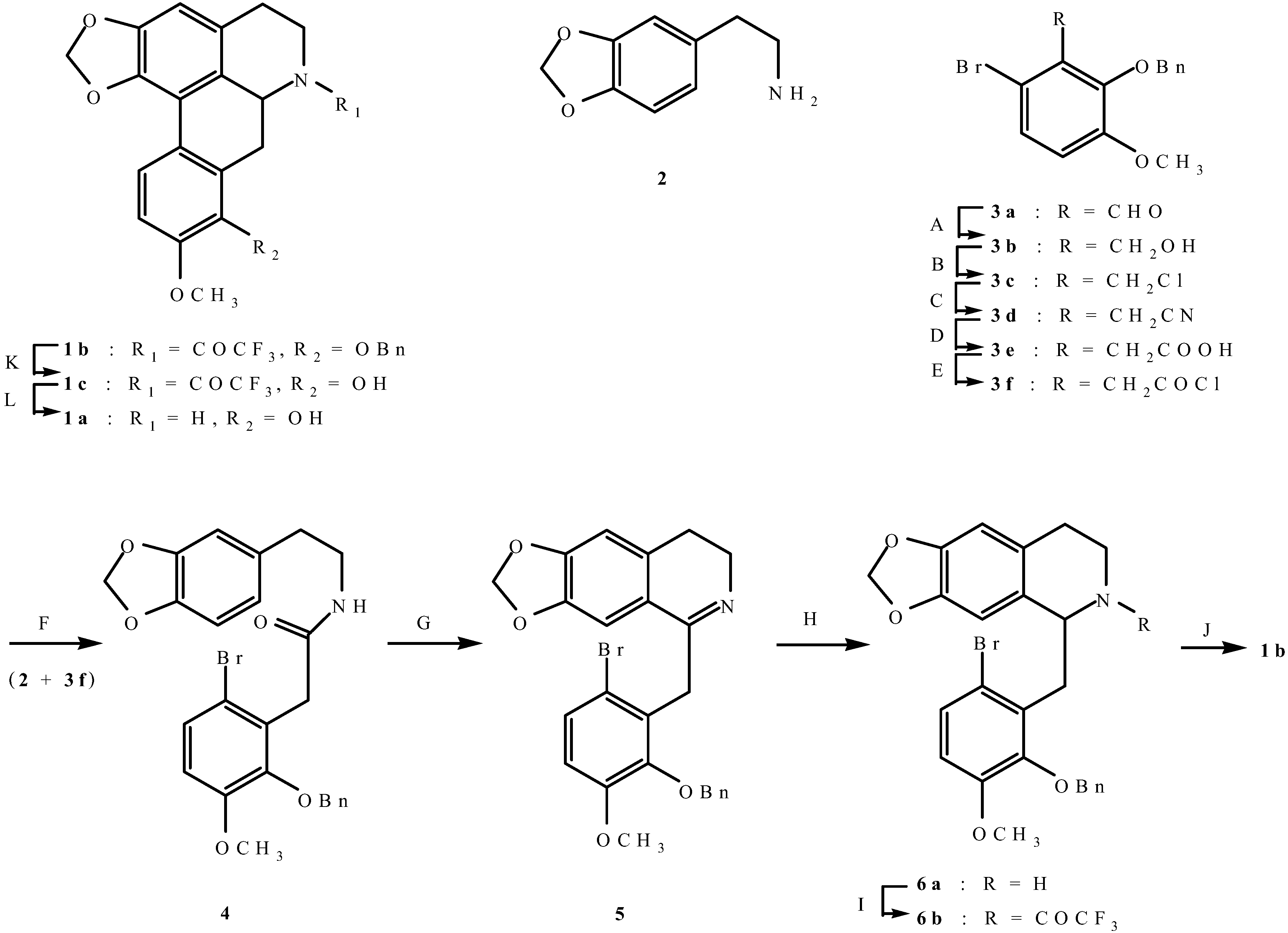
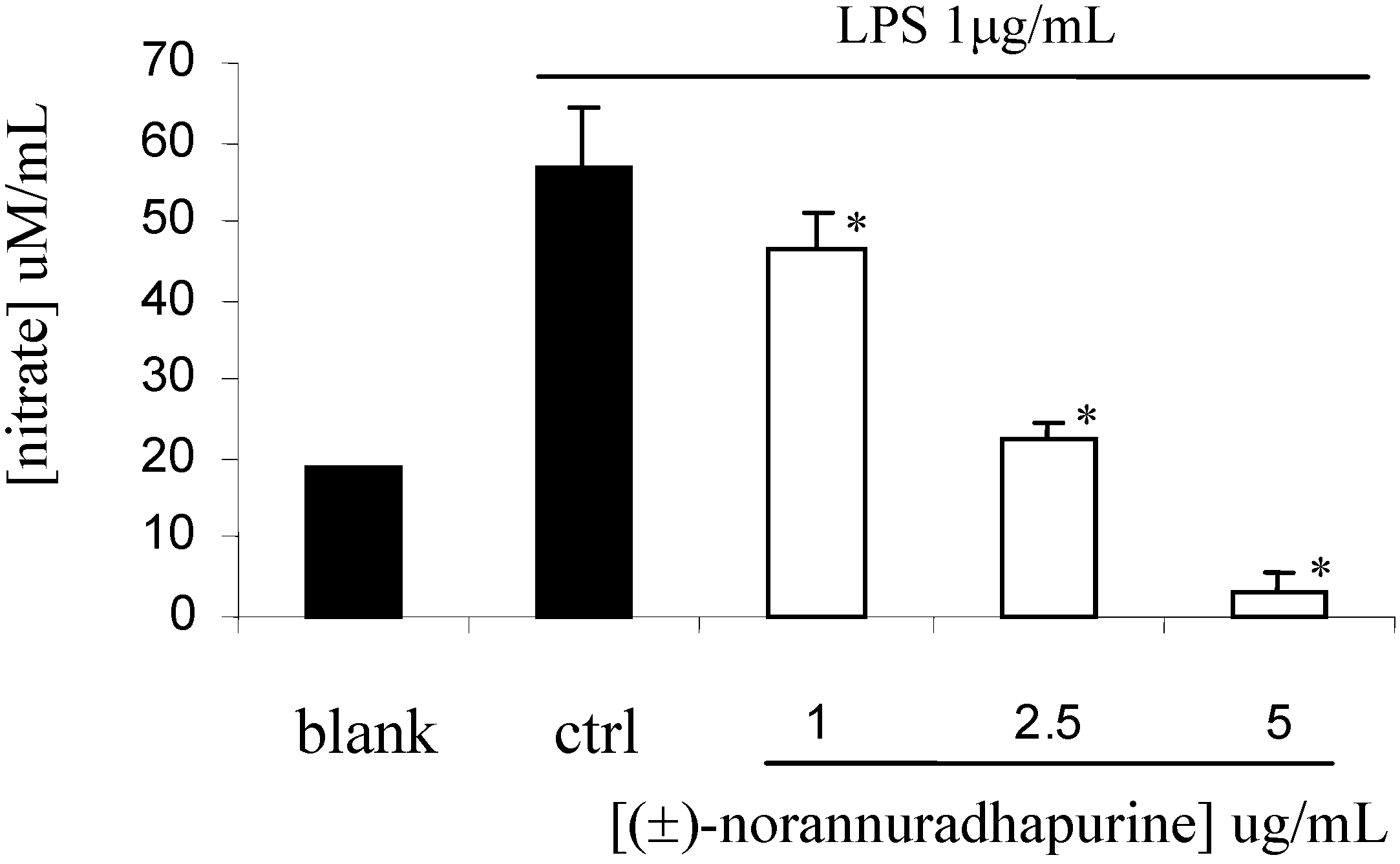
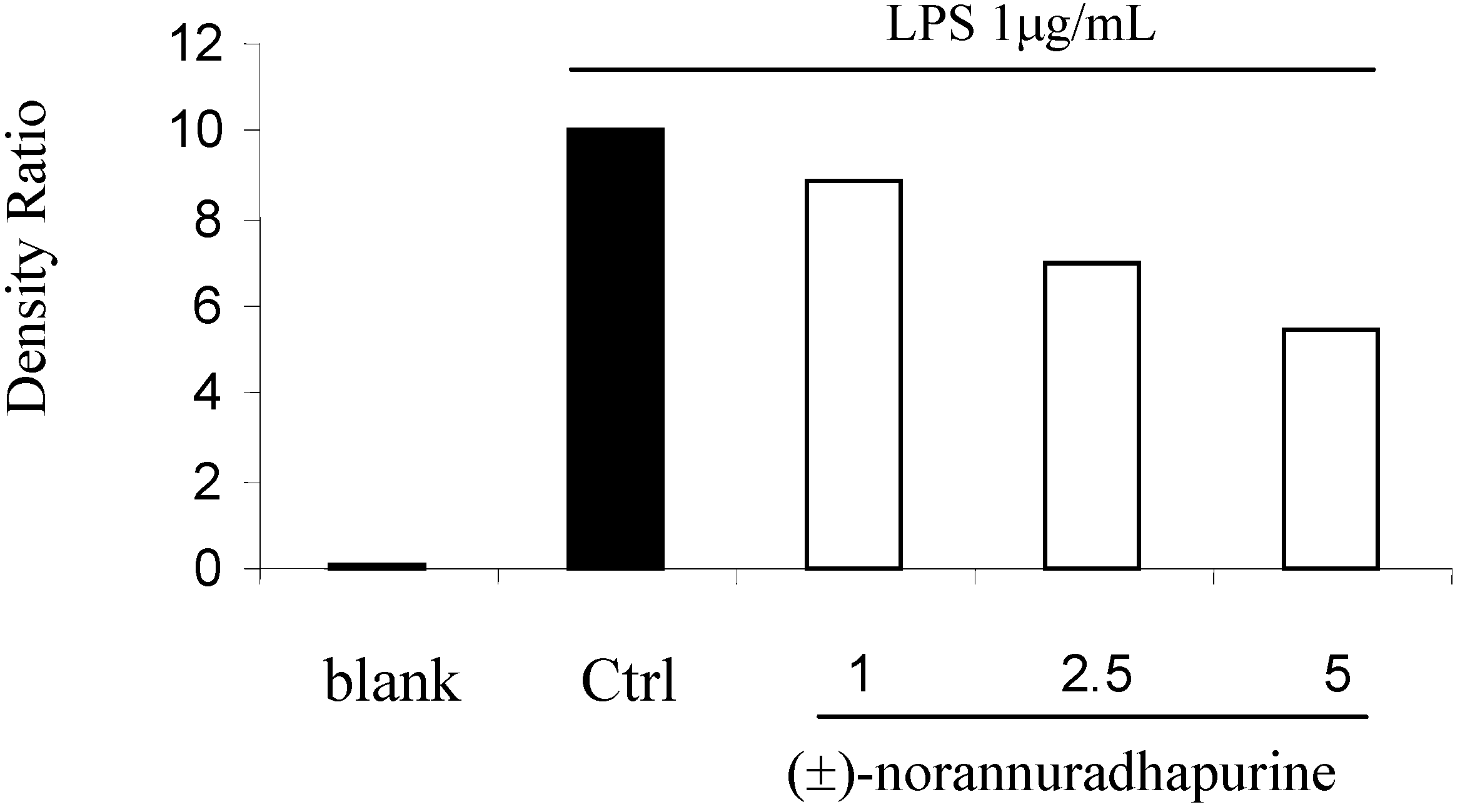
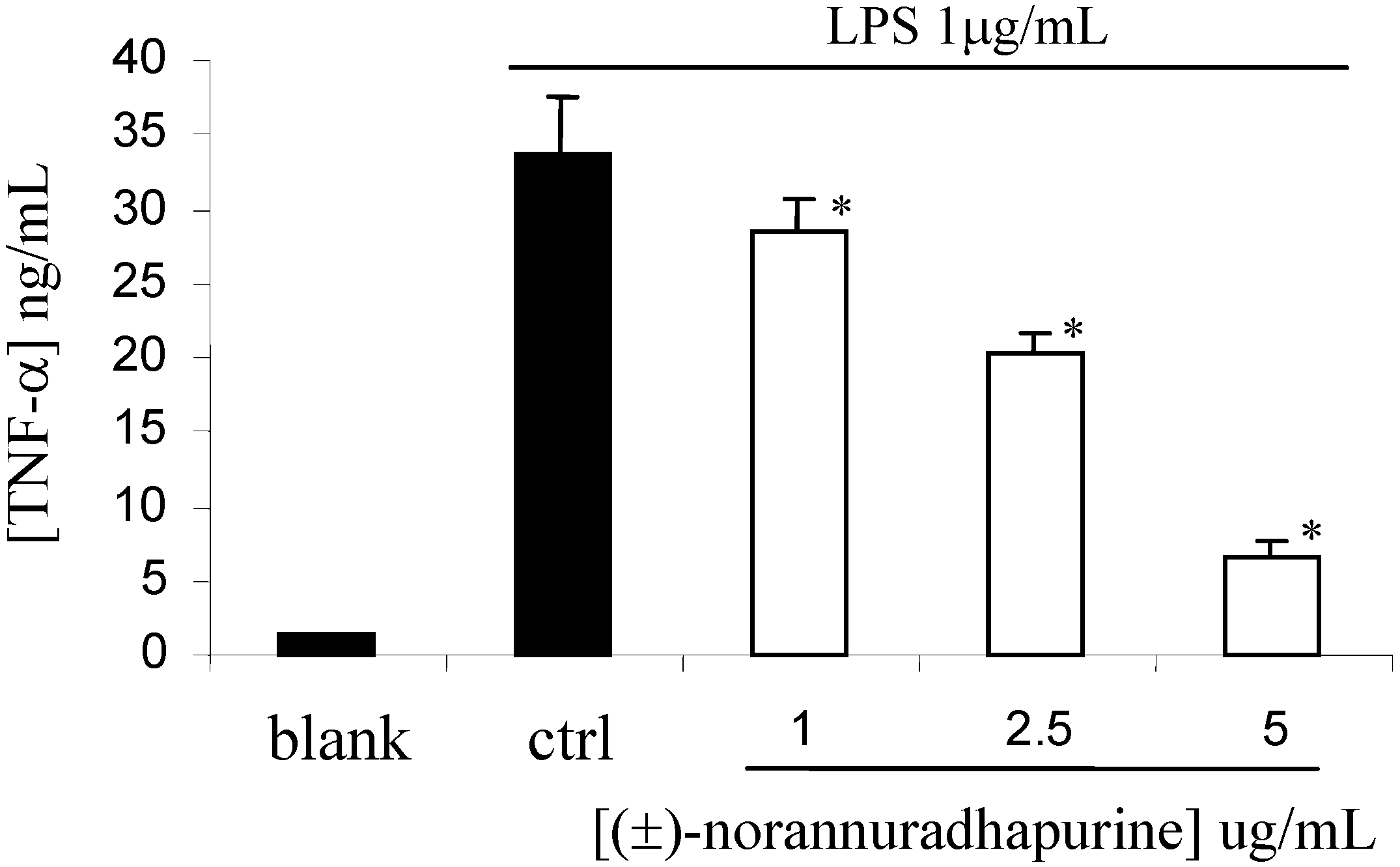
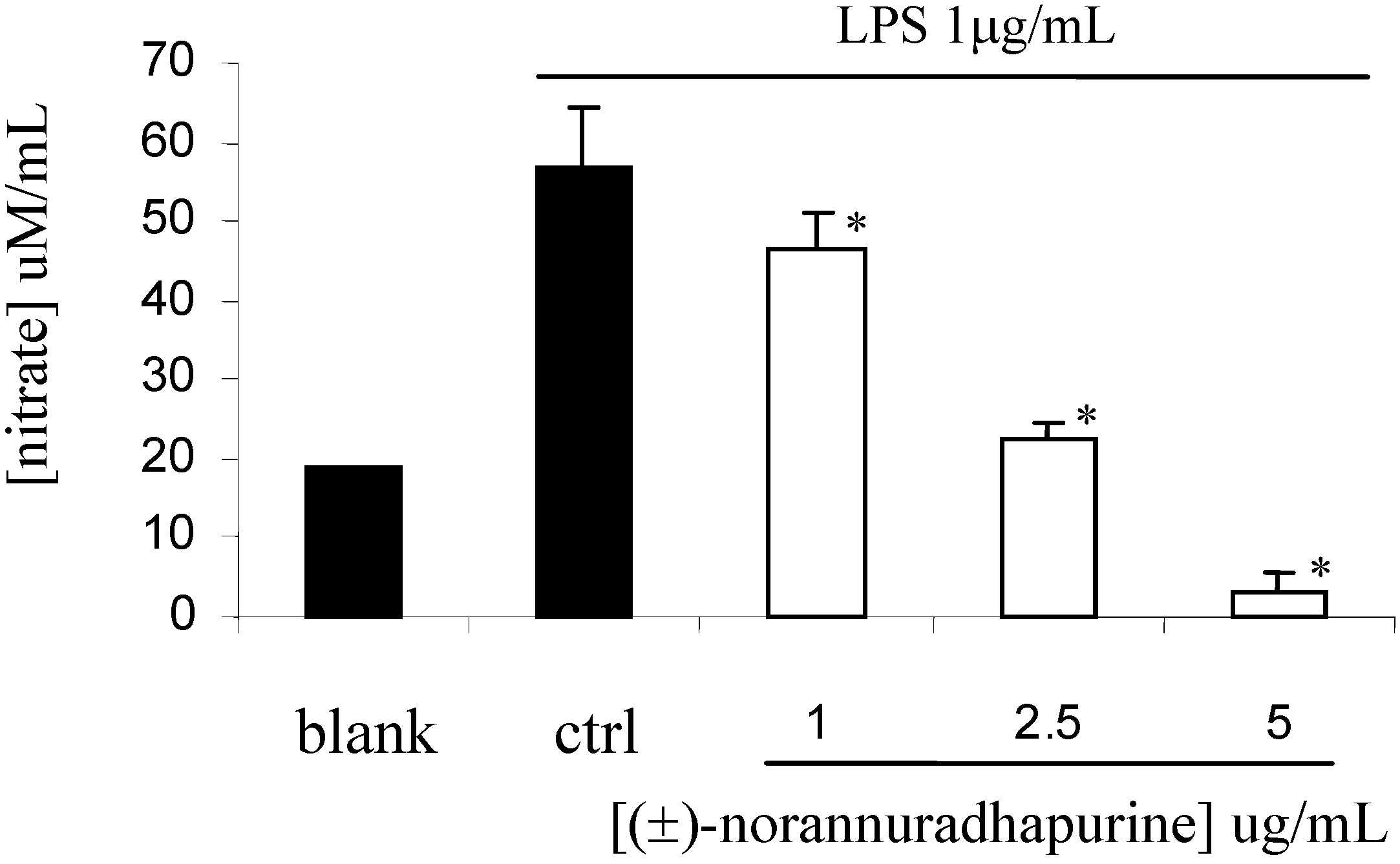
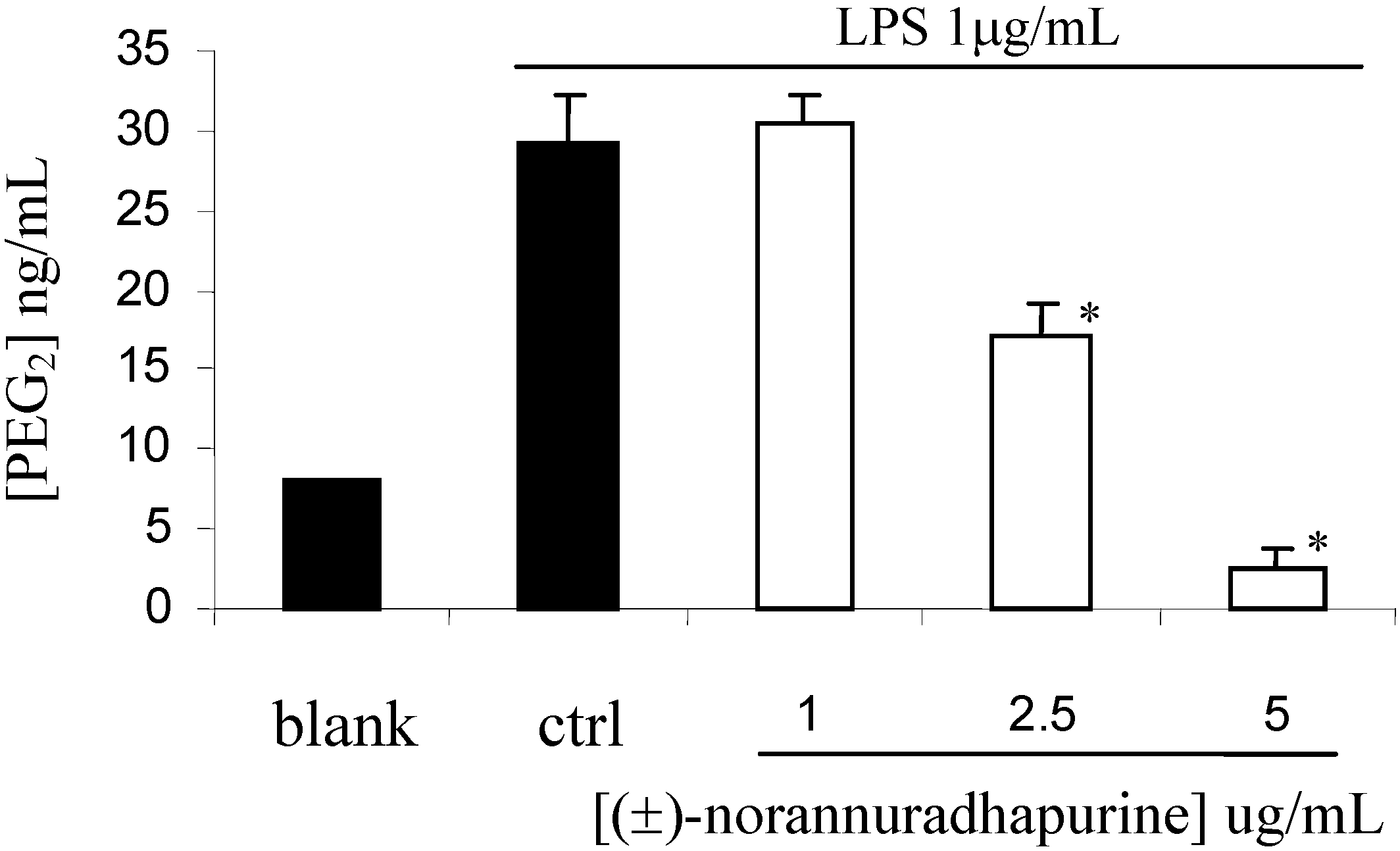
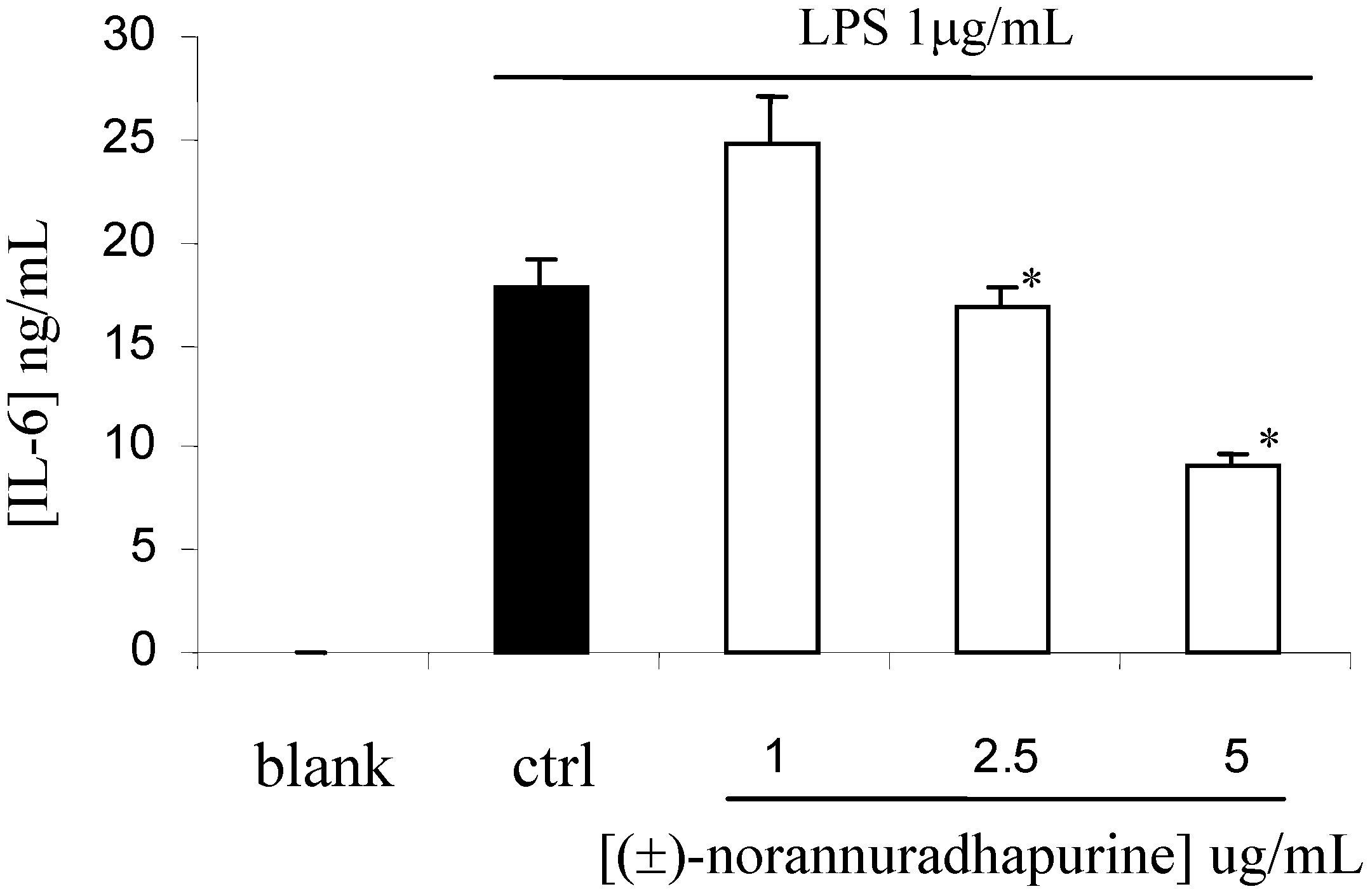
(±)-Norannuradhapurine (1a). Noraporphine 1c (0.77 g) was dissolved in methanol (140 mL) with heating. Potassium carbonate (1.46 g) in water (4.5 mL) was added and then the mixture was refluxed for 2.5 h. The solvent was removed under vacuum. The resulting residue was shaken with water (60 mL) and 10% sodium carbonate (40 mL), and extracted with chloroform (4 x 40 mL) and then dried. Removal of the solvent under vacuum followed by recrystallisation form ethanol gave (±)norannuradhapurine (1a) as purple needles (0.46 g, 78.2%) ; m.p. 207-209°C; UV λmax nm (log ε ) 217 (4.43), 283 (4.24), 298sh (4.09), 320sh (3.76); IR νmax (KBr): 3276 (OH), 2583, 1608, 1578, 1288, 1258, 1235, 1167, 1146, 1127, 1083, 1059, 1023, 991, 947, 909, 841, 792 cm-1; EI-MS(70 eV) m/z 311(M+, 54%), 310 (100), 278(17), 91(14); Anal. calc. for C18H17NO4: C, 69.4; H, 5.5; N, 4.5. Found: C, 69.6; H, 5.3; N, 4.6%; 1H-NMR: d 7.64 (1H, d, J = 8.40 Hz, H-11), 6.82 (1H, d, J = 8.40 Hz, H-10), 6.54 (1H, s, H-3), 6.00 (1H, AB q, J = 1.50 Hz, 2H, OCH2O), 3.92 (3H, s, OCH3), 3.88 (1H, dd, J = 13.80, 5.10 Hz, H-6a), 3.45-2.34 (6H, m, CH2); 13C-NMR (CDCl3): d 146.5 (C), 145.9 (C), 142.2 (C), 142.0 (C), 127.8 (C), 126.8 (C), 125.0 (C), 121.5 (C), 118.8 (CH), 116.4 (C), 108.4 (CH), 107.3 (CH), 100.5 (CH2), 56.0 (OCH3), 53.3 (CH), 43.5 (CH2), 29.6 (CH2), 29.2 (CH2); 13C NMR (d6-DMSO) d/ppm 147.0 (C), 145.9 (C), 142.7 (C), 141.4 (C), 128.0 (C), 126.9 (C), 124.2 (C), 122.6 (C), 118.1 (CH), 116.1 (C), 109.4 (CH) , 106.8 (CH), 100.1 (CH2), 55.8 (OCH3), 53.0 (CH), 43.0 (CH2), 29.2 (CH2), 29.1 (CH2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}