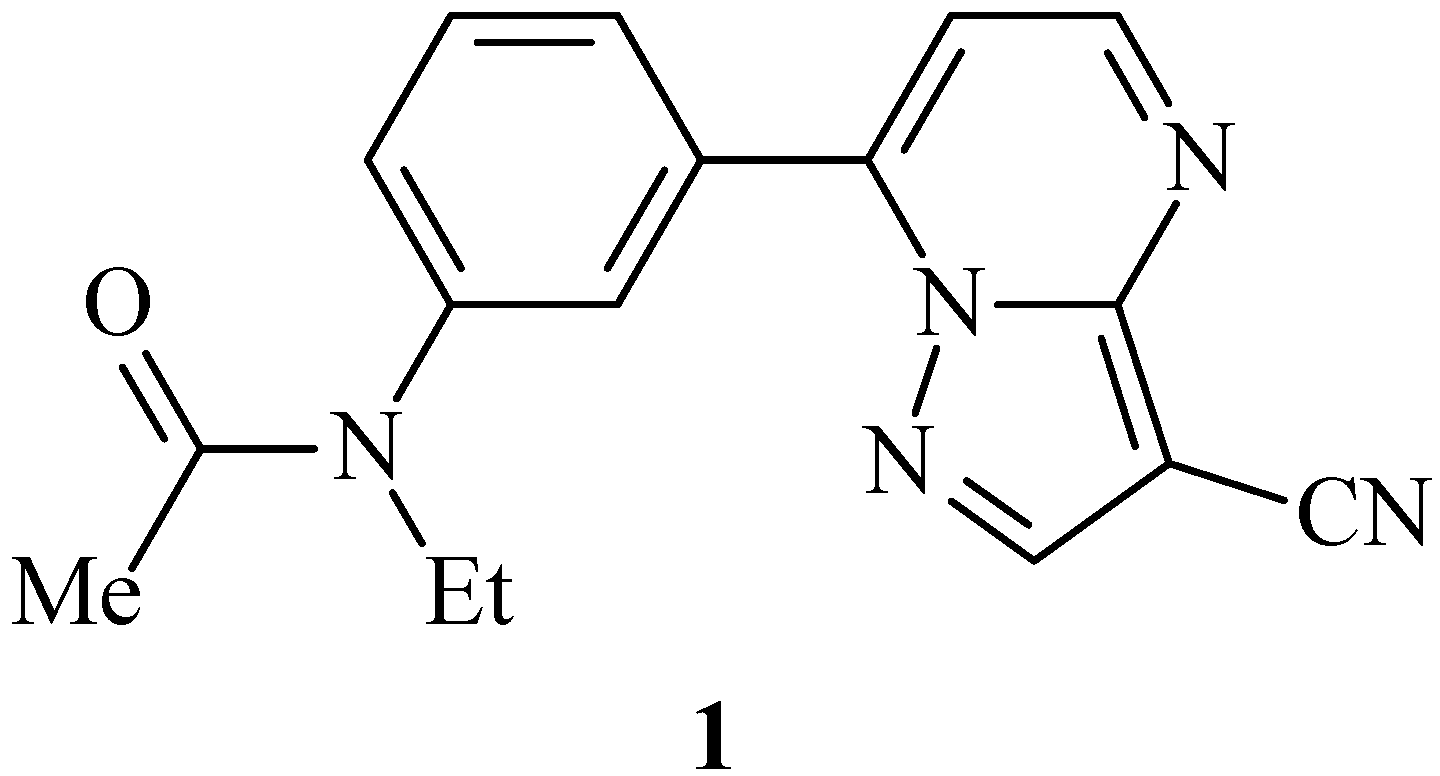
Results and Discussion
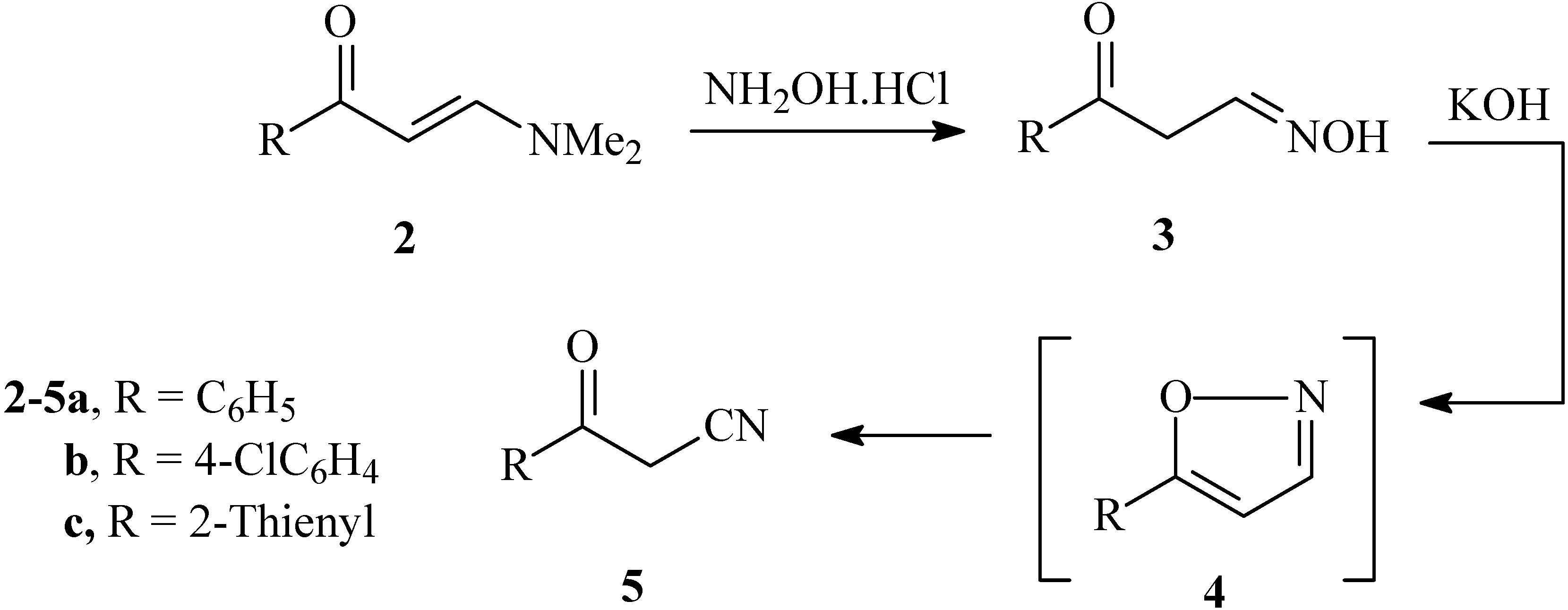
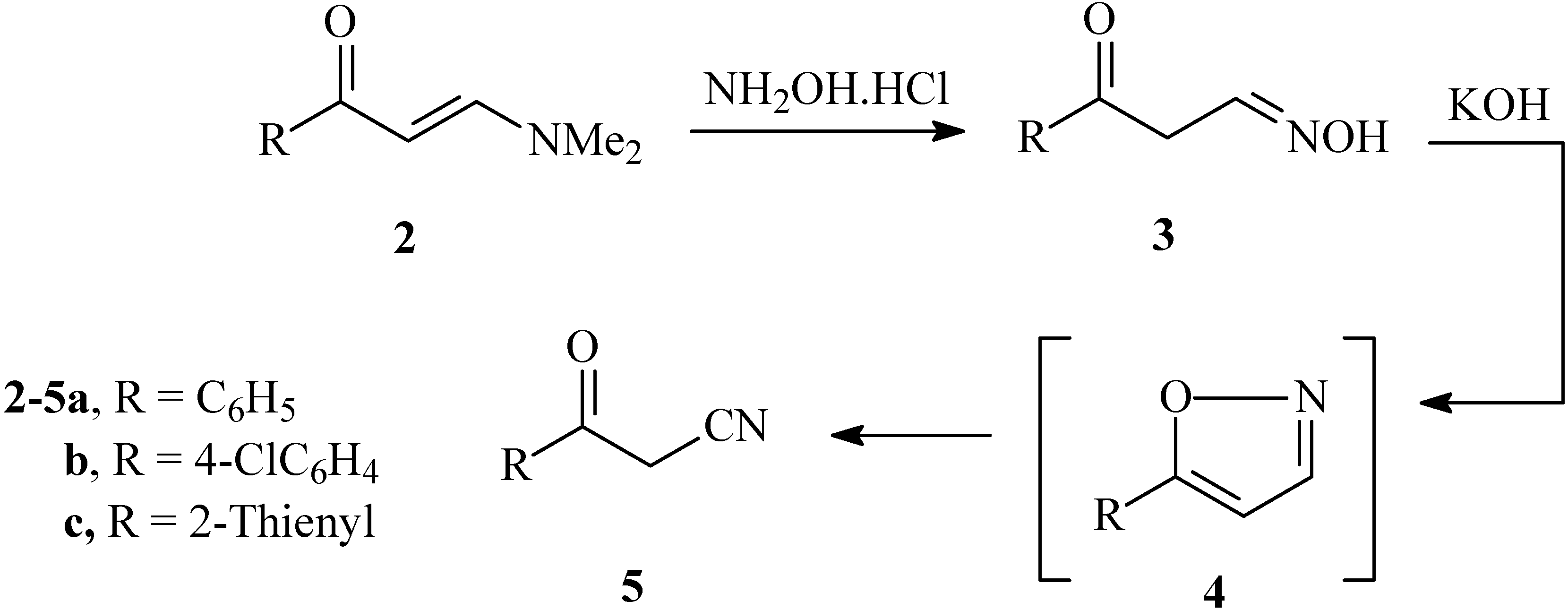
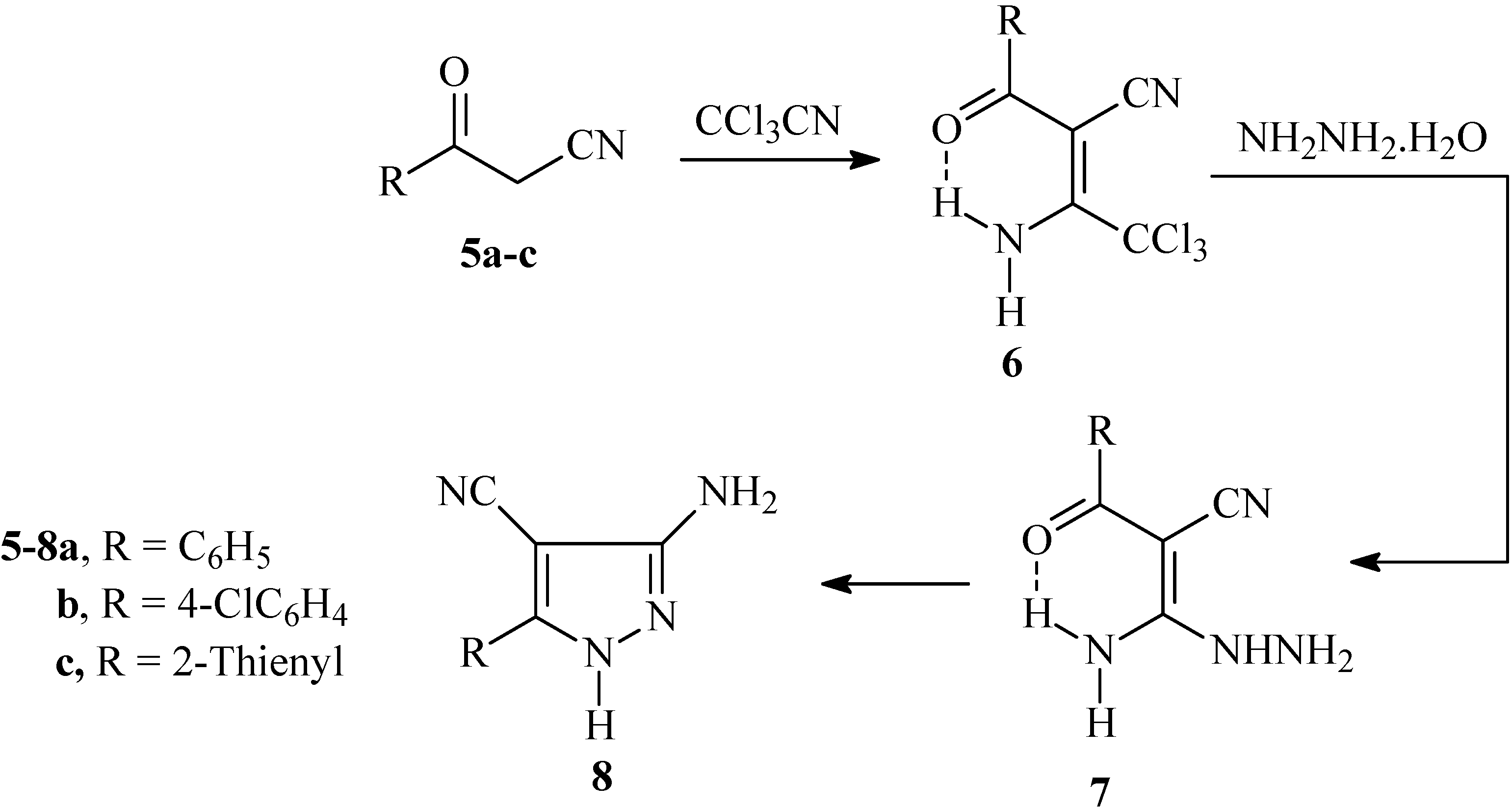
Recently we reported that the reaction of enaminones
2 with hydroxylamine hydrochloride gave the aldoximes
3 in good yields. These were converted to oxoalkanonitriles
5 via treatment with diethyl oxalate in the presence of sodium hydride [
8]. We now describe a second more efficient process for the preparation of
5 by addition of a solution of hydroxylamine hydrochloride to enaminones
2a-c in alcoholic KOH. Such a transformation apparently results
via initial formation of an isoxazole
4 that then undergoes base catalyzed ring opening furnishing
5a-c (cf.
Scheme 1).
Scheme 1.
Synthesis of 3-oxo-3-arylpropanenitriles 5a-c from enaminones 2a-c.
Scheme 1.
Synthesis of 3-oxo-3-arylpropanenitriles 5a-c from enaminones 2a-c.
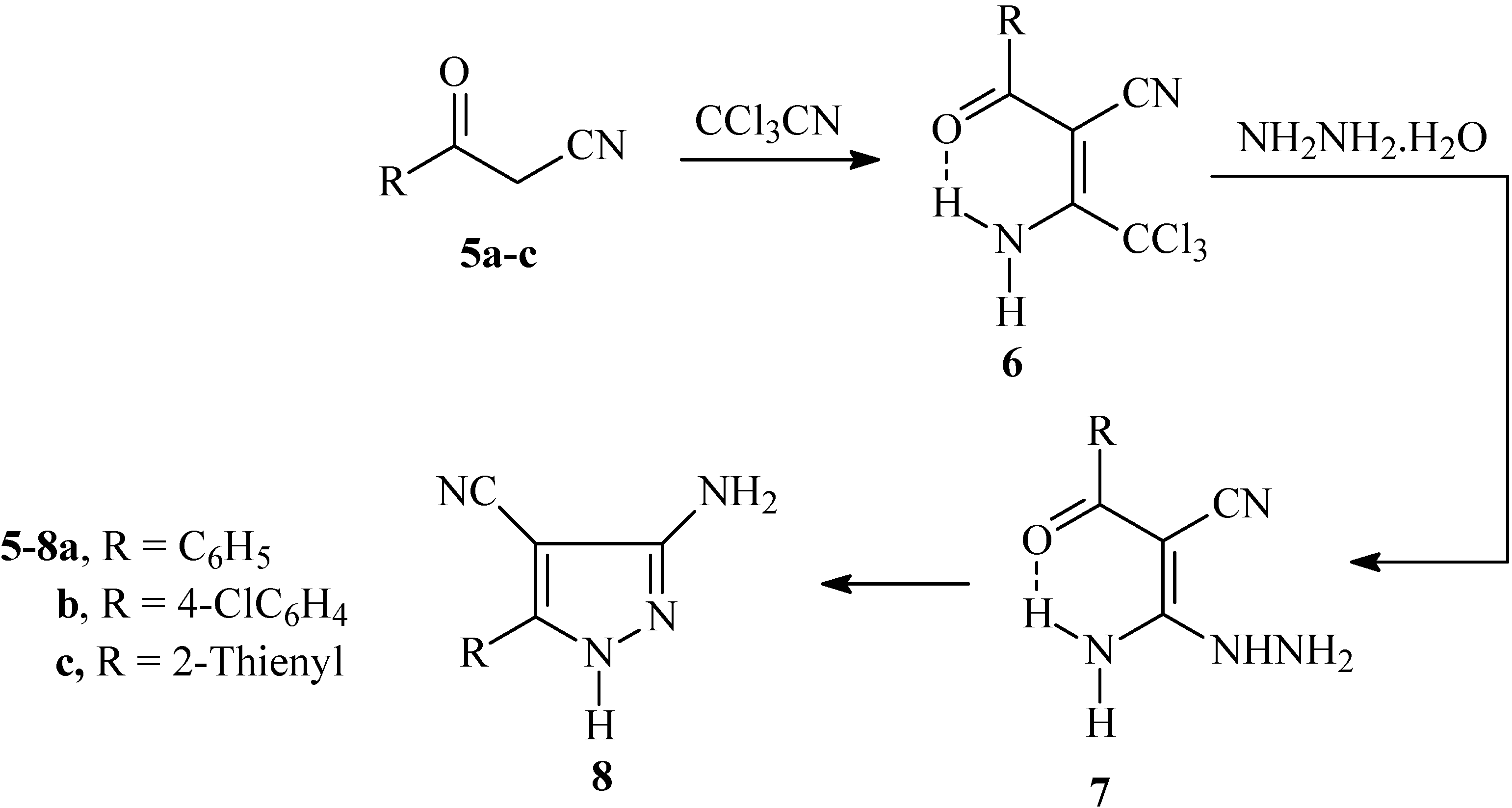
The oxoalkanonitrile derivatives
5a-c so obtained were reacted with trichloroacetonitrile to yield adducts
6a-c. Condensation of these adducts with hydrazine hydrate afforded
7a-c, that cyclized under reflux in dioxane yielding
8a-c in good yields (cf.
Scheme 2). The doublet splitting of amino group signals observed in the
1H-NMR spectra of adducts
6a-c at ca. δ
H = 10 and 12 ppm indicates a non-equivalence of the amino protons, which is probably related to the involvement of one amino proton in an intramolecular H-bond with the carbonyl moiety. Similar features were observed in
7a-c at ca. δ
H = 9 and 10 ppm. There are alternative procedures described in the literature for the preparation of
8a,b by treating arylmethylenemalononitrile with hydrazine [
9,
10] or
via transformation of isothiazoles and isoxazoles into pyrazoles
8a,b using hydrazines [
11]. In the present article, the condensation reaction of 3-amino-1
H-4-pyrazolecarbonitrile derivatives
8a-c with enaminone
2c and enaminonitrile
9, recently prepared in our laboratory [
12], is examined.
Scheme 2.
Synthesis of 3-amino-5-aryl-1H-4-pyrazolecarbonitrile 8a-c from 3-oxo-3-arylpropanenitrile 5a-c.
Scheme 2.
Synthesis of 3-amino-5-aryl-1H-4-pyrazolecarbonitrile 8a-c from 3-oxo-3-arylpropanenitrile 5a-c.
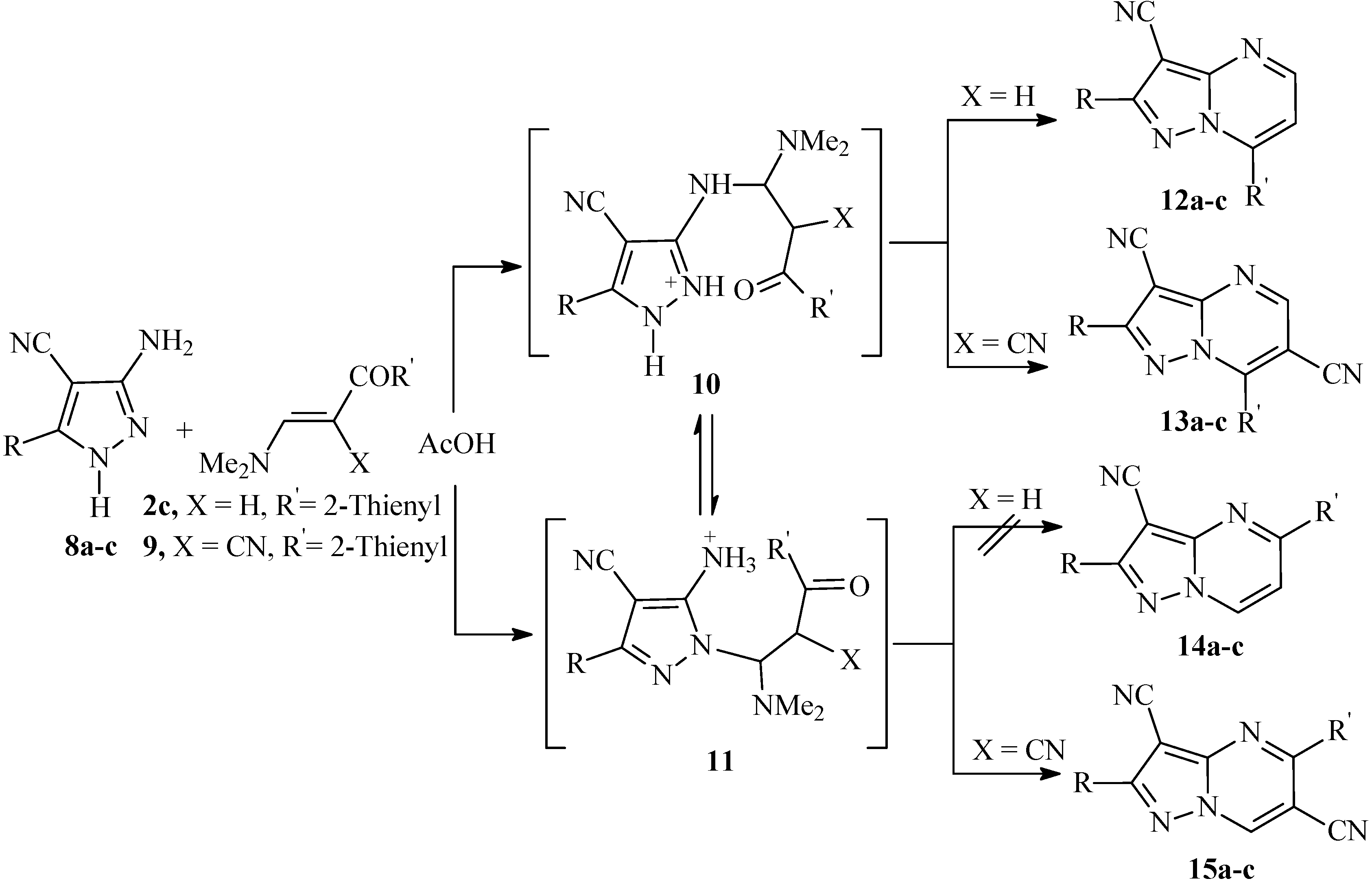
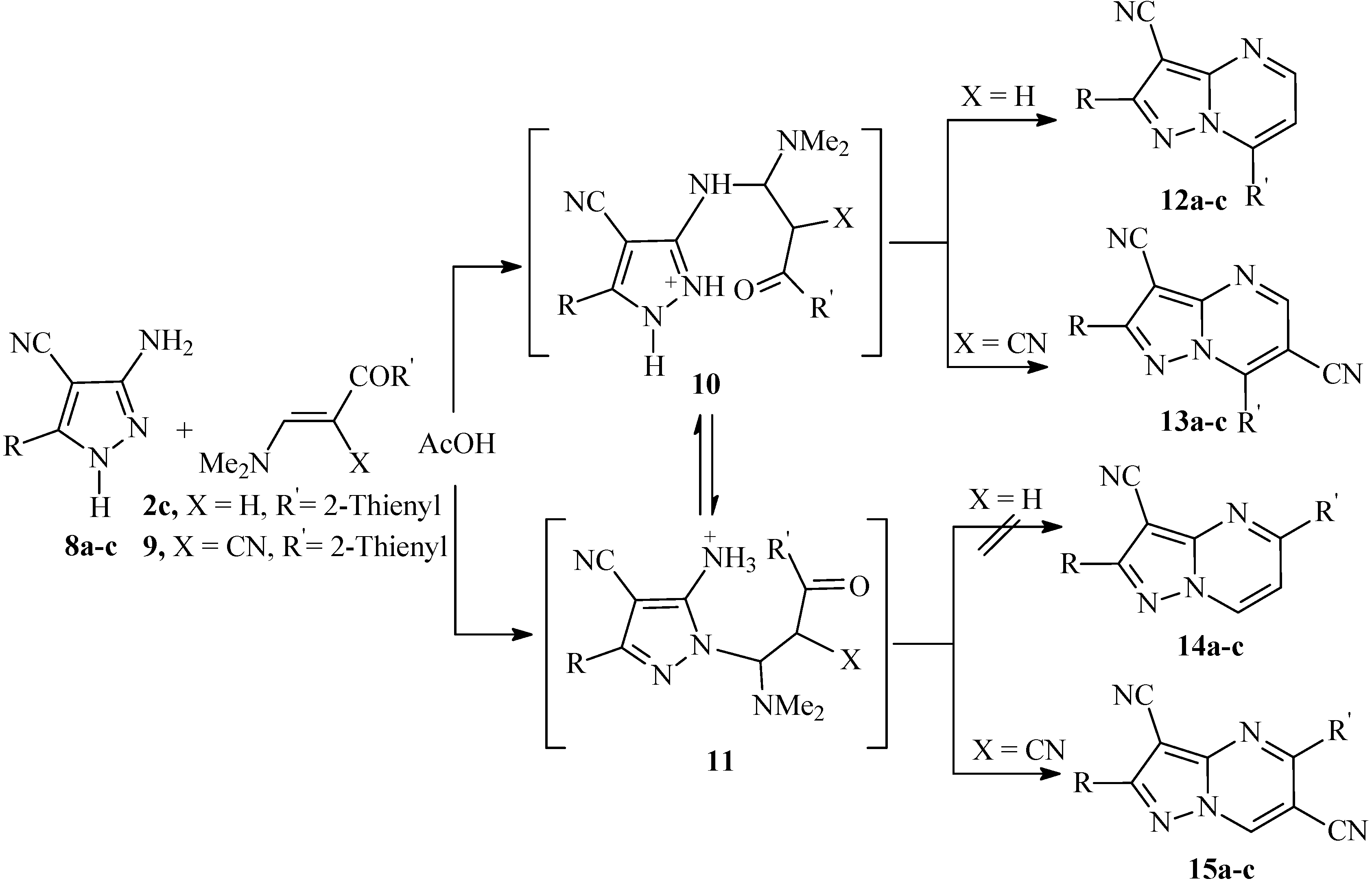
The site at which nucleophiles attack occurs on 1
H-3-aminopyrazole derivatives has been a subject of considerable debate in the past [
13,
14]. Reactions of unsymmetrical 1,3-diketones with 3(5)-aminopyrazoles often lead to the formation of inseparable mixtures of two regioisomeric pyrazolo[1,5-
a]pyrimidines due to comparable reactivity’s of the two electrophilic centers in the initial diketone. Recently, use of 1,3-dimethyluracil as the electrophile was reported to involve the attack on both endocyclic and exocyclic nitrogen affording either the pyrazolo[1,5-
a]pyrimidin-5-one or 7-one isomers, depending on the reaction conditions [
15], whereas the reaction of 1
H-3-aminopyrazole with benzylidenemalononitrile [
16] and with 1-arylbutane-1,3-diones [
17] are established to involve exocyclic amino group. It is thought that there is an equilibrium between possible initial attack at the ring nitrogen and the exocyclic amino group. Structures of the products resulting from reactions of a,b-unsaturated compounds with aminoazoles should be determined in each case, as the outcome of the reactions would be dependent on several factors, including steric consideration, relative basicities and solubility of both isomers in reaction medium. Although it is generally accepted that 3(5)-aminopyrazoles reacts with enaminones to yield the 7-substituted isomers, we noticed that this pattern is not always followed, as the reaction product proved to be dependent on both reaction conditions as well as nature of reagents. In the present article we have found that reaction of
8a-c with enaminonitrile
2c in acetic acid at reflux temperature over a long period of time resulted in selective formation of pyrazolo[1,5-
a]pyrimidines derivatives in good yields (cf.
Scheme 3).
Scheme 3.
Proposed mechanism for the formation of pyrazolo[1,5-a]pyrimidines 12a-c, 13a-c and 15a-c in acetic acid.
Scheme 3.
Proposed mechanism for the formation of pyrazolo[1,5-a]pyrimidines 12a-c, 13a-c and 15a-c in acetic acid.
The
1H-NMR spectra gave no reliable information on the structure of the products; on the other hand they indicate that only one of the possible isomeric structures,
12 or
14, are formed, rather than their mixture. The condensation products were assigned structures
12a-c on the basis of
1H-
15N HMBC measurements. For example compound
12a, showed chemical shifts for N-7a at δ (
15N) = 215 ppm, N-4 at δ (
15N) = 268 ppm. and N-1 at δ (
15N) = 285 ppm. Cross peak correlations for the coupling of the shielded proton H-6 at δ (
1H) = 8.05 ppm is observed with N-7a at δ (
15N) = 215 ppm
3J (H-6, N-7a), N-4 at δ (
15N) = 268 ppm
3J (H-6, N-4) and with N-1 at δ (
15N) = 285 ppm
4J (H-6, N-1). Coupling of the deshielded proton at δ (
1H) = 8.83 ppm with N-7a at δ (
15N) = 215 ppm
4J (H-5, N-7a) and with N-4 at δ (
15N) = 268 ppm
2J (H-5, N-4) are also observed. Alternative structure
14 would show coupling of the deshielded H-7 proton to be with N-1 in the spectrum at δ (
15N) = 285 ppm. The correlations in the
1H-
15N HMBC measurements for compounds
12b,c showed similar coupling correlations as
12a. Reaction of 3-amino-5-aryl-1
H-4-pyrazolecarbonitriles
8a-c with enaminonitrile
9 (X = CN) in acetic acid under the same reaction conditions afforded a mixture of (5)7-substituted pyrazolo[1,5-
a]pyrimidine derivatives
13a-c and
15a-c, which could not be separated by chromatographical means. Both isomers showed the same molecular weights in LC-MS. Moreover, the
1H-NMR spectra of the reaction products of
8a-c with
9 showed two singlets for two deshielded protons at ca. δ
H = 9.0 and 9.3 ppm. Based on these data, it is concluded that the compounds obtained are isomeric mixtures of
13 and
15, whose ratio (approximately 1:3) was estimated by integration of the deshielded protons in the corresponding
1H-NMR spectra. Apparently, in AcOH where the aminopyrazoles are most likely protonated, both adducts
10 and
11 are formed, as outlined in
Scheme 3. Competing cyclization leading to the 5-isomer would also be possible and mixtures are thus formed.
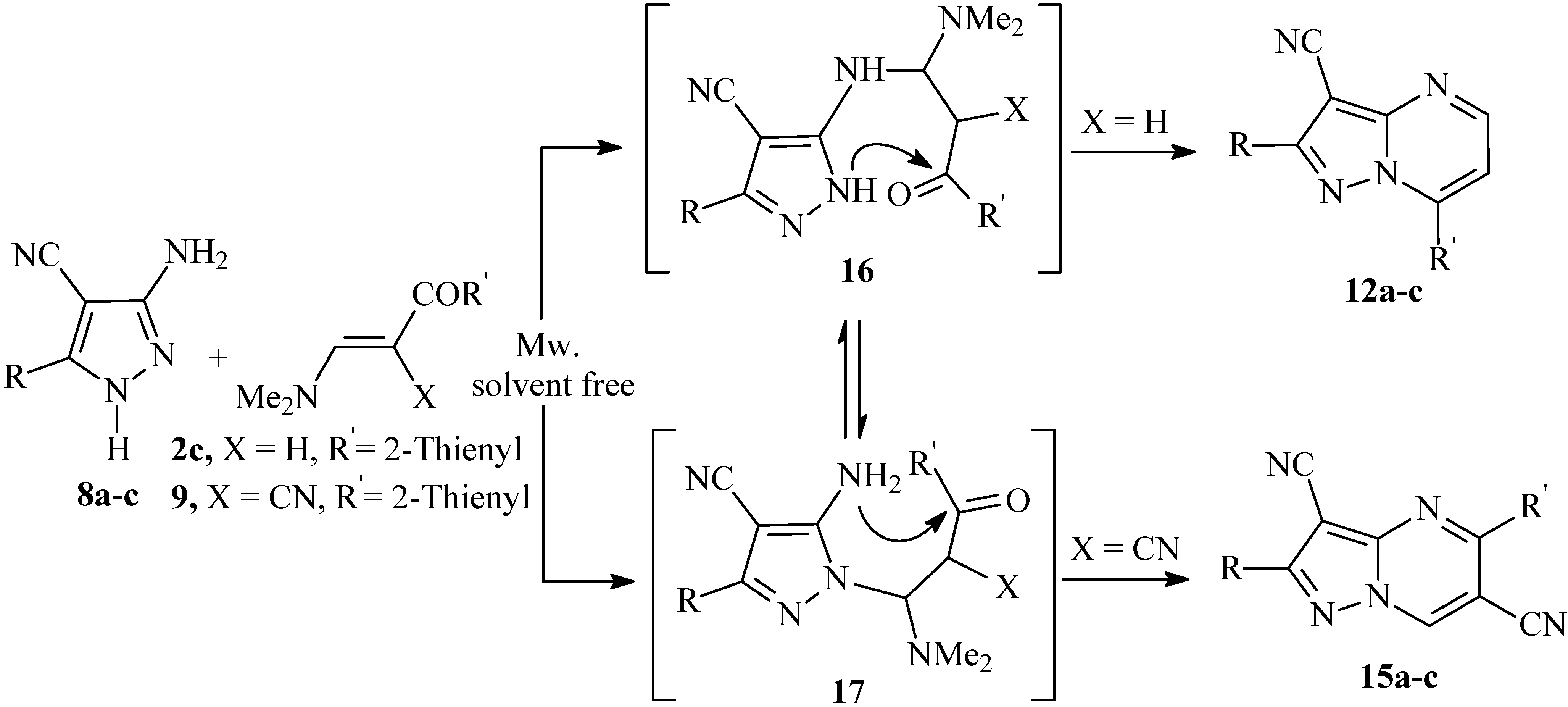
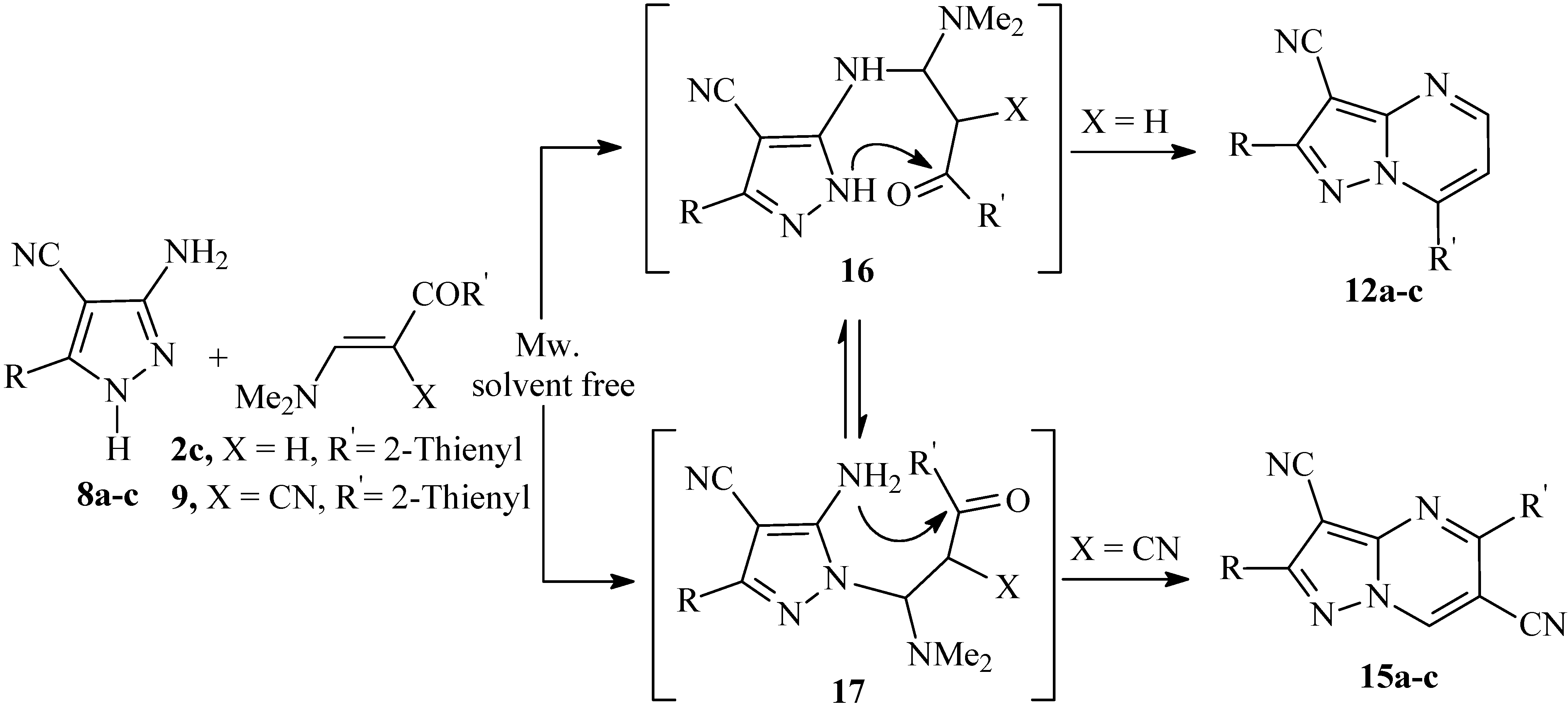
On the other hand, reaction of 3-amino-5-aryl-1
H-4-pyrazolecarbonitriles
8a-c with enaminonitrile
9 (X = CN) by heating in a direct beam microwave oven and under solvent free conditions proceeded regiospecifically to yield the 5-substituted pyrazolo[1,5-
a]pyrimidine-3-carbonitrile derivatives
15a-c in good yield. The addition reaction occurs in a manner different to the formation of the 7-substituted pyrazolo[1,5-
a]pyrimidine derivatives, as outlined in
Scheme 4. The structure
15 of the products obtained was assigned on the basis of
1H-
15N HMBC measurements. The major discrepancy between the two isomers
13 and
15 was the cross peak correlation observed between the deshielded proton at δ (
1H) = 9.06 ppm with N-1 at δ (
15N) = 283 ppm,
3J (H-7, N-1). Alternate structure
13 in which the deshielded proton is on C-5, would not show this correlation and thus structure
13 could be ruled out. There is no doubt that adducts at both exocyclic and endocyclic nitrogen atoms occur, but in this case, the latter cyclize more readily into the 5-substituted pyrazolo[1,5-
a]pyrimidines derivatives thus shifting the equilibrium. It should be noted that the isomers
12a-c were also obtained as sole products when the reactions of
8a-c with
2c was carried out by heating in a direct beam microwave oven and under solvent free conditions.
Scheme 4.
Cyclocondensation reaction of 8a-c with 2c and 9 by heating in a direct beam microwave oven and solvent free conditions resulted in selective formation of pyrazolo[1,5-a]pyrimidines 12a-c and 15a-c.
Scheme 4.
Cyclocondensation reaction of 8a-c with 2c and 9 by heating in a direct beam microwave oven and solvent free conditions resulted in selective formation of pyrazolo[1,5-a]pyrimidines 12a-c and 15a-c.
The behavior of
8a-c with
9 under microwave irradiation may be attributed to the formation of hot spots that affect the reaction selectivity due to the increase in heating rate. This may lead to the formation of thermodynamically stable products in preference to the kinetic ones [
18,
19]. One can thus conclude that this solvent-free reactions proceeds in a regiospecific fashion by the relative reactivity of exocyclic nitrogen and ring nitrogen atoms.
Experimental
General
Melting points were determined on a Shimadzu-Gallenkamp apparatus and are uncorrected. Microwave mediated chemistry was conducted in heavy-walled Pyrex tubes fitted with PCS cap. and performed with a single mode cavity Explorer Microwave Synthesizer (CEM Corporation, NC, USA), producing continuous irradiation and equipped with simultaneous external air-cooling system. Elemental analyses were obtained on a LECO CHNS-932 Elemental Analyzer. 1H-NMR spectra were obtained in DMSO-d6 on a Bruker DPX 400 MHz superconducting spectrometer in DMSO-d6 with TMS as an internal standard. Two-dimensional NMR spectra were determined on Bruker Avance II 600 MHz superconducting spectrometer in DMSO-d6 and FT-IR measurements were recorded in KBr disks on a Perkin Elmer 2000 FT-IR system. Mass spectrometric analyses were recorded on a VG-Autospec-Q high performance tri-sector GC/MS/MS.
General procedure for the preparation of compounds 5a-c
A mixture of each enaminone 2a-c (10 mmol) and NH2OH.HCl (0.69 g, 10 mmol) in EtOH (30 mL), was added a solution of KOH (5.60 g, 10 mmol) in H2O (8 mL). The reaction mixture was heated under reflux and deemed completed when the yellow color of the solution changed to brown (in 30-60 min.). The reaction mixtures were then poured onto water and neutralized with HCl. The solid products so obtained were collected by filtration and crystallized from benzene.
3-Oxo-3-phenylpropanenitrile (
5a): White crystals, yield (89 %, 1.30 g); mp 79-81
oC (Lit. [
12,
13] mp. 80-82); IR (cm
-1): 2255 (CN) and 1689 (CO); MS m/z (M)
+ = 145;
1H-NMR: d = 4.78 (s, 2H, CH
2), 7.57 (t, 2H,
J = 6.8 Hz, Ph-H), 7.72 (t, 1H,
J = 7.4 Hz, Ph-H), 7.94 (d, 2H,
J = 6.8 Hz, Ph-H); Anal. calcd. for C
9H
7NO: (145.16): C, 74.47; H, 4.86; N, 9.65. Found: C, 74.50; H, 4.75; N, 9.57.
3-(4-Chlorophenyl)-3-oxopropanenitrile (
5b): Yellow crystals, yield (86 %, 1.54 g); mp 126-128
oC (Lit. [
12,
13] mp. 128
oC); IR (cm
-1): 2255 (CN) and 1680 (CO); MS m/z (M)
+ = 179;
1H-NMR: d = 4.75 (s, 2H, CH
2), 7.60 (d, 2H,
J = 8.1 Hz, arom-H), 7.75 (d, 2H,
J = 8.3 Hz, arom-H); Anal. calcd. for C
9H
6Cl NO: (179.01): C, 60.19; H, 3.37; N, 7.80. Found: C, 59.87; H, 3.30; N, 7.67.
3-Oxo-3-(2-thienyl)propanenitrile 5c: Yellow crystals, yield (88 %, 1.32 g); mp 112-114
oC (Lit. [
12,
13] mp. 110-112
oC); IR (cm
-1): 3243 (NH
2), 2255 (CN) and 1666 (CO); MS m/z (M)
+ = 151;
1H-NMR: d = 4.72 (s, 2H, CH
2), 7.30 (t, 1H,
J = 4.4 Hz, thienyl H-4), 7.99 (d, 1H,
J = 4.8 Hz, thienyl H-3), 8.14 (d, 1H,
J = 4.8 Hz, thienyl H-5); Anal. calcd. for C
7H
5NOS: (151.19): C, 55.61; H, 3.33; N, 9.26; S, 21.21. Found: C, 55.72; H, 3.31; N, 9.28; S, 21.00.
General procedure for the preparation of compounds 6a-c
To a stirred mixture of each of oxoalkanonitrile 5a-c (10 mmol) in EtOH (20 mL) and in the presence of anhydrous NaOAc (1 g), was added 3,3,3-trichloropropanenitrile (10 mmol, 1.57 g). The resulting mixture was stirred for six hrs at r.t., then evaporated under vacuum to half its volume. The reaction mixture was then poured onto water. The solid product obtained was collected by filtration and crystallized from ethanol.
(Z)-3-Amino-2-benzoyl-4,4,4-trichloro-2-butenenitrile (6a): Pale yellow crystals, yield (82 %, 2.36 g); mp 182-184 oC; IR (cm-1): 3267 and 3250 (NH2), 2210 (CN) and 1613 (CO); MS m/z (M+-1) = 288; 1H-NMR: d = 7.48 (t, 2H, J = 7.8 Hz, Ph-H), 7.57 (t, 1H, J = 7.9 Hz, Ph-H), 7.68 (d, 2H, J = 7.8 Hz, Ph-H), 9.96 (s, 1H, NH2), 11.94 (s, 1H, NH2); Anal. calcd. for C11H7Cl3N2O: (289.55): C, 45.63; H, 2.44; N, 9.67. Found: C, 45.90; H, 2.41; N, 9.56.
(Z)-3-Amino-4,4,4-trichloro-2-(4-chlorobenzoyl)-2-butenenitrile (6b): Yellow crystals, yield (76 %, 2.46 g); mp 221-222 oC; IR (cm-1): 3260 (NH2), 2214 (CN) and 1610 (CO); MS m/z (M+-1) = 322; 1H-NMR: d = 7.58 (d, 2H, J = 8.0 Hz, arom-H), 7.71 (d, 2H, J = 8.2 Hz, arom-H), 10.05 (s, 1H, NH2), 11.87 (s, 1H, NH2); Anal. calcd. for C11H6Cl4N2O: (323.99): C, 40.78; H, 1.87; N, 8.65. Found: C, 40.90; H, 2.02; N, 8.80.
(Z)-3-Amino-4,4,4-trichloro-2-(thiophene-2-carbonyl)-2-butenenitrile (6c): Brown crystals, yield (85 %, 2.50 g); mp 168-170 oC; IR (cm-1): 3243 (NH2), 2216 (CN) and 1624 (CO); MS m/z (M+-1) = 294; 1H-NMR: d = 7.25 (t, 1H, J = 4.0 Hz, thienyl H-4), 8.01 (d, 1H, J = 5.2 Hz, thienyl H-3), 8.17 (d, 1H, J = 4.0 Hz, thienyl H-5), 9.96 (s, 1H, NH2), 12.01 (s, 1H, NH2); Anal. calcd. for C9H5Cl3N2OS: (295.57): C, 36.57; H, 1.71; N, 9.48; S, 10.85. Found: C, 36.25; H, 1.96; N, 9.25; S, 10.50.
Reaction of 6a-c with hydrazine hydrate for the preparation of 7a-c
To each compound 6a-c (10 mmol), excess hydrazine hydrate (3 mL) was added and stirred for 3 min. (exothermic reaction). The reaction mixture is then allowed to cool to ambient temperature. During time a precipitate is formed that was filtered off and recrystallized from ethanol.
(E)-3-Amino-2-benzoyl-3-hydrazino-2-propenenitrile (7a): This compound was obtained as white crystals, yield (93 %, 1.90 g); mp 143-145 oC; IR (cm-1): 3444, 3345 and 3209 (NH and NH2), 2182 (CN) and 1656 (CO); MS m/z (M)+ = 202; 1H-NMR: d = 4.72 (s, 2H, NH2), 7.38-7.46 (m, 3H, arom. H), 7.48 (br s, 1H, NH), 7.58 (d, 2H, J = 7.8 Hz, arom-H), 8.51 (s, 1H, NH2), 9.69 (s, 1H, NH2); Anal. calcd. for C10H10N4O: (202.21): C, 59.40; H, 4.98; N, 27.71. Found: C, 59.36; H, 5.2; N, 27.95.
(E)-3-Amino-2-(4-chlorobenzoyl)- 3-hydrazino-2-propenenitrile (7b): Beige crystals, yield (89 %, 2.10 g); mp 238-240 oC; IR (cm-1): 3387, 3350 and 3305 (NH and NH2), 2185 (CN) and 1646 (CO); MS m/z (M)+ = 236; 1H-NMR: d = 4.73 (s, 2H, NH2), 7.48 (d, 2H, J = 8.0 Hz, arom-H), 7.53 (br s, 1H, NH), 7.59 (d, 2H, J = 8.0 Hz, arom-H), 8.54 (s, 1H, NH2), 9.97 (s, 1H, NH2); Anal. calcd. for C10H9ClN4O: (236.66): C, 50.75; H, 3.83; N, 23.67. Found: C, 50.81; H, 3.92; N, 23.38.
(Z)-3-Amino-3-hydrazino-2-(2-thienylcarbonyl)-2-propenenitrile (7c): Beige crystals, yield (94 %, 1.95 g); mp 175-177 oC; IR (cm-1): 3449, 3331 and 3217 (NH and NH2), 2189 (CN) and 1659 (CO); MS m/z (M)+ = 208; 1H-NMR: d = 4.72 (s, 2H, NH2), 7.14 (t, 1H, J = 4.0 Hz, thienyl H-4), 7.55 (br s, 1H, NH), 7.76 (d, 1H, J = 4.8 Hz, thienyl H-3), 7.90 (d, 1H, J = 4.2 Hz, thienyl H-2), 8.54 (s, 1H, NH2), 9.98 (s, 1H, NH2); Anal. calcd. for C8H8N4OS: (208.24): C, 46.14; H, 3.87; N, 26.90; S, 15.40. Found: C, 45.89; H, 4.09; N, 26.59; S, 15.25.
General procedure for the preparation of compounds 8a-c
Each compound 7a-c (10 mmol) was refluxed in dioxane (20 mL) for 30 min. then left to cool at r.t. The target compounds separated as crystals that were collected by filtration and crystallized from the appropriate solvent.
3-Amino-5-phenyl-1H-4-pyrazolecarbonitrile (8a): Buff crystals from dioxane, yield (93 %, 1.71 g) mp 200-202 oC; IR (cm-1): 3348, 3303 (NH2), 3193 (NH) and 2230 (CN); MS m/z (M)+ = 184; 1H- NMR: d = 6.50 (s, 2H, NH2), 7.41-7.46 (m, 3H, arom. H), 7.80 (d, 2H, J = 7.2 Hz, arom-H), 12.16 (br s, 1H, NH); Anal. calcd. for C10H8N4: (184.20): C, 65.21; H, 4.38; N, 30.42. Found: C, 65.03; H, 4.57; N, 30.12.
3-Amino-5-(4-chlorophenyl)-1H-4-pyrazolecarbonitrile (8b): Brownish crystals from dioxane, yield (95 %, 2.07 g); mp 218-220 oC; IR (cm-1): 3348, 3302 (NH2), 3137 (NH) and 2223 (CN); MS m/z (M)+ = 218; 1H-NMR: d = 6.40 (s, 2H, NH2), 7.53 (d, 2H, J = 8.4 Hz, arom-H), 7.80 (d, 2H, J = 8.4 Hz, arom-H), 12.06 (br s, 1H, NH); Anal. calcd. for C10H7ClN4: (218): C, 54.93; H, 3.23; N, 25.62. Found: C, 55.07; H, 3.46; N, 25.54.
3-Amino-5-(2-thienyl)-1H-4-pyrazolecarbonitrile (8c): Beige crystals from n-propanol, yield (93 %, 1.76 g); mp 238-240 oC; IR (cm-1): 3325, 3298 (NH2), 3177 (NH) and 2228 (CN); MS m/z (M)+ = 190;1H-NMR: δ = 6.38 (s, 2H, NH2), 7.14 (t, 1H, J = 4.4 Hz, thienyl H-4), 7.70 (d, 1H, J = 4.4 Hz, thienyl H-3), 7.93 (d, 1H, J = 4.2 Hz, thienyl H-2), 12.00 (br s, 1H, NH); Anal. calcd. for C8H6N4S: (190.23): C, 50.51; H, 3.18; N, 29.45; S, 16.86. Found: C, 50.63; H, 3.02; N, 29.57; S, 17.02.
Reaction of 8a-c with enaminone 2c and with enaminonitrile 9
Procedure A: a mixture of each of compound 8a-c (10 mmol) and each of 2c or 9 (10 mmol) was heated under reflux in AcOH (15 mL) for 4h, during which time a precipitate is formed. The reaction mixture was filtered off and recrystallized from acetone.
Procedure B: a mixture of each of compound 8a-c (10 mmol) and each of 2c or 9 (10 mmol) was dissolved in ethanol (2 mL) in a small beaker. The reaction mixture was dried in air and the beaker was put in a domestic microwave oven 290 W and irradiate for 15 min. The progress of the reaction was monitored every 3 min. The product was extracted with acetone (2 x 10 mL). The solvent was removed by distillation under reduced pressure to obtain the crude product that was further recrystallized from the appropriate solvent.
2-Phenyl-7-(2-thienyl)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (12a): Yellow crystals from acetone, yield (89 %, 2.68 g); mp 237-239 oC; IR (cm-1): 2222 (CN); MS m/z (M)+ = 302; 1H-NMR: d = 7.45 (t, 1H, J = 4.2 Hz, thienyl H-4), 7.62-7.70 (m, 3H, arom. H), 8.06 (d, 1H, J = 4.8 Hz, H-6), 8.20 (d, 2H, J = 8.0 Hz, arom. H), 8.24 (d, 1H, J = 4.2 Hz, thienyl H-3), 8.63 (d, 1H, J = 4.2 Hz, thienyl H-5), 8.82 (d, 1H, J = 4.8 Hz, H-5); Anal. calcd. for C17H10N4S: (302.35): C, 67.53; H, 3.33; N, 18.53; S, 10.61. Found: C, 67.64; H, 3.47; N, 18.47; S, 10.43.
2-(4-Chlorophenyl)-7-(2-thienyl)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (12b): Brownish red crystals from ethanol, yield (86 %, 2.88 g); mp 252-254 oC; IR (cm-1): 2220 (CN); MS m/z (M)+ = 336; 1H-NMR: d = 7.36 (t, 1H, J = 4.2 Hz, thienyl H-4), 7.66 (d, 2H, J = 6.6 Hz, arom. H), 7.99 (d, 1H, J = 4.8 Hz, H-6), 8.12 (d, 2H, J = 6.6 Hz, arom. H), 8.16 (d, 1H, J = 4.2 Hz, thienyl H-3), 8.54 (d, 1H, J = 4.2 Hz, thienyl H-5), 8.74 (d, 1H, J = 4.8 Hz, H-5); Anal. calcd. for C17H9ClN4S: (336.8): C, 60.62; H, 2.69; N, 16.64; S, 9.52. Found: C, 60.32; H, 3.07; N, 16.87; S, 9.64.
2,7-Di(2-thienyl)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (12c): Beige crystals from acetone, yield (88 %, 2.71 g); mp 251-253 oC; IR (cm-1): 2222 (CN); MS m/z (M)+ = 308; 1H-NMR: d = 7.34 (t, 1H, J = 4.8 Hz, thienyl H-4), 7.45 (t, 1H, J = 4.8 Hz, thienyl H-4), 7.91 (d, 1H, J = 4.8 Hz, thienyl H-3), 7.96 (d, 1H, J = 4.8 Hz, thienyl H-3), 8.03 (d, 1H, J = 5.2 Hz, H-6), 8.25 (d, 1H, J = 4.8 Hz, thienyl H-5), 8.60 (d, 1H, J = 4.8 Hz, thienyl H-5), 8.79 (d, 1H, J = 5.2 Hz, H-5); Anal. calcd. for C15H8N4S2: (308.38): C, 58.42; H, 2.61; N, 18.17; S, 20.80. Found: C, 58.12; H, 2.67; N, 18.09; S, 20.63.
2-Phenyl-5-(2-thienyl)pyrazolo[1,5-a]pyrimidine-3,6-dicarbonitrile (15a): Grey crystals from acetone, yield (88 %, 2.87 g); mp 291-293 oC; IR (cm-1): 2226 (CN); MS m/z (M)+ = 327; 1H-NMR: d = 7.46 (t, 1H, J = 4.2 Hz, thienyl H-4), 7.56-7.60 (m, 3H, arom. H), 8.12 (d, 2H, J = 8.2 Hz, arom. H), 8.39 (d, 1H, J = 4.2 Hz, thienyl H-3), 8.67 (d, 1H, J = 4.2 Hz, thienyl H-5), 9.09 (s, 1H, H-7); Anal. calcd. for C17H10N4S: (327.36): C, 66.04; H, 2.77; N, 21.39; S, 9.79. Found: C, 66.25; H, 2.96; N, 21.17; S, 10.03.
2-(4-Chlorophenyl)-5-(2-thienyl)pyrazolo[1,5-a]pyrimidine-3,6-dicarbonitrile (15b): Brown crystals from acetone, yield (85 %, 3.06 g); mp 294-295 oC; IR (cm-1): 2230 (CN); MS m/z (M)+ = 361; 1H- NMR: d = 7.46 (t, 1H, J = 4.2 Hz, thienyl H-4), 7.67 (d, 2H, J = 8.2 Hz, arom. H), 8.08 (d, 2H, J = 8.2 Hz, arom. H), 8.39 (d, 1H, J = 4.2 Hz, thienyl H-3), 8.65 (d, 1H, J = 4.2 Hz, thienyl H-5), 9.08 (s, 1H, H-7); Anal. calcd. for C17H9ClN4S: (361.81): C, 59.75; H, 2.23; N, 19.36; S, 8.86. Found: C, 59.46; H, 2.49; N, 19.19; S, 8.82.
2,5-Di(2-thienyl)pyrazolo[1,5-a]pyrimidine-3,6-dicarbonitrile 15c: Brown crystals from acetone, yield (87 %, 2.89 g); mp 303-304 oC; IR (cm-1): 2227 (CN); MS m/z (M)+ = 333; 1H-NMR: d = 7.27 (t, 1H, J = 4.8 Hz, thienyl H-4), 7.45 (t, 1H, J = 4.8 Hz, thienyl H-4), 7.87 (d, 1H, J = 4.8 Hz, thienyl H-3), 7.92 (d, 1H, J = 4.8 Hz, thienyl H-3), 8.40 (d, 1H, J = 4.8 Hz, thienyl H-5), 8.63 (d, 1H, J = 4.8 Hz, thienyl H-5), 9.06 (s, 1H, H-7); Anal. calcd. for C15H8N4S2: (333.39): C, 57.64; H, 2.12; N, 21.01; S, 19.24. Found: C, 57.42; H, 2.43; N, 20.85; S, 19.57.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}