Results and Discussion
Our initial approach to produce 2-
sec-amino-4
H-3,1-benzothiazin-4-ones was the treatment of methyl 2-thioureidobenzoates
1 with concentrated sulphuric acid. This procedure was introduced to prepare 2-aminothieno[2,3-
d][1,3]thiazin-4-ones [
20] and successfully applied to other heterocyclic systems [
18,
19,
21,
22,
23]. Recently, Tarzia
et al. have prepared the benzothiazine analogue of URB754 that way [
24]. Ring closure to 4
H-3,1-benzothiazine-4-ones was also achieved by treatment of 2-benzoylaminothiobenzamide with concentrated sulphuric acid [
25].
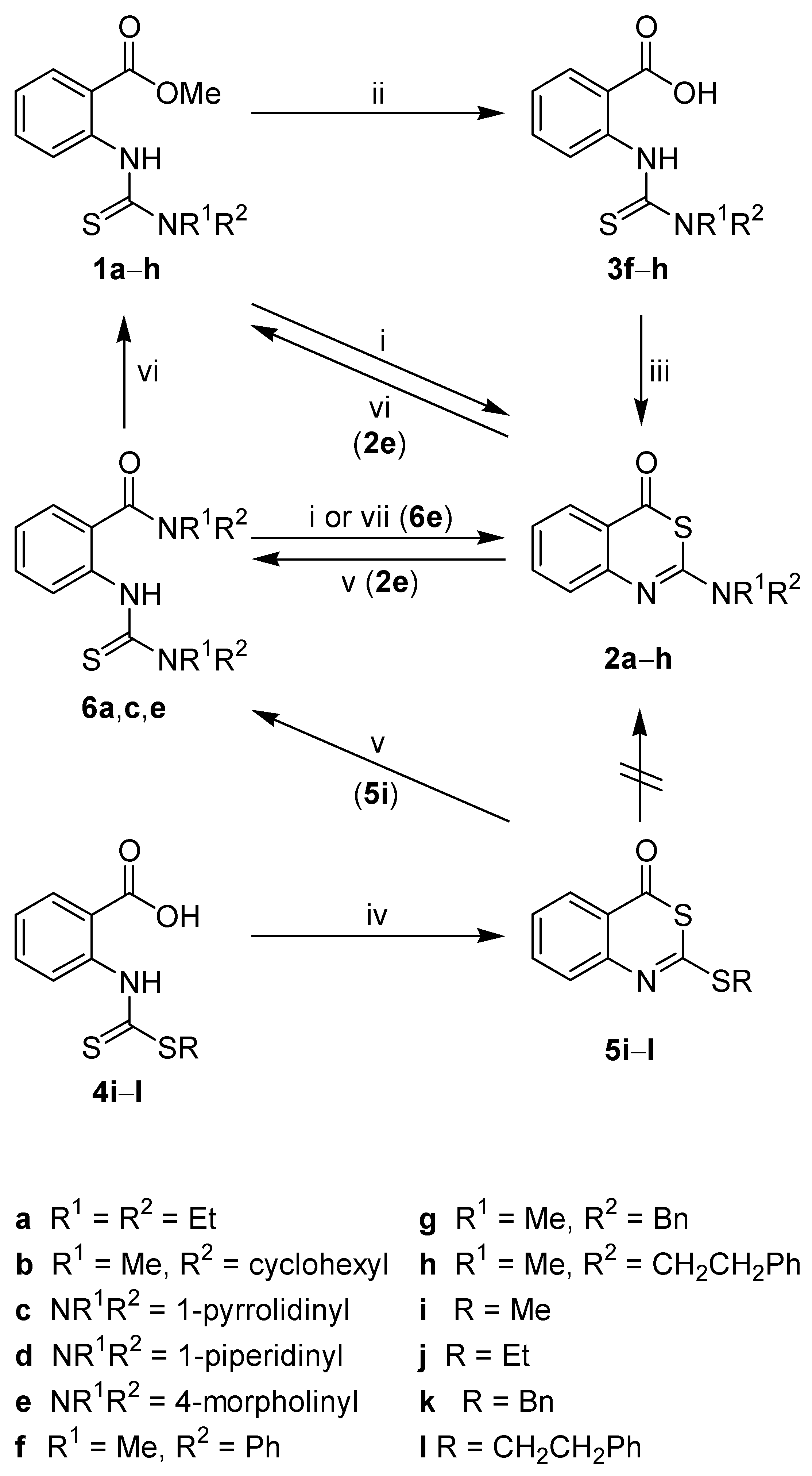
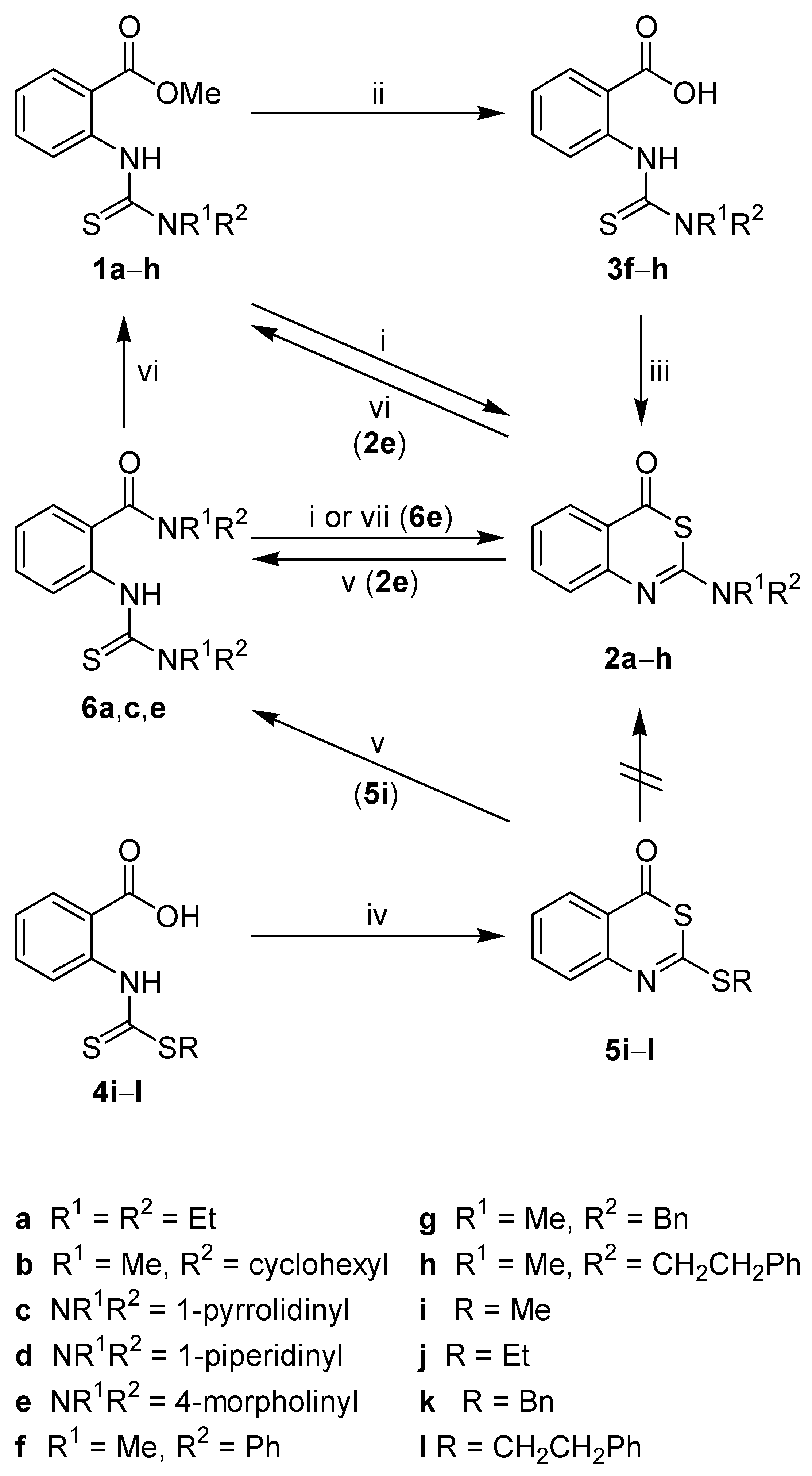
The new thioureas
1a–
h were obtained from methyl 2-isothiocyanatobenzoate and secondary amines (
Scheme 1). The treatment of
1a–
e with concentrated sulphuric acid at room temperature conveniently afforded the desired benzothiazinones
2a–
e. The benzyl(methyl)thiourea derivative
1g was not converted to
2g due to
N-debenzylation under the strong acidic conditions used. The methyl(phenyl)thiourea
1f gave the corresponding benzothiazinone
2f in only 20% yield, and the methyl(2-phenylethyl)thiourea
1h could not be transformed to
2h. Therefore, an extended synthetic route was chosen.
1f–
h were first hydrolyzed to the corresponding benzoic acid derivatives
3f–
h, and subsequently cyclised with acetic anhydride [
26,
27] to yield
2f–
h, thus allowing the facile introduction of aromatic structures within the 2-substituent of
2. Attempts to directly generate thioureidobenzoic acids
3 from anthranilic acid, 1,1’-thiocarbonyldiimidazole and secondary amines failed (data not shown).
Scheme 1.
Synthesis and interconversion of 2-amino- and 2-alkylthio-4H-3,1-benzothiazin-4-ones.
Scheme 1.
Synthesis and interconversion of 2-amino- and 2-alkylthio-4H-3,1-benzothiazin-4-ones.
Reagents and conditions: i) concd. H2SO4, r.t.; ii) NaOH, EtOH, H2O, reflux; iii) Ac2O, r.t.; iv) Ac2O, reflux; v) HNR1R2, acetone, r.t., then reflux; vi) HCl, MeOH, reflux, 3 h; vii) HCl, MeOH, reflux, 2 min.
A synthetic access to 2-alkylthio-4
H-3,1-benzothiazin-4-ones was envisaged
via dithiocarbamates
4i–
l, which were prepared from anthranilic acid, carbon disulfide and alkyl halides. These intermediates underwent an easy cyclocondensation upon treatment with acetic anhydride to furnish the new 2-alkylthio derivatives
5i–
l. Only one representative of this heterocyclic class,
i.e. 6,7-difluoro-2-(methylthio)-4
H-3,1-benzothiazin-4-one, has already been described by Mazuoka
et al. [
28].
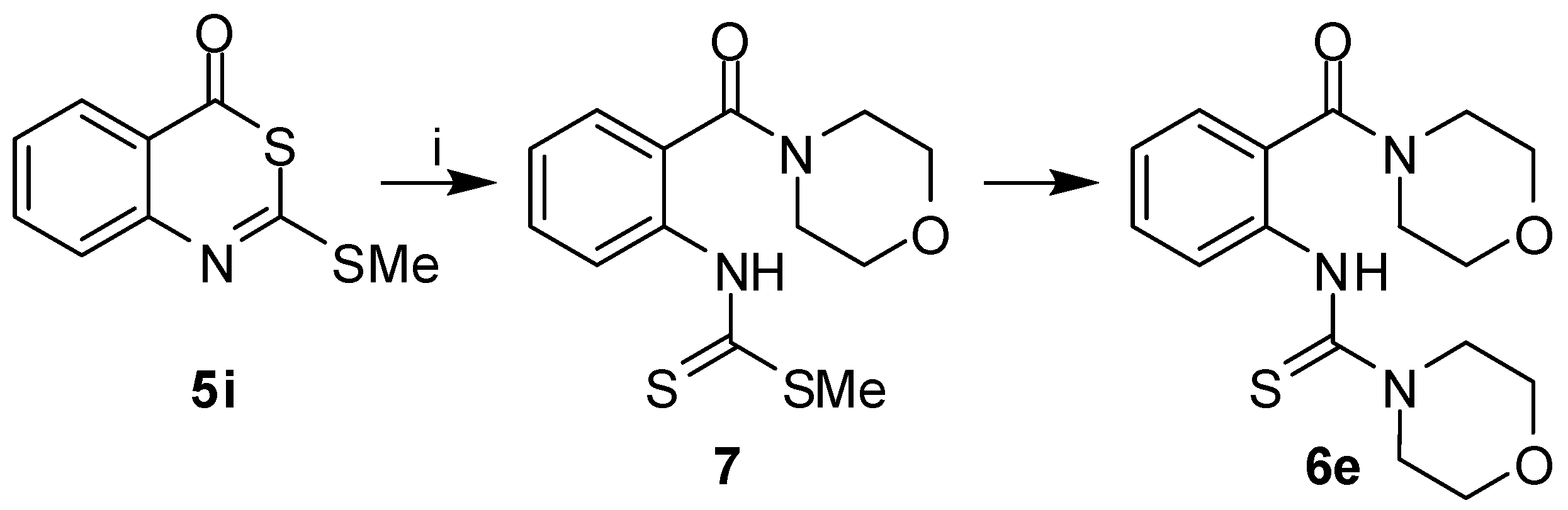
To explore an alternative entry to 2-
sec-amino-4
H-3,1-benzothiazin-4-ones, the
S-methyl derivative
5i was reacted with secondary amines. However, 2-aminobenzothiazinones
2 were not formed and instead, we obtained 2-thioureidobenzamides
6a,
c,
e. The attack of an amine on
5i might either occur at the C-2 or C-4 carbons. An attack at C-2 followed by C-2–S-3 bond breakage would not lead to
6. The nucleophilic substitution with the release of the methanethiol would generate 2-amino-benzothiazinones
2. Such intermediates could subsequently undergo ring cleavage due to the attack of the amine at C-4 to produce
6. When treating the 2-morpholinobenzothiazinone
2e with morpholine under the conditions used for the conversion of
5i to
6, compound
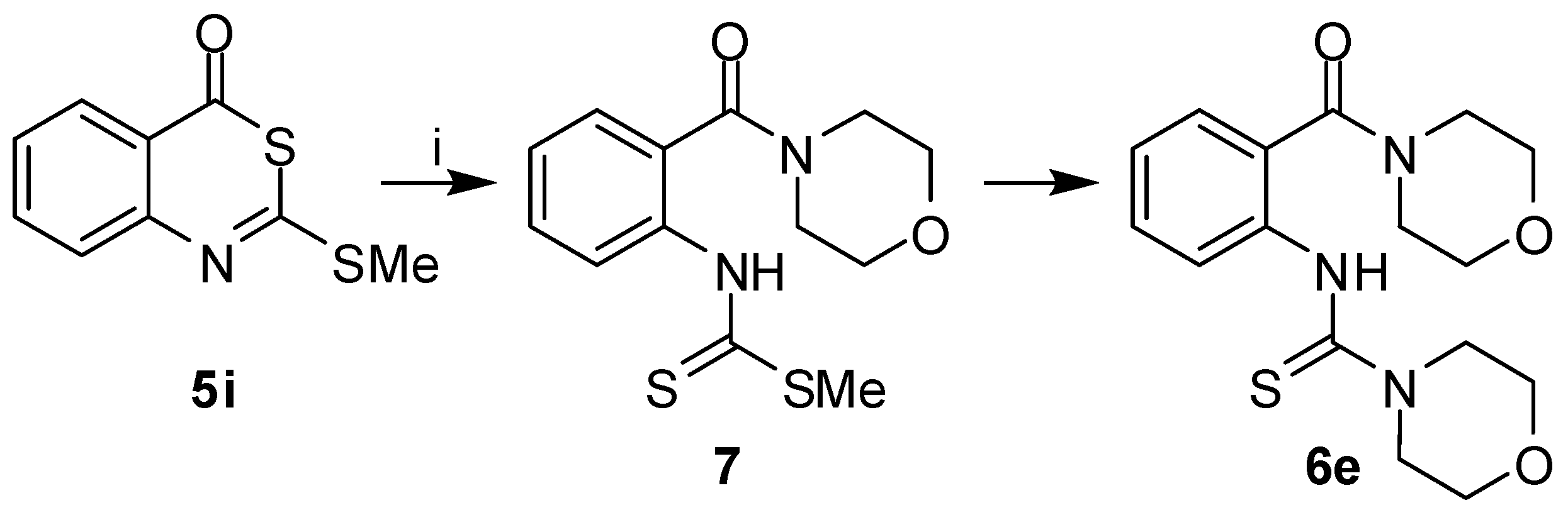
6e was indeed obtained. However, a different mechanism was proposed based on the isolation of the intermediate
7 in the reaction of
5i with morpholine (
Scheme 2). Hence, the secondary amine attacks the 2-alkylthiobenzothiazinones
5 at C-4, followed by ring opening and subsequent transformation of the dithiocarbamate substituent into a thiourea. Leistner and Wagner reported on a similar formation of 2-thioureido
thiobenzamides when reacting 2-(methylthio)-4
H-3,1-benzothiazin-4-
thione with secondary amines [
29].
With the novel 2-thioureidobenzamides
6 in hand, we also investigated their utility as precursors to
2. Indeed, the corresponding 2-aminobenzothiazinones
2a,
c,
e were obtained in quantitative yield and high purity by reacting the benzamide derivatives
6 with concentrated sulphuric acid (
Scheme 1).
Heating the 2-thioureidobenzamides
6a,
c,
e in methanolic hydrochloric acid yielded methyl thioureidobenzoates
1a,
c,
e. This transformation is formally an acid-catalyzed amide alcoholysis under conditions where a simple benzamide such as 4-benzoylmorpholine did not react [
30]. A ring closure–reopening mechanism operative in the conversion of
6 to
1 is initiated by the rapid cyclocondensation to intermediate 2-aminobenzothiazinones
2. This could be concluded as the product
2e was identified after short-time treatment of
6e with methanolic hydrochloric acid. Prolonged heating of
2e then led to the formation of the methyl thioureidobenzoate
1e.
Scheme 2.
Reaction pathway from 5i to 6e.
Scheme 2.
Reaction pathway from 5i to 6e.
Reagents and conditions: i) morpholine, acetone, r.t., 1 h.
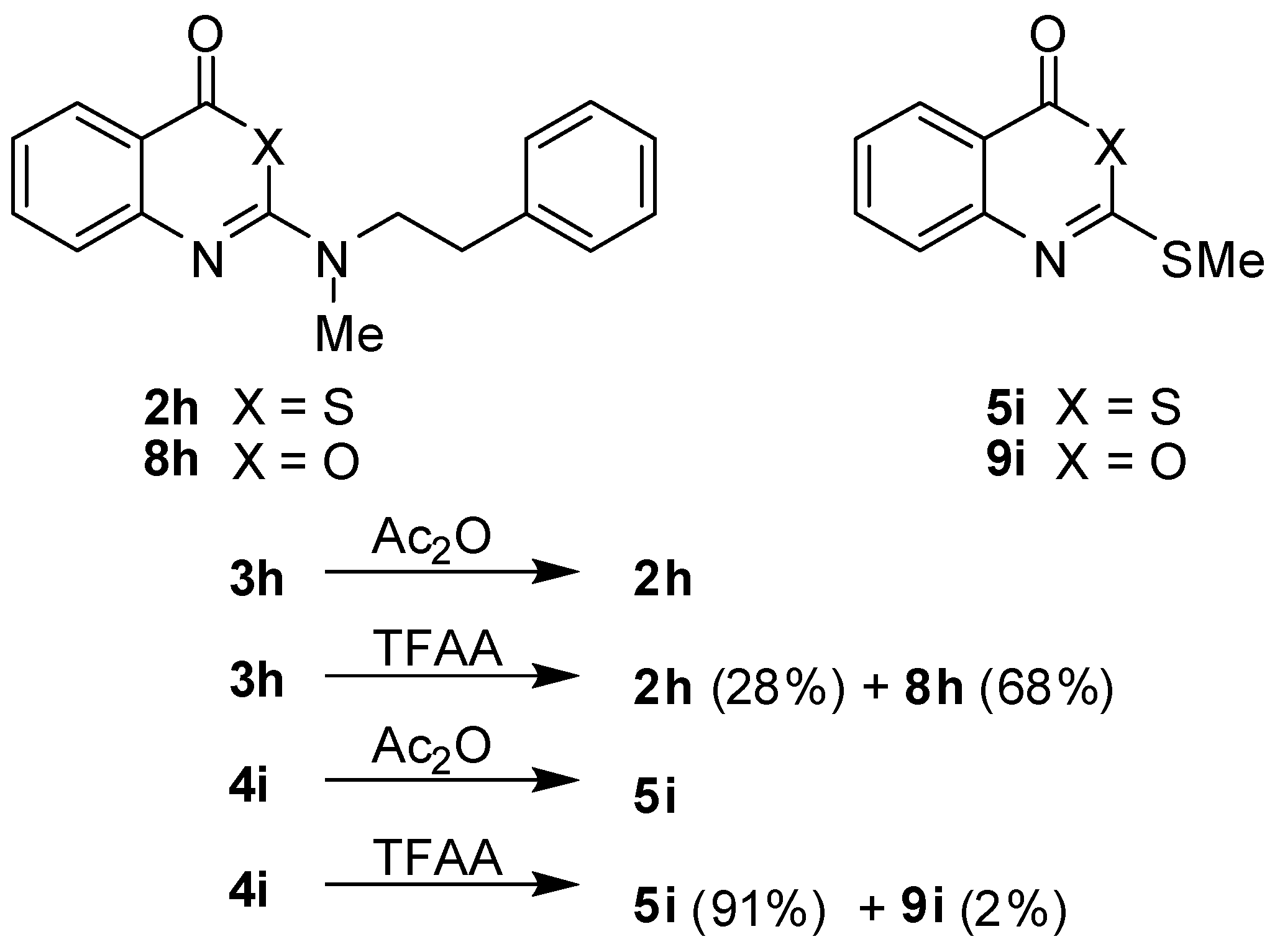
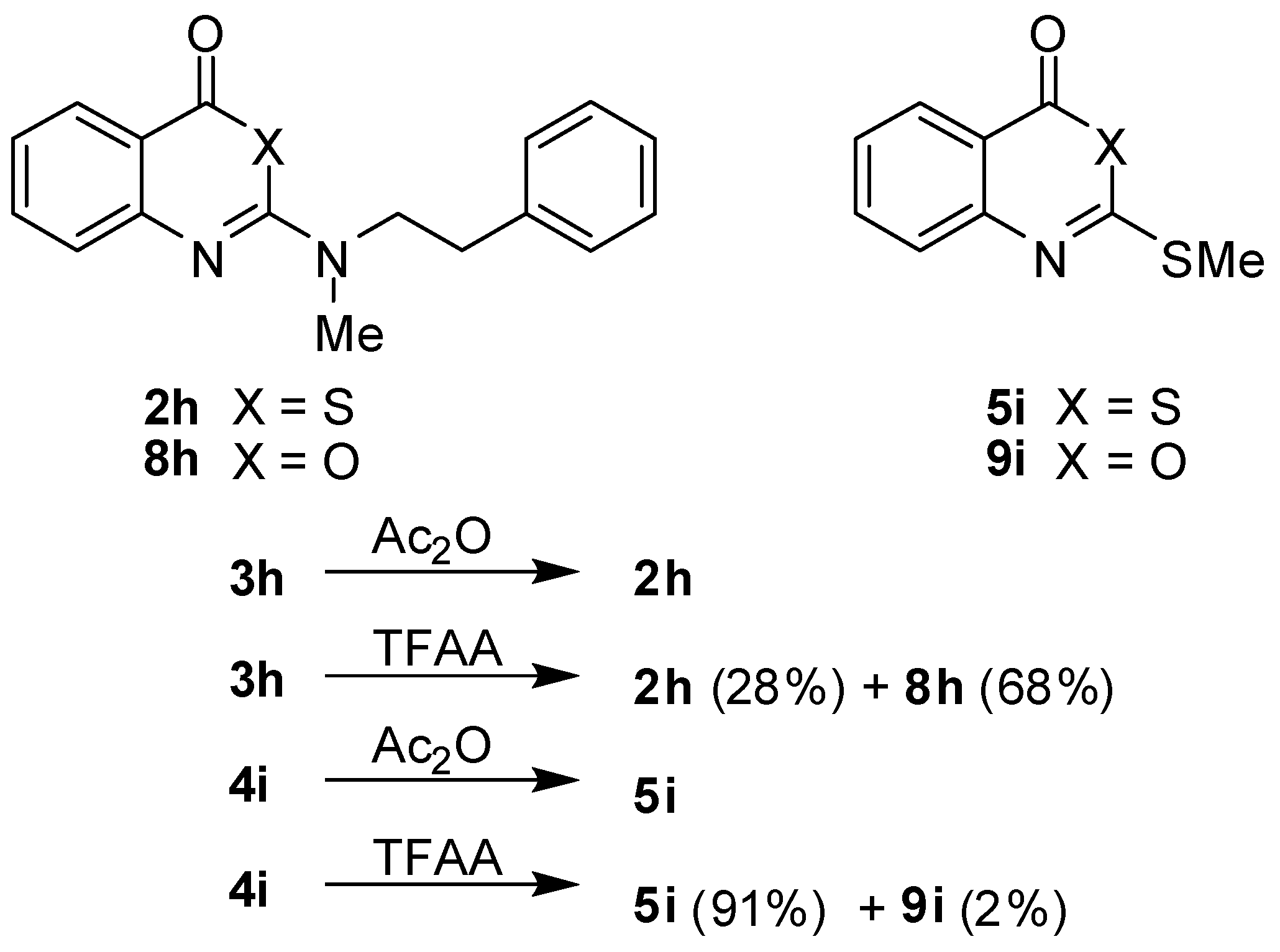
In the course of this study, acetic anhydride was successfully used in cyclocondensations to convert the benzoic acid derivatives
3 and
4 to benzothiazinones
2 and
5, respectively. Unexpectedly, the replacement of acetic anhydride by trifluoroacetic anhydride (TFAA) produced different results (
Scheme 3). The treatment of
3h with this reagent gave a mixture of the benzothiazinone
2h and the benzoxazinone
8h with the latter compound being the dominant product. On the other hand, the benzothiazinone
5i was the main product of the reaction of
4i with TFAA while the corresponding benzoxazinone
9i was only formed in traces. The formation of
8h is envisaged to occur by a nucleophilic attack of the carboxyl oxygen at the activated thiocarbonyl carbon [
31,
32,
33,
34,
35,
36]. Further investigations are needed to clarify the mechanism of this desulphurisation-cyclisation.
In the
13C-NMR spectra of the benzothiazinone representatives
2h and
5i the characteristic signals for C-2/C-4 appeared at 156/184 ppm (
2h) and 164/182 ppm (
5i). The other benzothiazinones had similar NMR data. The corresponding chemical shifts of the benzoxazinones were observed at 154/160 ppm (
8h) and 164/159 ppm (
9i). These values were in accordance with literature data for 4
H-3,1-benzoxazin-4-ones [
14,
32,
37,
38,
39]. A similar influence of the sulphur-oxygen exchange on the chemical shift of the C-4 carbon was observed for pairs of 2-thien-2-yl and 2-cyano substituted 4
H-3,1-benzothiazin(oxazin)-4-ones [
32,
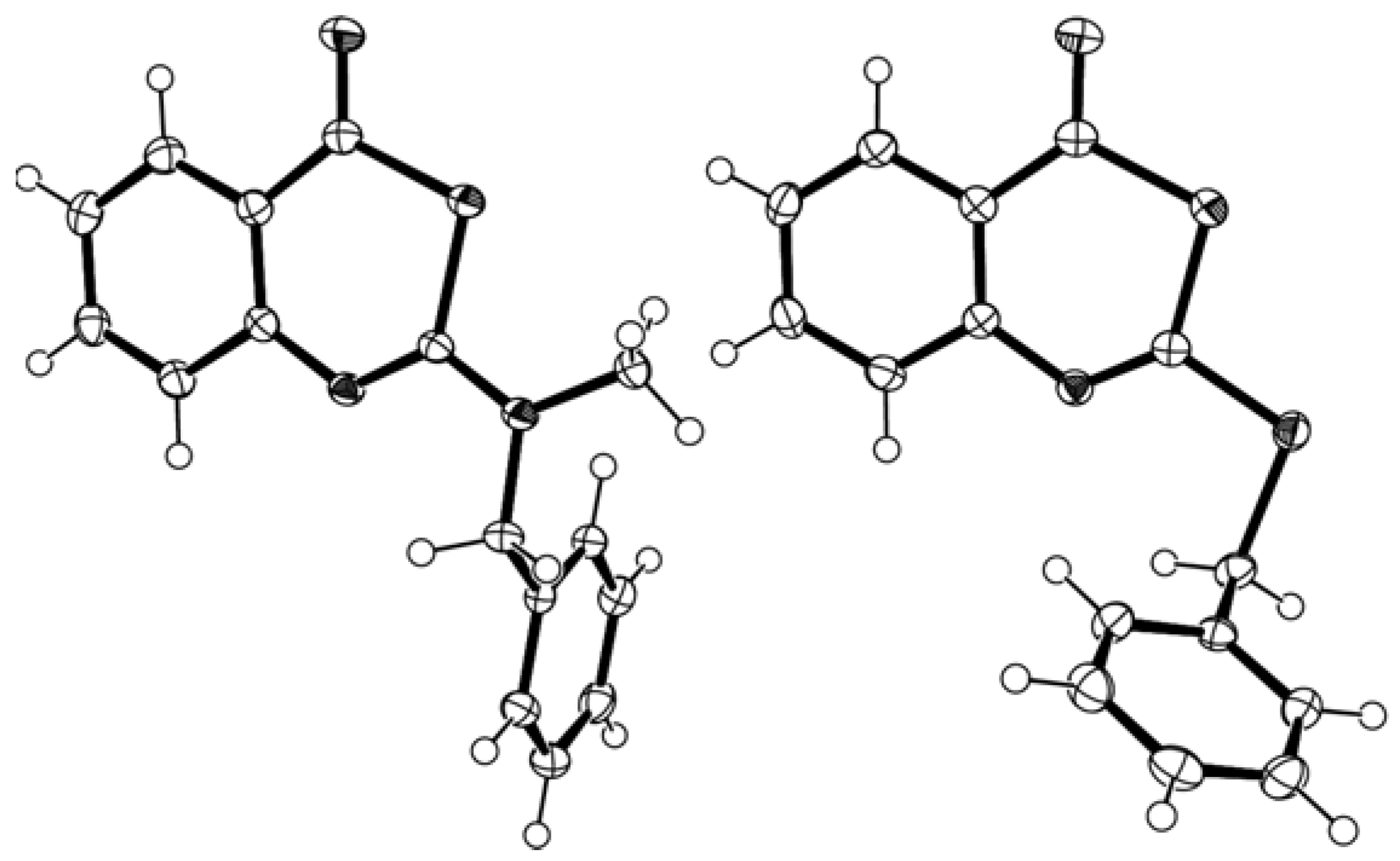
40]. The structure of the title compounds was furthermore confirmed by X-ray crystal structure analyses [
41] (
Figure 1).
The bond lengths within the thiazinone ring of the 2-aminobenzothiazinone
2g and the 2-alkylthio-benzothiazinone
5k were similar (see
Electronic Supplementary Information). The thiazinone rings adopt an almost planar conformation with the largest deviation from the least square planes defined by the six atoms of the heterocyclic ring being 0.022(1) Å (
2g) and 0.024(2) Å (
5k).
Scheme 3.
Cyclisation reactions of benzoic acid derivatives 3h and 4i with acetic anhydride and trifluoroacetic anhydride.
Scheme 3.
Cyclisation reactions of benzoic acid derivatives 3h and 4i with acetic anhydride and trifluoroacetic anhydride.
Figure 1.
X-ray crystal structure of 2-(N-benzyl-N-methylamino)-4H-3,1-benzothiazin-4-one 2g (left) and of 2-(benzylthio)-4H-3,1-benzothiazin-4-one 5k (right).
Figure 1.
X-ray crystal structure of 2-(N-benzyl-N-methylamino)-4H-3,1-benzothiazin-4-one 2g (left) and of 2-(benzylthio)-4H-3,1-benzothiazin-4-one 5k (right).
2-Aminobenzothiazinones
2a–
h and 2-alkylthiobenzothiazinones
5i–
l were evaluated as potential inhibitors of HLE [
42] (
Table 1). Other representative members of serine proteases (human cathepsin G, bovine chymotrypsin and bovine trypsin) were also investigated. The compounds were furthermore assessed towards the cysteine protease human cathepsin L and the metalloprotease angiotensin-converting enzyme (ACE). Two serine esterases, acetylcholinesterase (AChE) and cholesterol esterase (CEase), which share the acyl transfer mechanism with serine proteases were also included in the inhibition studies.
None of the investigated 2-aminobenzothiazinones inhibited HLE. As 2-aminosubstituted 4
H-3,1-benzoxazin-4-ones are potent inhibitors of HLE, a replacement of the ring oxygen by sulphur resulted in a loss of activity, which can be attributed to the increased intrinsic stability of the benzothiazinones. The second order rate constant for the alkaline hydrolysis of
2e (1.7 M
-1s
-1) was significantly lower than that of the analogous 2-(morpholin-4-yl)-4
H-3,1-benzoxazin-4-one (28 M
-1s
-1) [
43]. 2-(
N-Cyclohexyl-
N-methylamino)-4
H-3,1-benzothiazin-4-one (
2b) exhibited a remarkable inhibitory capacity against human cathepsin L [
44]. This compound was selective for cathepsin L with respect to the other enzymes investigated in this study. It might therefore serve as a lead structure for cysteine protease inhibitors. Further investigations are needed to inspect selectivity among cysteine proteases.
Two of the 2-alkylthiobenzothiazinones were identified as HLE inhibitors. The 2-methylthio and 2-ethylthio derivatives,
5i and
5j, exhibited IC
50 values in the low micromolar range. These compounds carry 2-substituents with the least steric demand among all the benzothiazinones tested. HLE has a primary substrate specificity for small aliphatic amino acid residues at P
1 position. It can therefore be assumed, that the alkylthio moiety is accommodated by the S
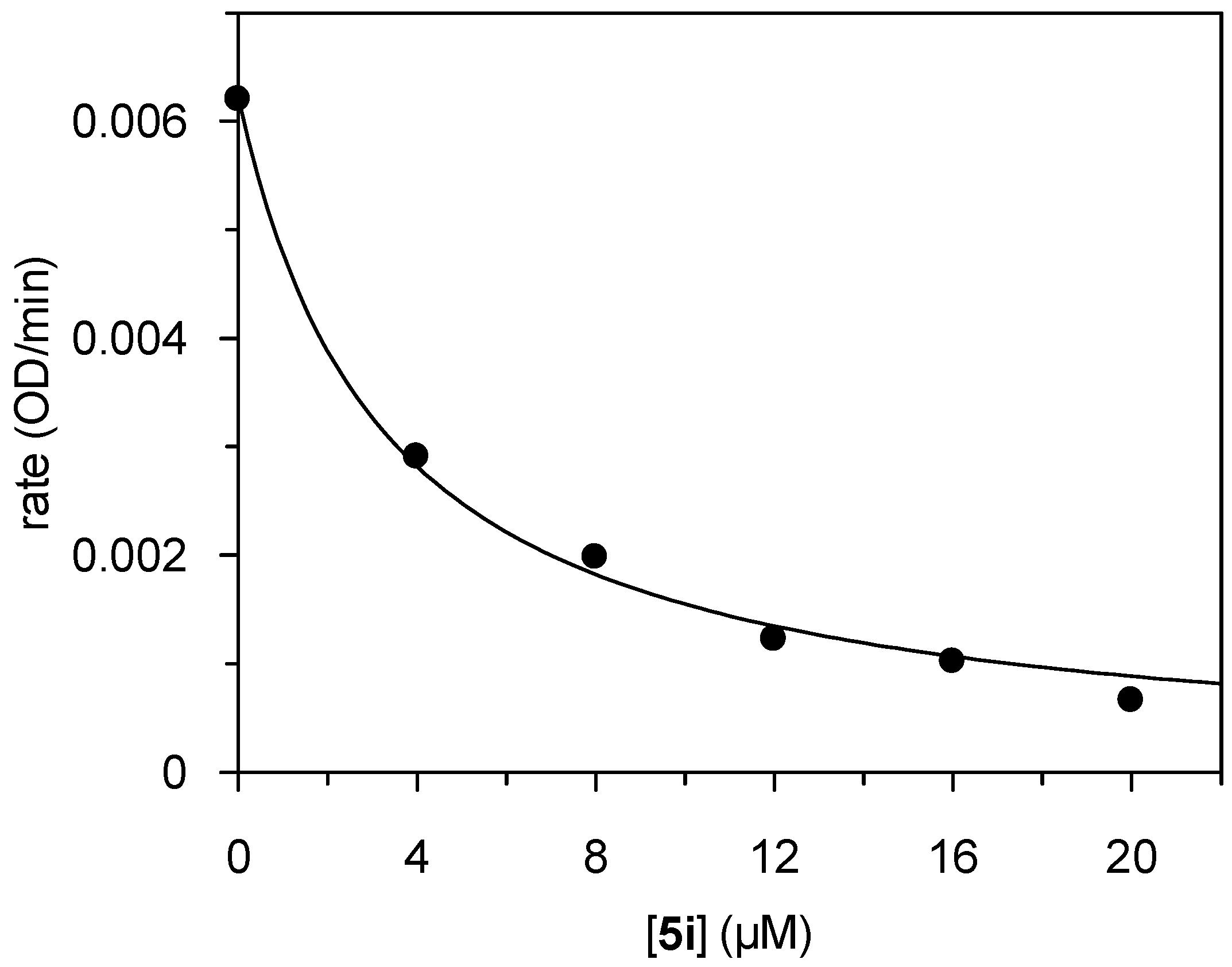
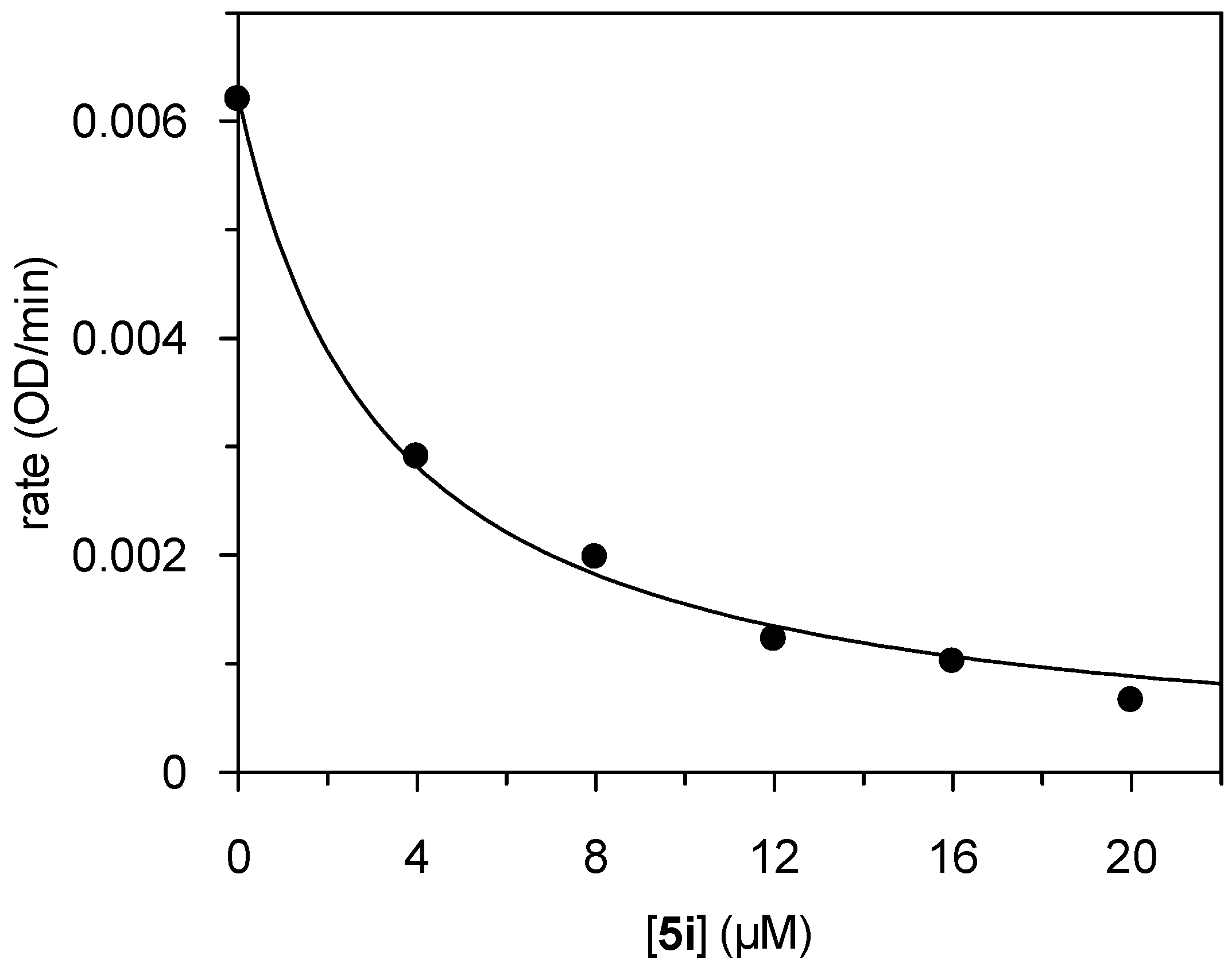
1 subsite of HLE. The concentration-dependent inhibition by
5i is presented in
Figure 2. The progress curves of the HLE-catalyzed substrate consumption were linear over the 10-min time course. Thus, the time-independent inhibition indicated a non-covalent interaction of
5i with HLE. Provided that
5i behaved kinetically as a competitive inhibitor, a
Ki value of 1.2 µM corresponds to the IC
50 value of 3.3 µM [
45]. Noteworthy, the 2-methylthiobenzothiazinone
5i did not inhibit any of the other enzymes studied here.
Table 1.
Enzyme inhibitory activities of 2-amino and 2-alkylthio-4H-3,1-benzothiazin-4-ones.
Table 1.
Enzyme inhibitory activities of 2-amino and 2-alkylthio-4H-3,1-benzothiazin-4-ones.
| | | | | IC50 values (µM)a | | | | |
|---|
| Compound | HLE | Cathepsin G | Chymotrypsin | Trypsin | Cathepsin L | ACE | AChE | CEase |
| 2a | > 100 | >100 | > 25 | > 100 | > 50 | > 100 | >25 | > 25 |
| 2b | > 100 | > 50 | > 100 | > 100 | 8.93 ± 1.58b | > 100 | > 50 | > 50 |
| 2c | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 50 | > 100 |
| 2d | > 100 | > 100 | > 25 | > 100 | > 50 | > 100 | > 25 | > 50 |
| 2e | > 100 | > 100 | > 50 | > 100 | > 25 | > 100 | > 25 | > 25 |
| 2f | > 25 | > 100 | 10.4 ± 0.5c | > 100 | > 50 | > 100 | > 100 | > 50 |
| 2g | > 25 | > 100 | 22d | > 100 | 22e | > 100 | > 50 | 25f |
| 2h | > 25 | > 100 | > 50 | > 100 | > 50 | > 100 | > 100 | > 50 |
| 5i | 3.31 ± 0.24g | > 100 | > 100 | > 100 | > 100 | > 100 | > 50 | > 25 |
| 5j | 8.11 ± 0.96b | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 25 |
| 5k | > 25 | > 100 | 18d | > 100 | > 100 | > 100 | > 50 | > 50 |
| 5l | > 50 | > 100 | > 25 | > 50 | > 100 | > 100 | 19f | > 50 |
Figure 2.
Plot of the steady-state rates versus inhibitor concentration for the inhibition of HLE by compound 5i.
Figure 2.
Plot of the steady-state rates versus inhibitor concentration for the inhibition of HLE by compound 5i.
Experimental
General
Solvents and reagents were obtained from Acros (Geel, Belgium), Fluka (Taufkirchen, Germany) or Sigma (Steinheim, Germany), if commercially available. Human leukocyte elastase (HLE), human cathepsin G, human cathepsin L and human angiotensin-converting enzyme (ACE) were obtained from Calbiochem, Darmstadt, Germany. MeOSuc-Ala-Ala-Pro-Val-pNA, Suc-Ala-Ala-Pro-Phe-pNA, Suc-Ala-Ala-Pro-Arg-pNA, Z-Phe-Arg-pNA, and 2-furanacryloyl-phenylalanylglycylglycine (FA-Phe-Gly-Gly) were purchased from Bachem (Bubendorf, Switzerland). Bovine chymotrypsin was purchased from Fluka (Deisenhofen, Germany). Trypsin from bovine pancreas, acetylcholinesterase (AChE) from
Electrophorus electricus, cholesterol esterase (CEase) from bovine pancreas, 5,5’
-dithio-bis-(2-nitrobenzoic acid) (DTNB), sodium taurocholate (TC), and
para-nitrophenylbutyrate (pNPB) were purchased from Sigma (Steinheim, Germany). Methyl 2-isothiocyanatobenzoate was prepared under the conditions reported by Carpenter
et al. [
46]. Thin-layer chromatography was carried out on Merck aluminum sheets, silica gel 60 F
254. Preparative column chromatography was performed on Merck silica gel 60, 70–230 mesh. Melting points were determined on a Boëtius melting point apparatus (PHMK, VEB Wägetechnik Rapido, Radebeul, Germany) and are uncorrected.
1H- and
13C-NMR spectra were acquired on a Bruker Avance DRX 500 spectrometer operating at 500 MHz for
1H and 125 MHz for
13C. Chemical shifts δ are given in ppm referring to the signal center using the solvent peaks for reference: CDCl
3 7.26 ppm/77.0 ppm and DMSO-
d6 2.49 ppm/39.7 ppm. The NMR signals were assigned by two-dimensional
1H,
1H COSY and
1H,
13C correlation spectra (HSQC, HMBC) using standard pulse sequences. Elemental analyses were carried out with a Vario EL apparatus. The spectrophotometric assays were done on Varian Cary 50 Bio and Varian Cary 100 Bio UV/VIS spectrometers with a cell holder equipped with a constant temperature water bath.
Methyl 2-(3,3-diethylthioureido)benzoate (1a)
Method 1: Diethylamine (0.476 g, 6.5 mmol) was added dropwise to a stirring solution of methyl 2-isothiocyanatobenzoate (0.966 g, 5.0 mmol) in CH2Cl2 (20 mL). The reaction mixture was stirred at r.t. for 3 h. The organic layer was washed with HCl (0.5 M, 2 × 5 mL), dried over Na2SO4, filtered, and evaporated to dryness. Recrystallisation from EtOH yielded 1a (0.946 g, 71%) as colourless needles, mp 85–87 °C (EtOH); 1H-NMR (CDCl3) δ 1.34 (t, J = 6.9 Hz, 6H, CH2CH3), 3.82 (q, J = 6.9 Hz, 4H, CH2CH3), 3.88 (s, 3H, CO2CH3), 7.03 (ddd, J = 8.2, 7.3, 1.3 Hz, 1H, 5-H), 7.48 (ddd, J = 8.6, 7.3, 1.6 Hz, 1H, 4-H), 7.93 (dd, J = 8.2, 1.6 Hz, 1H, 6-H), 8.73 (dd, J = 8.6, 1.3 Hz, 1H, 3-H), 10.66 (s, 1H, NH); 13C-NMR (CDCl3) δ 12.46 (CH2CH3), 45.68 (CH2CH3), 52.31 (CO2CH3), 116.86 (C-1), 122.22 (C-5), 123.62 (C-3), 130.22 (C-6), 132.82 (C-4), 143.28 (C-2), 168.99 (CO2CH3), 179.23 (NHCS); Anal. calcd. for C13H18N2O2S: C, 58.6; H, 6.8; N, 10.5. Found: C, 58.4; H, 6.8; N, 10.4.
Method 2: 2-(3,3-Diethylthioureido)-N,N-diethylbenzamide (6a, 0.307 g, 1.0 mmol) was heated to reflux in anhydrous methanolic HCl (0.25 M, 5 mL) for 3 h. The mixture was allowed to cool to r.t. and kept at -15 °C. The precipitate was removed by suction filtration to give 1a (0.169 g, 63%) as white needles.
Methyl 2-(3-cyclohexyl-3-methylthioureido)benzoate (1b)
According to the preparation of 1a (Method 1), 1b (1.50 g, 98%) was obtained from methyl 2-isothiocyanatobenzoate and N-methylcyclohexylamine as a semisolid crude material. 1H-NMR (CDCl3) δ 1.05–1.95 (m, 10H, 2’/3’/4’/5’/6’-H), 3.20 (s, 3H, NCH3), 3.88 (s, 3H, CO2CH3), 7.03 (ddd, J = 8.2, 7.1, 1.3 Hz, 1H, 5-H), 7.48 (ddd, J = 8.8, 7.1, 1.6 Hz, 1H, 4-H), 7.93 (dd, J = 8.2, 1.6 Hz, 1H, 6-H), 8.71(d, J = 8.2 Hz, 1H, 3-H), 10.70 (s, 1H, NH); 13C-NMR (CDCl3) δ 25.48 (C-4’), 25.54 (C-3’/5’), 30.01 (C-2’/6’), 32.57 (NCH3), 52.29 (CO2CH3), 59.23 (C-1’), 116.74 (C-1), 122.16 (C-5), 123.35 (C-3), 130.23 (C-6), 132.86 (C-4), 143.27 (C-2), 168.99 (CO2CH3), 179.90 (NHCS); Anal. calcd. for C16H22N2O2S: C, 62.7; H, 7.2; N, 9.1. Found: C, 62.7; H, 6.7; N, 9.0.
Methyl 2-[(1-pyrrolidinylthiocarbonyl)amino]benzoate (1c)
Method 1: According to the preparation of 1a (Method 1), 1c (1.32 g, 82%) was obtained from methyl 2-isothiocyanatobenzoate and pyrrolidine as colourless needles, mp 124–127 °C (EtOH); 1H-NMR (CDCl3) δ 1.87–2.16 (m, 4H, 3’/4’-H), 3.65–3.96 (m, 7H, CO2CH3, 2’/5’-H), 7.04 (ddd, J = 7.9, 7.3, 1.3 Hz, 1H, 5-H), 7.51 (ddd J = 8.5, 7.3, 1.6 Hz, 1H, 4-H), 7.95 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 9.00 (dd, J = 8.5, 1.0 Hz, 1H, 3-H), 10.82 (s, 1H, NH); 13C-NMR (CDCl3) δ 24.59, 26.20 (C-3’/4’), 48.30, 52.13 (C-2’/5’), 52.33 (CO2CH3), 116.40 (C-1), 122.26, 122.65 (C-3/5), 130.35 (C-6), 133.16 (C-4), 142.88 (C-2), 169.05 (CO2CH3), 176.47 (NHCS); Anal. calcd. for C13H16N2O2S: C, 59.1; H, 6.1; N, 10.6. Found: C, 59.1; H, 6.35; N, 10.5.
Method 2: According to the preparation of 1a (Method 2), 1c (0.222 g, 84%) was obtained from 6c as colourless needles.
Methyl 2-[(1-piperidinylthiocarbonyl)amino]benzoate (1d)
According to the preparation of 1a (Method 1), 1d (1.11 g, 80%) was obtained from methyl 2-isothiocyanatobenzoate and piperidine as colourless plates, mp 116–117 °C (EtOH); 1H-NMR (CDCl3) δ 1.68–1.73 (m, 6H, 3’/4’/5’-H), 3.88 (s, 3H, CO2CH3), 3.95–4.01 (m, 4H, 2’/6’-H), 7.02 (ddd, J = 8.2, 7.3, 1.3 Hz, 1H, 5-H), 7.48 (ddd, J = 8.8, 7.3, 1.6 Hz, 1H, 4-H), 7.93 (dd, J = 8.1, 1.8 Hz, 1H, 6-H), 8.53 (dd, J = 8.5, 1.3 Hz, 1H, 3-H), 10.75 (s, 1H, NH); 13C-NMR (CDCl3) δ 24.40 (C-4’), 25.67 (C-3’/5’), 49.68 (C-2’/6’), 52.33 (CO2CH3), 116.41 (C-1), 122.04 (C-5), 123.03 (C-3), 130.33 (C-6), 132.97 (C-4), 143.40 (C-2), 169.06 (CO2CH3), 179.60(NHCS); Anal. calcd. for C14H18N2O2S: C, 60.4; H, 6.5; N, 10.1. Found: C, 60.6; H, 6.55; N, 10.1.
Methyl 2-[(4-morpholinylthiocarbonyl)amino]benzoate (1e)
Method 1: According to the preparation of
1a,
1e (1.11 g, 80%) was obtained from methyl 2-isothiocyanatobenzoate and morpholine as a white solid, mp 103–107 °C (EtOH), lit. [
47] 106–110 °C);
1H-NMR (CDCl
3) δ 3.79 (t,
J = 4.9 Hz, 4H, 2’/6’-H), 3.89 (s, 3H, CO
2CH
3), 4.04 (t,
J = 4.9 Hz, 4H, 3’/5’-H), 7.06 (ddd,
J = 8.2, 6.9, 1.3 Hz, 1H, 5-H), 7.51 (ddd,
J = 8.6, 7.3, 1.6 Hz, 1H, 4-H), 7.96 (dd,
J = 8.1, 1.6 Hz, 1H, 6-H), 8.67 (dd,
J = 8.5, 1.0 Hz, 1H, 3-H), 10.97 (s, 1H, NH);
13C-NMR (CDCl
3) δ 48.25 (C-3’/5’), 52.48 (CO
2CH
3), 66.29 (C-2’/6’), 116.57 (C-1), 122.58, 122.93 (C-3/5), 130.43 (C-6), 133.19 (C-4), 142.95 (C-2), 169.19 (
CO
2CH
3), 180.72 (NHCS); Anal. calcd. for C
13H
16N
2O
3S: C, 55.7; H, 5.75; N, 10.0. Found: C, 56.0; H, 5.9; N, 9.8.
Method 2: According to the preparation of 1a (Method 2), 1e (0.229 g, 82%) was obtained from 6e as a light yellow solid.
Method 3: 2-(Morpholin-4-yl)-4H-3,1-benzothiazin-4-one (2e, 0.160 g, 0.64 mmol) was heated to reflux in anhydrous methanolic HCl (0.25 M, 3 mL) for 3 h. The mixture was allowed to cool to r.t. and kept at -15 °C. The precipitate was removed by suction filtration to give 1e (0.151 g, 84%) as a light yellow solid.
Methyl 2-(3-methyl-3-phenylthioureido)benzoate (1f)
According to the preparation of 1a (Method 1), 1f (1.28 g, 84%) was obtained from methyl 2-isothiocyanatobenzoate and N-methylaniline as colourless needles, mp 70–71 °C (EtOH); 1H-NMR (DMSO-d6) δ 3.61 (s, 3H, NCH3), 3.70 (s, 3H, CO2CH3), 7.15–7.17 (m, 1H, 5-H), 7.39–7.45 (m, 3H, 2’/4’/6’-H), 7.50–7.55 (m, 3H, 4/3’/5’-H), 7.77 (dd, J = 8.2, 1.6 Hz, 1H, 6-H), 8.24 (dd, J = 8.4, 1.0 Hz, 1H, 3-H), 9.67 (s, 1H, NH); 13C-NMR (DMSO-d6) δ 43.28 (NCH3), 52.44 (CO2CH3), 120.31 (C-1), 123.65 (C-5), 125.06 (C-3), 127.01 (C-2’/6’), 128.27 (C-4’), 130.01 (C-6), 130.26 (C-3’/5’), 132.46 (C-4), 141.32 (C-2), 143.71 (C-1’), 167.08 (CO2CH3), 180.48 (NHCS); Anal. calcd. for C16H16N2O2S: C, 64.0; H, 5.4; N, 9.3. Found: C, 63.7; H, 5.4; N, 9.3.
Methyl 2-(3-benzyl-3-methylthioureido)benzoate (1g)
According to the preparation of 1a (Method 1), 1g (1.45 g, 92%) was obtained from methyl 2-isothiocyanatobenzoate and N-benzylmethylamine as white plates, mp 88–92 °C (EtOH); 1H-NMR (CDCl3) δ 3.30 (s, 3H, NCH3), 3.87 (s, 3H, CO2CH3), 5.25 (s, 2H, CH2Ph), 7.08 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H, 5-H), 7.26–7.28 (m, 1H, 4’-H), 7.29–7.34 (m, 4H, 2’/3’/5’/6’-H), 7.53 (ddd, J = 8.5, 7.3, 1.6 Hz, 1H, 4-H), 7.96 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.87 (d, J = 8.6 Hz, 1H, 3-H), 10.93 (s, 1H, NH); 13C-NMR (CDCl3) δ 37.63 (NCH3), 52.36 (CO2CH3), 56.78 (CH2Ph), 117.06 (C-1), 122.64 (C-5), 123.42 (C-3), 127.52 (C-2’/6’), 127.58 (C-4’), 128.72 (C-3’/5’), 130.31 (C-6), 133.01 (C-4), 136.43 (C-1’), 142.98 (C-2), 168.95 (CO2CH3), 180.94 (NHCS); Anal. calcd. for C17H18N2O2S: C, 64.9; H, 5.8; N, 8.9. Found: C, 64.9; H, 6.05; N, 8.9.
Methyl 2-[3-methyl-3-(2-phenylethyl)thioureido]benzoate (1h)
According to the preparation of 1a (Method 1), 1h (1.47 g, 90%) was obtained from methyl 2-isothiocyanatobenzoate and N-methyl phenethylamine as a semisolid crude material; 1H-NMR (CDCl3) δ 3.07 (t, J = 7.9 Hz, 2H, CH2CH2Ph), 3.28 (s, 3H, NCH3), 3.90 (s, 3H, CO2CH3), 4.05–4.14 (m, 2H, CH2CH2Ph), 7.06 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, 5-H), 7.19–7.23 (m, 1H, 4’-H), 7.27–7.31 (m, 4H, 2’/3’/5’/6’-H), 7.51 (ddd, J = 8.6, 7.2, 1.6 Hz, 1H, 4-H), 7.96 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.80 (d, J = 8.6 Hz, 1H, 3-H), 10.84 (s, 1H, NH); 13C-NMR (CDCl3) δ 33.38 (CH2CH2Ph), 39.22 (NCH3), 52.35 (CO2CH3), 55.82 (CH2CH2Ph), 116.80 (C-1) 122.41 (C-5), 123.27 (C-3), 126.52 (C-4’), 128.60 (C-2’/6’), 128.91 (C-3’/5’), 130.27 (C-6), 132.99 (C-4), 138.55 (C-1’), 143.05 (C-2), 169.02 (CO2CH3), 179.87 (NHCS); Anal. calcd. for C18H20N2O2S: C, 65.8; H, 6.1; N, 8.5. Found: C, 64.9; H, 5.8; N, 8.9.
2-(Diethylamino)-4H-3,1-benzothiazin-4-one (2a)
Method 1: Methyl 2-(3,3-diethylthioureido)benzoate (
1a, 0.799 g, 3.0 mmol) was kept in concd. H
2SO
4 (12 mL) at r.t. for 24 h. The solution was poored into a mixture of ice–water (100 mL) and EtOAc (100 mL). After neutralization, the aqueous layer was further extracted with EtOAc (2 × 100 mL). The combined organic layers were dried over Na
2SO
4, filtered, and evaporated to dryness. Recrystallisation from MeOH yielded
2a (0.505 g, 72%) as colourless needles, mp 74–75 °C (MeOH), lit. [
43] 72–74 °C;
1H-NMR (CDCl
3) δ 1.24 (t,
J = 7.3 Hz, 6H, CH
2C
H3), 3.59 (q,
J = 7.3 Hz, 4H, C
H2CH
3), 7.10 (ddd,
J = 8.2, 7.6, 1.3 Hz, 1H, 6-H), 7.37 (dd,
J = 7.9, 1.3 Hz, 1H, 8-H), 7.55 (ddd,
J = 8.5, 6.9, 1.6 Hz, 1H, 7-H), 8.00 (dd,
J = 8.0, 1.6 Hz, 1H, 5-H);
13C-NMR (CDCl
3) δ 13.03 (CH
2CH
3), 43.35 (
CH
2CH
3), 116.29 (C-4a), 122.93 (C-6), 124.71 (C-5), 128.21 (C-8), 135.61 (C-7), 151.50 (C-8a), 155.43 (C-2), 184.52 (C-4); Anal. calcd. for C
12H
14N
2OS: C, 61.5; H, 6.0; N, 12.0. Found: C, 61.5; H, 6.0; N, 12.0.
Method 2: 2-(3,3-Diethylthioureido)-N,N-diethylbenzamide 6a (0.615 g, 2.0 mmol) was treated with concd. H2SO4 (8 mL) as described under Method 1 obtaining 2a (0.449 g, 96%) as a white solid.
2-(N-Cyclohexyl-N-methylamino)-4H-3,1-benzothiazin-4-one (2b)
According to the preparation of 2a (Method 1), 2b (0.607 g, 74%) was obtained from 1b as colourless plates, mp 111–114 °C (EtOH); 1H-NMR (CDCl3) δ 1.05–1.88 (m, 10H, 2’/3’/4’/5’/6’-H), 3.05 (s, 3H, NCH3), 4.22 (br s, 1H, 1’-H), 7.10 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, 6-H), 7.38 (dd, J = 8.2, 1.3 Hz, 1H, 8-H), 7.56 (ddd, J = 8.5, 6.9, 1.6 Hz, 1H, 7-H), 8.00 (dd, J = 8.2, 1.6 Hz, 1H, 5-H); 13C-NMR (CDCl3) δ 25.40 (C-4’), 25.73 (C-3’/5’), 30.14 (C-2’/6’), 30.45 (NCH3), 56.82 (C-1’), 116.50 (C-4a), 123.01 (C-6), 124.75 (C-5), 128.18 (C-8), 135.64 (C-7), 151.36 (C-8a), 156.68 (C-2), 184.48 (C-4); Anal. calcd. for C15H18N2OS: C, 65.7; H, 6.6; N, 10.2. Found: C, 65.4; H, 6.6; N, 10.1.
2-(Pyrrolidin-1-yl)-4H-3,1-benzothiazin-4-one (2c)
Method 1: According to the preparation of 2a (Method 1), 2c (0.514 g, 74%) was obtained from 1c as white needles, mp 105–107 °C (EtOH); 1H-NMR (CDCl3) δ 1.97–2.04 (m, 4H, 3’/4’-H), 3.58 (br s, 4H, 2’/5’-H), 7.10 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, 6-H), 7.39 (dd, J = 8.2, 1.0 Hz, 1H, 8-H), 7.56 (ddd, J = 8.5, 7.3, 1.6 Hz, 1H, 7-H), 8.01 (dd, J = 8.4, 1.6 Hz, 1H, 5-H); 13C-NMR (CDCl3) δ 24.97 (C-3’/4’), 47.67 (C-2’/5’), 116.58 (C-4a), 122.87 (C-6), 124.86 (C-5), 128.06 (C-8), 135.65 (C-7), 151.57 (C-8a), 154.62 (C-2), 184.22 (C-4); Anal. calcd. for C12H12N2OS: C, 62.0; H, 5.2; N, 12.1. Found: C, 61.9; H, 5.3; N, 11.9.
Method 2: According to the preparation of 2a (Method 2), 2c (0.435 g, 94%) was obtained from 6c as a light yellow solid.
2-(Piperidin-1-yl)-4H-3,1-benzothiazin-4-one (2d)
According to the preparation of 2a (Method 1), 2d (0.594 g, 80%) was obtained from 1d as white needles, mp 87–88 °C (EtOH); 1H-NMR (CDCl3) δ 1.60–1.72 (m, 6H, 3’/4’/5’-H), 3.67–3.72 (m, 4H, 2’/6’-H), 7.12 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, 6-H), 7.36 (dd, J = 8.2, 1.0 Hz, 1H, 8-H), 7.56 (ddd, J = 8.5, 6.9, 1.6 Hz, 1H, 7-H), 8.00 (dd, J = 7.9, 1.6 Hz, 1H, 5-H); 13C-NMR (CDCl3) δ 24.64 (C-4’), 25.62 (C-3’/5’), 46.84 (C-2’/6’), 116.36 (C-4a), 123.28 (C-6), 124.81 (C-5), 128.13 (C-8), 135.69 (C-7), 151.22 (C-8a), 156.27 (C-2), 184.29 (C-4); Anal. calcd. for C13H14N2OS: C, 63.4; H, 5.7; N, 11.4. Found: C, 63.35; H, 5.9; N, 11.3.
2-(Morpholin-4-yl)-4H-3,1-benzothiazin-4-one (2e)
Method 1: According to the preparation of
2a (Method 1),
2e (0.395 g, 53%) was obtained from
1e as colourless needles, mp 137–138 °C (EtOH), lit. [
48] 136–137 °C;
1H-NMR (CDCl
3) δ 3.71–3.79 (m, 8H, 2’/3’/5’/6’-H), 7.18 (ddd,
J = 8.0, 6.9, 1.3 Hz, 1H, 6-H), 7.38 (dd,
J = 8.2, 1.3 Hz, 1H, 8-H), 7.60 (ddd,
J = 8.5, 6.9, 1.6 Hz, 1H, 7-H), 8.02 (dd,
J = 8.1, 1.7 Hz, 1H, 5-H);
13C-NMR (CDCl
3) δ 45.90 (C-3’/5’), 66.40 (C-2’/6’), 116.74 (C-4a), 124.08 (C-6), 124.95 (C-5), 128.32 (C-8), 135.88 (C-7), 150.45 (C-8a), 156.75 (C-2), 183.44 (C-4); Anal. calcd. for C
12H
12N
2O
2S: C, 58.05; H, 4.9; N, 11.3. Found: C, 58.1; H, 4.9; N, 11.2.
Method 2: According to the preparation of 2a (Method 2), 2e (0.453 g, 92%) was obtained from 6e as light yellow needles.
Method 3: N-[2-(Morpholin-4-ylcarbonyl)phenyl]morpholine-4-carbothioamide (6e, 0.711 g, 2.0 mmol) was heated under reflux in anhydrous methanolic HCl (0.25 M, 10 mL) for 2 min. After cooling to r.t., the precipitate was removed by suction filtration, washed with H2O (30 mL), dried under vacuo to give 2e (0.380 g, 77%) as light yellow needles.
2-(N-Methyl-N-phenylamino)-4H-3,1-benzothiazin-4-one (2f)
Method 1: According to the preparation of 2a (Method 1), 2f (0.160 g, 20%) was obtained from 1f as colourless needles, mp 78–79 °C (EtOH); 1H-NMR (CDCl3) δ 3.59 (s, 3H, NCH3), 7.18 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, 6-H), 7.25–7.29 (m, 2H, 2’/6’-H), 7.37–7.42 (m, 1H, 4’-H), 7.42–7.47 (m, 2H, 3’/5’-H), 7.51 (dd, J = 8.2, 1.0 Hz, 1H, 8-H), 7.62 (ddd, J = 8.5, 6.9, 1.6 Hz, 1H, 7-H), 8.02 (dd, J = 8.0, 1.6 Hz, 1H, 5-H); 13C-NMR (CDCl3) δ 39.93 (NCH3), 117.07 (C-4a), 123.93 (C-6), 125.00 (C-5), 128.26 (C-2’/6’), 128.29 (C-8), 128.76 (C-4’), 130.16 (C-3’/5’), 135.72 (C-7), 142.20 (C-1’), 150.50 (C-8a), 156.97 (C-2), 184.20 (C-4); Anal. calcd. for C15H12N2OS: C, 67.1; H, 4.5; N, 10.4. Found: C, 67.0; H, 4.6; N, 10.4.
Method 2: 2-(3-Methyl-3-phenylthioureido)benzoic acid (3f, 0.859 g, 3.0 mmol) and Ac2O (7.0 mL) were kept at r.t. for 12 h. The solvent was removed under reduced pressure. Recrystallisation from EtOH gave 2f (0.346 g, 43%).
2-(N-Benzyl-N-methylamino)-4H-3,1-benzothiazin-4-one (2g)
2-(3-Benzyl-3-methylthioureido)benzoic acid (3g, 0.150 g, 0.50 mmol) and Ac2O (1.0 mL) were kept at r.t. for 8 h. The resulting crystals were removed by suction filtration to obtain 2g (0.109 g, 77%) as colourless needles, mp 70–71 °C; 1H-NMR (CDCl3) δ 3.13 (s, 3H, NCH3), 4.87 (s, 2H, CH2Ph), 7.15 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H, H-6), 7.26–7.36 (m, 5H, H-2’/3’/4’/5’/6’), 7.42 (dd, J = 8.2, 1.0 Hz, 1H, H-8), 7.59 (ddd, J = 8.4, 6.9, 1.6 Hz, 1H, H-7), 8.03 (dd, J = 8.0, 1.6 Hz, 1H, H-5); 13C-NMR (CDCl3) δ 35.95 (NCH3), 53.56 (CH2Ph), 116.34 (C-4a), 123.42 (C-6), 124.84 (C-5), 127.58 (C-2’/6’), 127.73 (C-4’), 128.30 (C-8), 128.79 (C-3’/5’), 135.76 (C-7), 136.31 (C-1’), 151.04 (C-8a), 157.07 (C-2), 183.95 (C-4); Anal. calcd. for C16H14N2OS: C, 68.1; H, 5.0; N, 9.9. Found: C, 67.7; H, 5.2; N, 9.8.
2-[N-Methyl-N-(2-phenylethyl)amino]-4H-3,1-benzothiazin-4-one (2h)
Method 1: According to the preparation of 2f (Method 2), 2h (0.578 g, 65%) was obtained from 3h as a white solid, mp 72–75 °C (EtOH); 1H-NMR (CDCl3) δ 2.96 (t, J = 7.6 Hz, 2H, CH2CH2Ph), 3.08 (s, 3H, NCH3), 3.81 (t, J = 7.6 Hz, 2H, CH2CH2Ph), 7.14 (ddd, J = 8.2, 7.1, 1.3 Hz, 1H, 6-H), 7.20–7.33 (m, 5H, 2’/3’/4’/5’/6’-H), 7.44 (dd, J = 8.2, 1.0 Hz, 1H, 8-H), 7.59 (ddd, J = 8.2, 6.9, 1.6 Hz, 1H, 7-H), 8.03 (dd, J = 7.9, 1.6 Hz, 1H, 5-H); 13C-NMR (CDCl3) δ 33.76 (CH2CH2Ph), 37.07 (NCH3), 53.03 (CH2CH2Ph), 116.23 (C-4a), 123.38 (C-6), 124.83 (C-5), 126.68 (C-4’), 128.18 (C-8), 128.70 (C-2’/6’), 128.84 (C-3’/5’), 135.77 (C-7), 138.32 (C-1’), 150.90 (C-8a), 156.35 (C-2), 183.91 (C-4); Anal. calcd. for C17H16N2OS: C, 68.9; H, 5.4; N, 9.45. Found: C, 68.9; H, 5.4; N, 9.5.
Method 2: 2-[3-Methyl-3-(2-phenylethyl)thioureido]benzoic acid (3h, 0.940 g, 3.0 mmol) and TFAA (7.0 mL) were kept at r.t. for 12 h. After removal of the solvent, the resulting crude material was purified by column chromatography on silica using petroleum ether/EtOAc (8+1) as eluent to give 2h (0.249 g, 28%) as a yellowish solid.
2-(3-Methyl-3-phenylthioureido)benzoic acid (3f)
A mixture of methyl 2-(3-methyl-3-phenylthioureido)benzoate (1f, 0.601 g, 2.0 mmol), aqueous NaOH (1 M, 10 mL) and EtOH (10 mL) was heated to reflux for 1 h. The reaction was allowed to cool to r.t. and H2O (30 mL) was added. After filtration and cooling to 0 °C, the solution was slowly acidified with concd. HCl. The precipitate was removed by suction filtration and washed with H2O (50 mL) to obtain 3f (0.378 g, 66%) as a white solid, mp 135–138 °C; 1H-NMR (DMSO-d6) δ 3.61 (s, 3H, NCH3), 7.08 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, 5-H), 7.38–7.41 (m, 3H, 2’/4’/6’-H), 7.48–7.51 (m, 3H, 4/3’/5’-H), 7.80 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.68 (dd, J = 8.5, 1.0 Hz, 1H, 3-H), 10.54 (s, 1H, NH), 13.33 (br s, 1H, CO2H); 13C-NMR (DMSO-d6) δ 43.23 (NCH3), 118.36 (C-1), 122.75, 122.98 (C-3/5), 127.12 (C-2’/6’), 128.45 (C-4’), 130.32 (C-3’/5’), 130.40 (C-6), 132.43 (C-4), 142.13, 143.53 (C-2/1’), 169.33 (CO2H), 179.74 (NHCS); Anal. calcd. for C15H14N2O2S: C, 62.9; H, 4.9; N, 9.8. Found: C, 62.7; H, 5.1; N, 9.7.
2-(3-Benzyl-3-methylthioureido)benzoic acid (3g)
According to the preparation of 3f, compound 3g (0.365 g, 61%) was obtained from 1g as a white solid, mp 117–119 °C; 1H-NMR (DMSO-d6) δ 3.24 (s, 3H, NCH3), 5.19 (s, 2H, CH2Ph), 7.17 (ddd, J = 8.2, 7.3, 1.0 Hz, 1H, 5-H), 7.25–7.29 (m, 5H, 2’/3’/4’/5’/6’-H), 7.54 (ddd, J = 8.5, 7.3, 1.3 Hz, 1H, 4-H), 7.92 (dd, J = 7.9, 1.3 Hz, 1H, 6-H), 8.44 (dd, J = 8.5, 1.0 Hz, 1H, 3-H), 10.75 (s, 1H, NH), 13.46 (br s, 1H, CO2H); 13C-NMR (DMSO-d6) δ 37.78 (NCH3), 55.92 (CH2Ph), 120.41 (C-1), 123.34 (C-5), 124.50 (C-3), 127.26 (C-2’/6’), 127.32 (C-4’), 128.66 (C-3’/5’), 130.54 (C-6), 132.42 (C-4), 137.11 (C-2), 142.55 (C-1’), 169.42 (CO2H), 180.53 (NHCS); Anal. calcd. for C16H16N2O2S: C, 64.0; H, 5.4; N, 9.3. Found: C, 63.8; H, 5.5; N, 9.5.
2-[3-Methyl-3-(2-phenylethyl)thioureido]benzoic acid (3h)
According to the preparation of 3f, compound 3h (0.509 g, 81%) was obtained from 1h as a light yellow solid, mp 130–133 °C; 1H-NMR (DMSO-d6) δ 2.97 (t, J = 7.9 Hz, 2H, CH2CH2Ph), 3.25 (s, 3H, NCH3), 4.02 (t, J = 7.6 Hz, 2H, CH2CH2Ph), 7.12–7.16 (m, 1H, 5-H), 7.19–7.23 (m, 1H, 4-H’), 7.28–7.31 (m, 4H, 2’/3’/5’/6’-H), 7.53 (ddd, J = 8.5, 7.3, 1.6 Hz, 1H, 4-H), 7.92 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.46 (d, J = 7.9 Hz, 1H, 3-H), 10.71 (s, 1H, NH), 13.52 (br s, 1H, CO2H); 13C-NMR (DMSO-d6) δ 32.69 (CH2CH2Ph), 54.78 (CH2CH2Ph), 119.45 (C-1), 122.91 (C-5), 123.95 (C-3), 126.42 (C-4’), 128.54, 128.91 (C-2’/3’/5’/6’), 130.52 (C-6), 132.45 (C-4), 138.83 (C-2), 142.69 (C-1’), 169.69 (CO2H), 179.32 (NHCS); Anal. calcd. for C17H18N2O2S: C, 64.9; H, 5.8; N, 8.9. Found: C, 64.6; H, 6.1; N, 8.7.
2-[(Methylthio)thiocarbonylamino]benzoic acid (4i)
Triethylamine (1.70 g, 16.8 mmol) was added dropwise to an ice-cooled solution of anthranilic acid (0.960 g, 7.0 mmol) and carbon disulfide (1.07 g, 14.0 mmol) in 1,4-dioxane (30 mL). The cooled mixture was stirred for 5.5 h, followed by a dropwise addition of methyl iodide (1.09 g, 7.7 mmol) in 1,4-dioxane (20 mL). After stirring for further 1.5 h in the ice-bath, the reaction mixture was allowed to warm to r.t. and stirred for 21 h under light protection. The solvent was removed under reduced pressure, and the crude material was partionated between EtOAc (100 mL) and HCl (0.2 M, 100 mL). The aqueous phase was further extracted with EtOAc (2 × 200 mL). The combined organic layers were dried (Na2SO4), filtered and evaporated to dryness. Recrystallisation from PhMe gave 4i (1.22 g, 77%) as light yellow needles, mp 148–150 °C (PhMe); 1H-NMR (DMSO-d6) δ 2.59 (s, 3H, SCH3), 7.36 (td, J = 7.9, 1.3 Hz, 1H, 5-H), 7.61 (ddd, J = 8.2, 7.0, 1.6 Hz, 1H, 4-H), 7.94 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.12 (d, J = 7.9 Hz, 1H, 3-H), 11.95 (br s, 1H, NH); 13C-NMR (DMSO-d6) δ 18.11 (SCH3), 124.15 (C-1), 125.71 (C-3), 126.37 (C-5), 131.06 (C-6), 132.95 (C-4), 139.93 (C-2), 167.96 (CO2H), 198.06 (NHCS); Anal. calcd. for C9H9NO2S2: C, 47.6; H, 4.0; N, 6.2. Found: C, 47.3; H, 4.3; N, 6.2.
2-[(Ethylthio)thiocarbonylamino]benzoic acid (4j)
According to the preparation of 4i, compound 4j (1.42 g, 84%) was obtained from ethyl iodide as light yellow needles, mp 132–135 °C (PhMe); 1H-NMR (DMSO-d6) δ 1.26 (t, J = 7.3 Hz, 3H, CH2CH3), 3.20 (q, J = 7.3 Hz, 2H, CH2CH3), 7.36 (td, J = 7.6, 1.3 Hz, 1H, 5-H), 7.61 (ddd, J = 8.2, 6.9, 1.6 Hz, 1H, 4-H), 7.93 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.11 (d, J = 8.2 Hz, 1H, 3-H), 11.97 (br s, 1H, NH); 13C-NMR (DMSO-d6) δ 14.06 (CH2CH3), 29.08 (CH2CH3), 124.31 (C-1), 125.73 (C-3), 126.35 (C-5), 131.04 (C-6), 132.90 (C-4), 139.88 (C-2), 167.93 (CO2H), 197.11 (NHCS); Anal. calcd. for C10H11NO2S2: C, 49.8; H, 4.6; N, 5.8. Found: C, 49.7; H, 4.7; N, 5.8.
2-[(Benzylthio)thiocarbonylamino]benzoic acid (4k)
According to the preparation of 4i, compound 4k (1.28 g, 60%) was obtained from benzyl bromide as a yellow solid, mp 142–144 °C (EtOAc); 1H-NMR (DMSO-d6) δ 4.54 (s, 2H, CH2Ph), 7.25 (td, J = 6.6, 1.6 Hz, 1H, H-4’), 7.30–7.34 (m, 2H, 2’/6’-H), 7.35–7.40 (m, 3H, 5/3’/5’-H), 7.62 (ddd, J = 8.3, 7.1, 1.6 Hz, 1H, 4-H), 7.94 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.03 (d, J = 8.5 Hz, 1H, 3-H), 11.98 (br s, 1H, NH); 13C-NMR (DMSO-d6) δ 39.79 (CH2Ph), 124.71 (C-1), 126.10 (C-3), 126.63 (C-5), 127.43 (C-4’), 128.64 (C-2’/6’), 129.15 (C-3’/5’), 131.07 (C-6), 132.95 (C-4), 136.64 (C-1’), 139.75 (C-2), 167.78 (CO2H), 196.67 (NHCS); Anal. calcd. for C15H13NO2S2: C, 59.4; H, 4.3; N, 4.6. Found: C, 59.6; H, 4.3; N, 4.65.
2-{[(2-Phenylethyl)thio]thiocarbonylamino}benzoic acid (4l)
According to the preparation of 4i, compound 4l (0.527 g, 24%) was obtained from 2-phenylethyl bromide as a yellow solid, mp 125–128 °C (CHCl3); 1H-NMR (DMSO-d6) δ 2.94 (t, J = 8.2 Hz, 2H, CH2CH2Ph), 3.47 (t, J = 7.9 Hz, 2H, CH2CH2Ph), 7.19–7.22 (m, 1H, 4’-H), 7.26–7.32 (m, 4H, 2’/3’/5’/6’-H), 7.37 (td, J = 7.9, 1.3 Hz, 1H, 5-H), 7.61 (td, J = 7.6, 1.6 Hz, 1H, 4-H), 7.94 (dd, J = 7.9, 1.6 Hz, 1H, 6-H), 8.04 (d, J = 7.9 Hz, 1H, 3-H), 11.94 (br s, 1H, NH); 13C-NMR (DMSO-d6) δ 34.67 (CH2CH2Ph), 36.00 (CH2CH2Ph), 124.64 (C-1), 126.04 (C-3), 126.49 (C-5/4’), 128.54 (C-2’/6’), 128.67 (C-3’/5’), 131.04 (C-6), 133.83 (C-4), 139.81 (C-1’), 140.13 (C-2), 167.84 (CO2H), 196.98 (NHCS); Anal. calcd. for C16H15NO2S2: C, 60.5; H, 4.8; N, 4.4. Found: C, 60.2; H, 4.8; N, 5.0.
2-(Methylthio)-4H-3,1-benzothiazin-4-one (5i)
Method 1: 2-[(Methylthio)thiocarbonylamino]benzoic acid (4i, 0.909 g, 4.0 mmol) was heated to reflux in Ac2O (10 mL) for 30 min. The solvent was removed under reduced pressure, and the crude material was recrystallised from MeOH to obtain 5i (0.782 g, 93%) as colourless needles, mp 54–56 °C (MeOH); 1H-NMR (DMSO-d6) δ 2.72 (s, 3H, SCH3), 7.58 (ddd, J = 7.9, 7.3, 1.3 Hz, 1H, 6-H), 7.72 (dd, J = 8.0, 1.2 Hz, 1H, 8-H), 7.92 (ddd, J = 8.5, 7.3, 1.9 Hz, 1H, 7-H), 8.06 (dd, J = 8.2, 1.9 Hz, 1H, 5-H); 13C-NMR (DMSO-d6) δ 13.92 (SCH3), 118.65 (C-4a), 124.68 (C-5), 128.33 (C-6), 129.86 (C-8), 136.84 (C-7), 147.50 (C-8a), 163.47 (C-2), 182.33 (C-4); Anal. calcd. for C9H7NOS2: C, 51.65; H, 3.4; N, 6.7. Found: C, 51.7; H, 3.4; N, 6.7.
Method 2: 2-[(Methylthio)thiocarbonylamino]benzoic acid (4i, 0.682 g, 3.0 mmol) and TFAA (7.0 mL) were kept at r.t. for 12 h. After removal of the solvent, the resulting crude material was purified by column chromatography on silica using petroleum ether/EtOAc (8+1) as eluent to give 5i (0.571 g, 91%) as yellowish needles.
2-(Ethylthio)-4H-3,1-benzothiazin-4-one (5j)
According to the preparation of 5i (Method 1), 5j (0.792 g, 89%) was obtained from 4j as yellow needles, mp 53–54 °C (EtOH); 1H-NMR (DMSO-d6) δ 1.38 (t, J = 7.3 Hz, 3H, SCH2CH3), 3.35 (q, J = 7.3 Hz, 2H, SCH2CH3), 7.58 (td, J = 7.6, 1.3 Hz, 1H, 6-H), 7.71 (dd, J = 8.1, 1.0 Hz, 1H, 8-H), 7.92 (ddd, J = 8.1, 6.8, 1.9 Hz, 1H, 7-H), 8.06 (dd, J = 7.9, 1.6 Hz, 1H, 5-H); 13C-NMR (DMSO-d6) δ 14.47 (SCH2CH3), 25.83 (SCH2CH3), 118.77 (C-4a), 124.66 (C-5), 128.36 (C-6), 129.89 (C-8), 136.82 (C-7), 147.52 (C-8a), 162.62 (C-2), 182.44 (C-4); Anal. calcd. for C10H9NOS2: C, 53.8; H, 4.1; N, 6.3. Found: C, 54.1; H, 4.2; N, 6.2.
2-(Benzylthio)-4H-3,1-benzothiazin-4-one (5k)
According to the preparation of 5i (Method 1), 5k (1.01 g, 88%) was obtained from 4k as white needles, mp 69–72 °C (EtOH); 1H-NMR (DMSO-d6) δ 4.64 (s, 2H, CH2Ph), 7.24–7.27 (m, 1H, 4’-H), 7.31–7.33 (m, 2H, 3’/5’-H), 7.49–7.50 (m, 2H, 2’/6’-H), 7.59 (ddd, J = 7.7, 7.6, 1.3 Hz, 1H, 6-H), 7.80 (dd, J = 8.5, 1.3 Hz, 1H, 8-H), 7.94 (ddd, J = 8.2, 7.3, 1.6 Hz, 1H, 7-H), 8.06 (dd, J = 7.9, 1.3 Hz, 1H, 5-H); 13C-NMR (DMSO-d6) δ 34.88 (CH2Ph), 118.73 (C-4a), 124.74 (C-5), 127.63 (C-4’), 128.50 (C-6), 128.66 (C-2’/6’), 129.41 (C-3’/5’), 129.88 (C-8), 136.83 (C-7), 136.90 (C-1’), 147.34 (C-8a), 162.03 (C-2), 182.25 (C-4); Anal. calcd. for C15H11NOS2: C, 63.1; H, 3.9; N, 4.9. Found: C, 63.2; H, 3.95; N, 4.9.
2-[(2-Phenylethyl)thio]-4H-3,1-benzothiazin-4-one (5l)
According to the preparation of 5i (Method 1), 5l (0.671 g, 56%) was obtained from 4l as pink blocks, mp 63–65 °C (twice from cyclohexane); 1H-NMR (CDCl3) δ 3.08 (t, J = 7.6 Hz, 2H, CH2CH2Ph), 3.55 (t, J = 7.9 Hz, 2H, CH2CH2Ph), 7.23–7.34 (m, 5H, 2’/3’/4’/5’/6’-H), 7.44 (td, J = 7.4, 1.3 Hz, 1H, 6-H), 7.68 (dd, J = 8.7, 1.6 Hz, 1H, 8-H), 7.76 (ddd, J = 8.4, 7.0, 1.6 Hz, 1H, 7-H), 8.15 (dd, J = 7.9, 1.6 Hz, 1H, 5-H); 13C-NMR (CDCl3) δ 32.71 (CH2CH2Ph), 35.66 (CH2CH2Ph), 119.46 (C-4a), 125.04 (C-5), 126.73 (C-4’), 127.56 (C-6), 128.62 (C-2’/6’), 128.64 (C-3’/5’), 129.81 (C-8), 135.78 (C-7), 139.69 (C-1’), 148.06 (C-8a), 163.21 (C-2), 183.33 (C-4); Anal. calcd. for C16H13NOS2: C, 64.2; H, 4.4; N, 4.7. Found: C, 64.3; H, 4.4; N, 4.7.
2-(3,3-Diethylthioureido)-N,N-diethylbenzamide (6a)
Diethylamine (0.914 g, 12.5 mmol) was added dropwise to a solution of 2-(methylthio)-4H-3,1-benzothiazin-4-one (5i, 1.05 g, 5.0 mmol) in acetone (15 mL). After stirring for 1 h, the mixture was heated to reflux for 1 h, and allowed to cool to r.t. The solvent was removed under reduced pressure. Recrystallisation from EtOH gave 6a (0.998 g, 65%) as colourless prisms, mp 116–117 °C (EtOH); 1H-NMR (DMSO-d6) δ 1.01–1.08 (m, 6H, CH2CH3), 1.14 (t, J = 7.1 Hz, 6H, 2 × CH2CH3), 3.20 (q, J = 6.9 Hz, 2H, CH2CH3), 3.37 (q, J = 6.9 Hz, 2H, CH2CH3), 3.67 (q, J = 6.9 Hz, 4H, CH2CH3), 7.20–7.26 (m, 2H, 5/6-H), 7.33–7.38 (m, 1H, 4-H), 7.41 (d, J = 7.9 Hz, 1H, 3-H), 8.67 (s, 1H, NH); 13C-NMR (DMSO-d6) δ 12.44, 12.64, 13.91 (3 × CH2CH3), 38.20, 43.09, 44.88 (3 × CH2CH3), 125.37, 125.84 (C-5/6), 128.58 (C-4), 130.00 (C-3), 134.59 (C-1), 138.17 (C-2), 168.26 (CON), 179.34 (NHCS); Anal. calcd. for C16H25N3OS: C, 62.5; H, 8.2; N, 13.7. Found: C, 62.8; H, 8.1; N, 13.8.
N-[2-(Pyrrolidin-1-ylcarbonyl)phenyl]pyrrolidine-1-carbothioamide (6c)
According to the preparation of 6a, compound 6c (1.13 g, 74%) was obtained from 5i and pyrrolidine as light yellow prisms, mp 160–162 °C (EtOH); 1H-NMR (DMSO-d6) δ 1.71–2.08 (m, 8H, 3/4/3’’/4’’-H), 3.38–3.70 (m, 8H, 2/5/2’’/5’’-H), 7.16 (td, J = 7.6, 1.3 Hz, 1H, 4’-H), 7.38 (td, J = 7.9, 1.6 Hz, 1H, 5’-H), 7.42 (dd, J = 7.7, 1.4 Hz, 1H, 3’-H), 7.88 (d, J = 7.9 Hz, 1H, 6’-H), 9.35 (s, 1H, NH); 13C-NMR (DMSO-d6) δ 24.02, 25.91 (C-3/4/3’’/4’’), 46.03, 49.13 (C-2/5/2’’/5’’), 124.03 (C-4’), 126.53 (C-6’), 127.20 (C-3’), 129.26 (C-5’), 130.52 (C-2’), 138.44 (C-1’), 167.39 (CON), 176.74 (NHCS); Anal. calcd. for C16H21N3OS: C, 63.3; H, 7.0; N, 13.85. Found: C, 63.5; H, 7.1; N, 13.8.
N-[2-(Morpholin-4-ylcarbonyl)phenyl]morpholine-4-carbothioamide (6e)
Method 1: According to the preparation of 6a, compound 6e (1.51 g, 90%) was obtained from 5i and morpholine as colourless prisms, mp 170–173 °C (EtOH); 1H-NMR (DMSO-d6) δ 3.48–3.63 (m, 12H, 2/6/2’/3’/5’/6’-H) 3.85 (t, J = 4.6 Hz, 4H, 3/5-H), 7.23–7.29 (m, 3H, 3’/4’/6’-H), 7.39 (ddd, J = 7.8, 6.9, 1.9 Hz, 1H, 5’-H), 9.31 (s, 1H, NH); 13C-NMR (DMSO-d6) δ 41.70, 47.48, 48.85, 66.06, 66.12, 66.30 (C-2/3/5/6/2’’/3’’/5’’/6’’), 125.75 (C-3’), 127.26 (C-4’), 129.29 (C-5’), 129.35 (C-6’), 133.38 (C-2’), 138.38 (C-1’), 167.06 (CON), 181.93 (NHCS); Anal. calcd. for C16H21N3O3S: C, 57.3; H, 6.3; N, 12.5. Found: C, 57.5; H, 6.35; N, 12.35.
Method 2: Morpholine (0.392 g, 4.5 mmol) was added dropwise to a solution of 2-(morpholin-4-yl)-4H-3,1-benzothiazin-4-one (2e, 0.497 g, 2.0 mmol) in acetone (6 mL). After stirring for 1 h, the mixture was heated to reflux for 1 h, and allowed to cool to r.t. The formed precipitate was removed by suction filtration and washed with cold acetone (5 mL) to give 6e (0.490 g, 73%) as white solid.
Methyl 2-(morpholin-4-ylcarbonyl)phenyldithiocarbamate (7)
Morpholine (0.392 g, 4.5 mmol) was added dropwise to a solution of 2-(methylthio)-4H-3,1-benzothiazin-4-one (5i, 0.418 g, 2.0 mmol) in acetone (6 mL). After stirring for 1 h, the resulting precipitate was removed by suction filtration and washed with cold acetone (5 mL) to obtain 7 (0.299 g, 50%) as a white solid, mp 143–145 °C; 1H-NMR (DMSO-d6) δ 2.55 (s, 3H, SCH3), 3.15–3.22, 3.45–3.64 (m, 8H, 2’/3’/5’/6’-H), 7.35–7.49 (m, 4H, 3/4/5/6-H), 11.52 (s, 1H, NH); 13C-NMR (DMSO-d6) δ 18.24 (SCH3), 41.90, 47.39 (C-3’’/5’’), 66.09, 66.13 (C-2’’/6’’), 127.50, 127.98 (C-3’/4’), 128.65, 129.77 (C-5’/6’), 133.22 (C-2’), 136.57 (C-1’), 168.40 (CO), 199.94 (NHCS); Anal. calcd. for C13H16N2O2S2: C, 52.7; H, 5.4; N, 9.45. Found: C, 52.8; H, 5.45; N, 9.5.
2-[N-Methyl-N-(2-phenylethyl)amino]-4H-3,1-benzoxazin-4-one (8h)
In the course of the preparation of
2h (Method 2) using TFAA, purification of the crude material by column chromatography on silica gave
8h (0.570 g, 68%) as a white solid, mp 68–70 °C, lit. [
8] 68.5–69 °C;
1H-NMR (CDCl
3) δ 2.94 (t,
J = 7.3 Hz, 2H, CH
2C
H2Ph), 3.05 (s, 3H, NCH
3), 3.75 (t,
J = 7.3 Hz, 2H, C
H2CH
2Ph), 7.07–7.12 (m, 1H, 6-H), 7.18–7.30 (m, 6H, 8/2’/3’/4’/5’/6’-H), 7.58 (ddd,
J = 7.8, 7.4, 1.8 Hz, 1H, 7-H), 7.97 (dd,
J = 7.9, 1.6 Hz, 1H, 5-H);
13C-NMR (CDCl
3) δ 34.01 (CH
2CH
2Ph), 35.68 (NCH
3), 51.35 (
CH
2CH
2Ph), 112.06 (C-4a), 123.00, 124.15, 128.61 (C-5/6/8), 126.54 (C-4’), 128.57 (C-2’/6’), 128.82 (C-3’/5’), 136.56 (C-7), 138.42 (C-1’), 150.93 (C-8a), 153.84 (C-2), 159.90 (C-4); Anal. calcd. for C
17H
16N
2O
2: C, 72.8; H, 5.75; N, 10.0. Found: C, 72.4; H, 6.05; N, 9.7.
2-(Methylthio)-4H-3,1-benzoxazin-4-one (9i)
In the course of the preparation of
5i (Method 2) using TFAA, purification of the crude material by column chromatography on silica yielded
9i (0.010 g, 2%) as a white solid, mp 103–105 °C, lit. [
3] 108–109 °C;
1H-NMR (CDCl
3) δ 2.58 (s, 3H, SCH
3), 7.40 (ddd,
J = 8.2, 7.2, 1.3 Hz, 1H, 6-H), 7.45 (d,
J = 7.9 Hz, 1H, 8-H), 7.74 (ddd,
J = 7.9, 7.3, 1.6 Hz, 1H, 7-H), 8.11 (dd,
J = 8.0, 1.6 Hz, 1H, 5-H);
13C-NMR (CDCl
3) δ 14.16 (SCH
3), 115.59 (C-4a), 125.59, 127.22, 128.77 (C-5/6/8), 136.76 (C-7), 146.89 (C-8a), 158.80 (C-4), 164.00 (C-2).
HLE inhibition assay
Human leukocyte elastase was assayed spectrophotometrically at 405 nm at 25 °C [
49]. Assay buffer was 50 mM sodium phosphate buffer, 500 mM NaCl, pH 7.8. An enzyme stock solution of 50 µg/mL was prepared in 100 mM sodium acetate buffer, pH 5.5 and diluted with assay buffer. Inhibitor stock solutions were prepared in DMSO. A stock solution of the chromogenic substrate MeOSuc-Ala-Ala-Pro-Val-pNA was prepared in DMSO and diluted with assay buffer. The final concentration of HLE was 50 ng/mL, of the chromogenic substrate MeOSuc-Ala-Ala-Pro-Val-pNA was 100 µM, and of DMSO was 5.5%. Into a cuvette containing 870 µL assay buffer, 50 µL of an inhibitor solution and 50 µL of the substrate solution were added and thoroughly mixed. The reaction was initiated by adding 50 µL of the HLE solution and was followed over 10 min. IC
50 values were calculated from the linear steady-state turnover of the substrate.
Cathepsin G inhibition assay
Human cathepsin G was assayed spectrophotometrically at 405 nm at 25 °C [
7,
8]. Assay buffer was 20 mM Tris HCl buffer, 150 mM NaCl, pH 8.4. Inhibitor stock solutions were prepared in DMSO. An enzyme stock solution of 200 mU/mL was prepared in 50 mM sodium acetate buffer, 150 mM NaCl, pH 5.5. A 50 mM stock solution of the chromogenic substrate Suc-Ala-Ala-Pro-Phe-pNA in DMSO was diluted with assay buffer. The final concentration of cathepsin G was 2.5 mU/mL, of the substrate Suc-Ala-Ala-Pro-Phe-NHNp was 500 µM, and of DMSO was 1.5%. Into a cuvette containing 882.5 µL assay buffer, 5 µL of an inhibitor solution and 100 µL of a substrate solution were added and thoroughly mixed. The reaction was initiated by adding 12.5 µL of the cathepsin G solution and was followed over 10 min. IC
50 values were calculated from the linear steady-state turnover of the substrate.
Chymotrypsin inhibition assay
Bovine chymotrypsin was assayed spectrophotometrically at 405 nm at 25 °C. Assay buffer was 20 mM Tris HCl buffer, 150 mM NaCl, pH 8.4. Inhibitor stock solutions were prepared in DMSO. An enzyme stock solution was prepared in 1 mM HCl and diluted with assay buffer. A 40 mM stock solution of the chromogenic substrate Suc-Ala-Ala-Pro-Phe-pNA in DMSO was diluted with assay buffer. The final concentration of chymotrypsin was 12.5 ng/mL, of the substrate Suc-Ala-Ala-Pro-Phe-NHNp was 200 µM, and of DMSO was 6%. Into a cuvette containing 845 µL assay buffer, 55 µL of an inhibitor solution and 50 µL of a substrate solution were added and thoroughly mixed. The reaction was initiated by adding 50 µL of a chymotrypsin solution and was followed over 12.5 min. IC50 values were calculated from the linear steady-state turnover of the substrate.
Trypsin inhibition assay
Trypsin from bovine pancreas was assayed spectrophotometrically at 405 nm at 25 °C. Assay buffer was 20 mM Tris HCl buffer, 150 mM NaCl, pH 8.4. An enzyme stock solution of 10 µg/mL was prepared in 1 mM HCl and diluted with assay buffer. Inhibitor stock solutions were prepared in DMSO. A 40 mM stock solution of the chromogenic substrate Suc-Ala-Ala-Pro-Arg-pNA in DMSO was diluted with assay buffer. The final concentration of trypsin was 12.5 ng/mL, of the substrate Suc-Ala-Ala-Pro-Arg-pNA was 200 µM, and of DMSO was 6%. Into a cuvette containing 845 µL assay buffer, 55 µL of an inhibitor solution and 50 µL of a substrate solution were added and thoroughly mixed. The reaction was initiated by adding 50 µL of the trypsin solution and was followed over 12.5 min. IC50 values were calculated from the linear steady-state turnover of the substrate.
Cathepsin L inhibition assay
Human cathepsin L was assayed spectrophotometrically at 405 nm at 37 °C [
50]. Assay buffer was 100 mM sodium phosphate buffer, pH 6.0, 100 mM NaCl, 5 mM EDTA, 0.01% Brij 35. An enzyme stock solution of 50 µg/mL in 20 mM sodium acetate buffer, pH 5.0, 100 mM NaCl, 10 mM trehalose, 1 mM EDTA, 50% glycerol was diluted 1:100 with assay buffer containing 5 mM DTT and incubated for 30 min at 37 °C. This enzyme solution was diluted 1:5 with assay buffer containing 5 mM DTT. Inhibitor stock solutions were prepared in DMSO. A 10 mM stock solution of the chromogenic substrate Z-Phe-Arg-pNA was prepared with DMSO. The final concentration of cathepsin L was 4 ng/mL, of the substrate Z-Phe-Arg-pNA was 100 µM, and of DMSO was 5%. Into a cuvette containing 910 µL assay buffer, 40 µL of an inhibitor solution and 10 µL of a substrate solution were added and thoroughly mixed. The reaction was initiated by adding 40 µL of the cathepsin L solution and was followed over 10 min. IC
50 values were calculated from the linear steady-state turnover of the substrate.
ACE inhibition assay
Human ACE was assayed spectrophotometrically at 352 nm at 37 °C [
51]. Assay buffer was 50 mM Tris HCl buffer, 300 mM NaCl, pH 7.5. An enzyme stock solution of 434 µg/mL in 12.5 mM HCl, pH 7.5, 75 mM NaCl, 500 nM ZnCl
2, 40% glycerol was diluted 1:100 with assay buffer. After incubation for 10 min at 37 °C, the enzyme solution was stored at 0 °C and used within 90 min. Inhibitor stock solutions were prepared in DMSO. A 300 mM stock solution of the chromogenic substrate FA-Phe-Gly-Gly was prepared in DMSO. The final concentration of ACE was 86.8 ng/mL, of the substrate FA-Phe-Gly-Gly was 3 mM, and of DMSO was 3%. Into a cuvette containing 950 µL assay buffer, 20 µL of an inhibitor solution and 10 µL of a substrate solution were added and thoroughly mixed. The reaction was initiated by adding 20 µL of the ACE solution and was followed over 20 min. IC
50 values were calculated from the linear steady-state turnover of the substrate.
AChE inhibition assay
Acetylcholinesterase inhibition was assayed spectrophotometrically at 412 nm at 25 °C [
52,
53,
54]. Assay buffer was 100 mM sodium phosphate, 100 mM NaCl, pH 7.3. The enzyme stock solution (~100 U/mL) in assay buffer was kept at 0 °C. Appropriate dilutions were prepared immediately before starting the measurement. ATCh (10 mM) and DTNB (7 mM) were dissolved in assay buffer and kept at 0 °C. Stock solutions of the test compounds were prepared in acetonitrile. The final concentration of AChE was ~30 mU/mL, of ATCh was 500 µM, of DTNB was 350 µM, and of acetonitrile was 6%. Into a cuvette containing 830 µL assay buffer, 50 µL of the DTNB solution, 50 µL acetonitrile, 10 µL of a solution of the test compound, and 10 µL of an enzyme solution (~3 U/mL) were added and thoroughly mixed. After incubation for 15 min at 25 °C, the reaction was initiated by adding 50 µL of the ATCh solution and was followed over 5 min. IC
50 values were calculated from the linear steady-state turnover of the substrate.
CEase inhibition assay
Cholesterol esterase inhibition was assayed spectrophotometrically at 405 nm at 25 °C [
55,
56]. Assay buffer was 100 mM sodium phosphate, 100 mM NaCl, pH 7.0. A stock solution of CEase was prepared in 100 mM sodium phosphate buffer, pH 7.0 and kept at 0 °C. A 1:122 dilution was done immediately before starting the measurement. TC (12 mM) was dissolved in assay buffer and kept at 25 °C. Stock solutions of all test compounds and of pNPB (20 mM) were prepared in acetonitrile. The final concentration of CEase was 10 ng/mL, of the substrate pNPB was 200 µM, of TC was 6 mM, and of acetonitrile was 6%. Into a cuvette containing 430 µL assay buffer, 500 µL of the TC solution, 40 µL acetonitrile, 10 µL of the pNPB solution, and 10 µL of a solution of the test compound were added and thoroughly mixed. After incubation for 5 min at 25 °C, the reaction was initiated by adding 10 µL of the enzyme solution (1 µg/mL). IC
50 values were calculated from the linear steady-state turnover of the substrate.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}