Abstract
The complete characterization of two compounds obtained from the acetone extract of Populus tremula knotwood has been was achieved using LC-DAD-MS, MS/MS, IR and NMR. The new compounds were unequivocally identified as a mixture of the ester isomers of the (E) and (Z) p-coumarate of 1-O-rutinose. The isomers showed the capacity to inhibit lipid peroxidation induced by tert-butylhydroperoxide and to trap peroxyl radicals, as determined by a chemiluminescence method. These new phenyl disaccharides also showed a significant ORAC (oxygen radical absorbance capacity) value, i.e. 11.7 μM TE (Trolox Equivalents).
Introduction
The genus Populus, which includes aspens and cottonwoods, is one of the several genera of the family Salicaceae. The chemical composition of Populus tremula (European aspen) wood has been studied by several research groups and its chemical constituents were first reported some eighty years ago []. Wood and bark of Populus species and their extracts were also extensively investigated a few decades ago [,]. In the last years, it has been found that tree knots, frequently discarded in the wood and paper industry, represent a rich raw material for producing polyphenols. The major polyphenols in aspen knotwood are known [], and the individual flavonoids have recently been isolated []. Polyphenols in knotwood constitute a large potential resource of natural antioxidants for the food and pharmaceutical industries [,,]. In the present work, we describe the identification of two new phenyl disaccharides from aspen knotwood, which might represent possible precursors in the biosynthetic pathway of plant biomolecules. This may suggest a new and different pathway for secondary plant metabolites in Populus tremula knotwood that may differ from that one existing in other members of Salicaceae. The present work is complementary to a previous publication [], in which chemical characterization and determination of the antioxidant capacity of the major polyphenolic compounds from Populus tremula knotwood was reported. Here a detailed structural elucidation of a new phenyl disaccharide, which was found in solution as a pair of geometrical isomers in equilibrium, is provided. The capacities of inhibition of lipid peroxidation, trapping of peroxyl radicals, as well as the ORAC value (oxygen radical absorbance capacity) for the new compound were determined.
Results and Discussion
Two unknown compounds, here named F1 and F2 were isolated from a Populus tremula knotwood extract by preparative HPLC using a previously described protocol [].The total extract and the unknown compounds were then analyzed by HPLC-MS. The analysis of the pure compounds showed two peaks, corresponding to the same unknown fractions found earlier by preparative-HPLC. These peaks corresponded to F1 (Rt: 23.7 min) and F2 (Rt: 28.3 min), according to data derived from the analytical HPLC chromatogram of the whole extract. In addition, further HPLC analysis of the fractions isolated from the extract again yielded two peaks: the F1 fraction gave F1-Z and F2-E peaks (left inset, Figure 1), and the F2 fraction led to F1-Z and F2-E peaks (right inset, Figure 1).
The UV spectra of the isolated fractions had almost identical shapes, showing an absorption band maximum at 315 nm (Figure 2). This absorption maximum is characteristic for a p-coumaroyl ester derivative []. The extinction coefficient of 28100±100 (M-1cm-1) was determined for the mixture of F1-Z and F2-E in ethanol at 315 nm. The IR spectra of the solid fractions for F1 and F2 showed the presence of a p-coumaroyl ester derivative with C=O stretching vibration signals at 1712 cm-1 (Figure 2), corresponding to carbonyl compounds with aryl and/or α,β-unsaturated ester [,,].
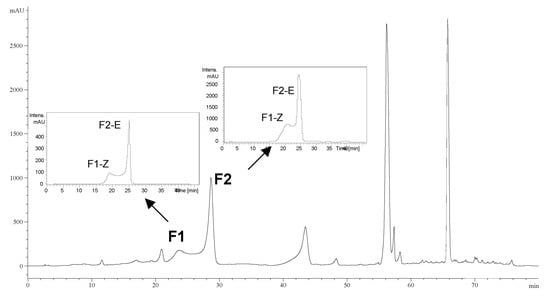
Figure 1.
Analytical HPLC chromatogram of Populus tremula knotwood extract recorded at 314 nm. Inserts represent the chromatograms of the fractions F1 and F2 isolated using preparative HPLC.
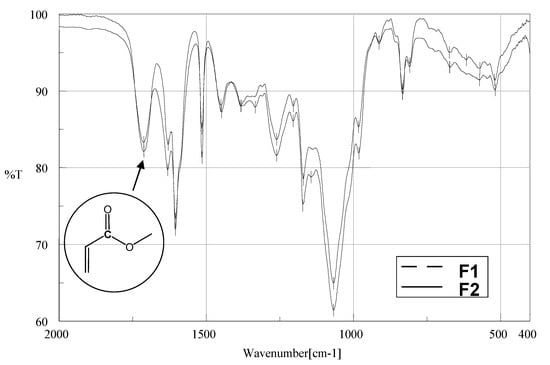
Figure 2.
UV spectra of the F1-Z and F2-E from fraction F1 obtained with the HPLC DAD detector and FTIR spectra of fractions F1 and F2, in KBr pellets.
MS and MS/MS analyses of the two fractions F1 and F2 showed identical molecular masses and fragmentation patterns, leading to the hypothesis that the fractions consisted of two isomeric compounds (Figure 3 A and Figure 3 B).

Figure 3.
MS and MS-MS spectra of peaks (F1-Z, F2-E) at m/z 471 from fraction F1 (A); MS and MS-MS spectra of peaks (F1-Z, F2-E) at m/z 471 from fraction F2 (B).
The relative intensities of the fragments obtained for the molecular ion (MS/MS spectra of Z isomer) are slightly different due to the purity differences of this isomer in the two fractions. LC-ESI/MS analysis showed the presence of a deprotonated compound at m/z 471 [M –H+]- and a fragment, m/z 162.8, for both compounds. This supports the presence of a p-coumaric acid moiety in the structure of the compound [,]. Moreover, some other fragments indicated the loss of specific sugar moieties from the original compound, i.e. a fragment with m/z 309 corresponds to a [p-coumaric-glucose–H2O]-, while the fragment at m/z 144.7, the presence of [rhamnose–H2O]-. High-resolution mass spectrometry gave the same formula for the TMS-ether derivatives, m/z 904.3964 (calculated for C39H76O12Si6, 904.395249), compatible with a structure including six hydroxyl groups accessible in the proposed structure.
1H- and 13C-NMR data prove and fully elucidate the structure of the unknown fractions and suggests that F1 and F2 are the (Z) and (E) p-coumarate of 1-O-rutinose isomers, respectively (see Figure 4). The same signals were observed in the proton NMR analysis of each fraction, but with slightly different chemical shifts, which suggests that a possible mixing of the compounds may take place, like a mixture previously analyzed by Cogne et al. []. Figure 5 shows the 1H-NMR spectrum in deuterated methanol, in which a good correlation of the aromatic signals was found with a pair of geometric isomers for the p-coumaroyl moiety (four proton signals, δ 5.84, 6.37, 6.94, and 7.73) and two sugar residues, as seen by the anomeric proton signals (δ 5.55, 5.52, and 4.72).
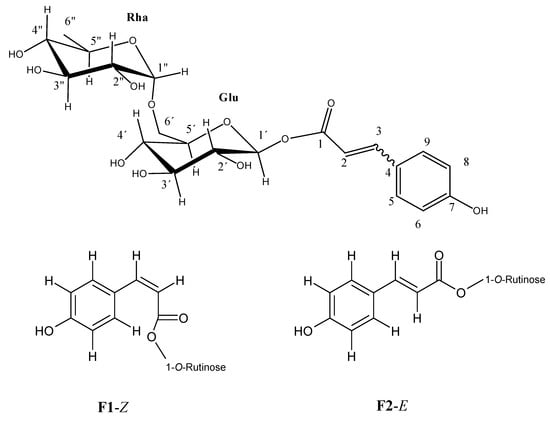
Figure 4.
Proposed structures for identified compounds F1-Z and F2-E.
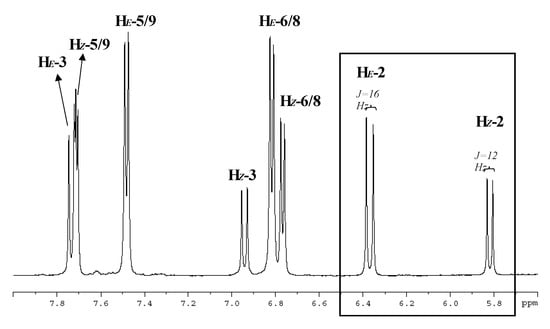
Figure 5.
Aromatic region of 1H-NMR spectrum of F2 in CD3OD at 500 MHz. Signal assignments are indicated in Figure 4 and characteristic vicinal H-H coupling constants in the insert below.
It can also be observed that two different proton signals are observed for the p-coumaroyl moiety, corresponding to the two different isomers, as follows: H5/H9 (Z) (δ 7.71), and H5/H9 (E), (δ 7.48) respectively; H6/H8 (Z)/(E), (δ 6.81 and 6.77) (Figure 5). In the HSQC spectra, a correlation was shown between the proton signals at δ 7.73 (assigned to the p-coumaroyl 3-position) and the carbon signal at δ 148.0 for the E isomer (C3), while the next correlation can be observed for the proton signals at δ 6.94 (assigned to the p-coumaroyl 3-position) and the carbon signal at δ 147.1 for the Z isomer (C3). A correlation for the two isomers in neighbouring position of the double bond was also observed as follows: the correlation between the proton signals at δ 6.37 (assigned to the p-coumaroyl 2-position) and the carbon signal at δ 115.4 for the Z isomer (C2) and the correlation between the proton signals at δ 5.84 (assigned to the p-coumaroyl 3-position), and the carbon signal at δ 114.4 for the E isomer (C2). The coupling constants for the protons signals were different for the two isomers: 12 Hz for the Z form and 16 Hz for the E isomer, which is in agreement with the results of Bergman et al. [].
The coupling constants of the anomeric proton signals at δ 5.55 and 5.52 were 7.7 (Z isomer) and 8.2 Hz (E isomer), respectively, thus suggesting that one glucose unit was of the β-D-glucopyranoside type []. In the HSQC spectra, correlations appeared between the anomeric proton signal at δ 5.55 (assigned to the 1´-position of glucose) and the carbon signal at δ 95.7 for the Z isomer, and the anomeric proton signal at δ 5.52 (assigned to the 1’-position of glucose) also correlates to the carbon signal at δ 95.5 for the E isomer. The anomeric proton signal at δ 4.72 corresponds to a rhamnose unit (assigned to the 1”-position of rhamnose) and correlates to the carbon signal at δ 102.2, suggesting that the bond between the two sugars is at the position 1”-6’, [,]. This notion has been confirmed by a Tollen´s test, since no reducing sugars were found. After the integration of the signals corresponding to vinyl protons (H-2), the percentage of each isomer in solution was determined (data calculated for F1 and F2); ratio (E)/(Z)= [δ ΗE (6.37ppm)]/[δ HZ (5.84ppm)] = 1.8; these areas were selected because of less interferences compared to other signals (see box in Figure 5). This ratio corresponds to a proportion close to the 35% of the Z isomer which has previously been proposed for photoisomerization of p-coumaric acid []. In addition, this result was identical to that derived from the integrated areas of the corresponding chromatogram peaks of each fraction and to that one obtained with the whole extract, which also confirms the instability of the chemical equilibrium in this conjugated system.
The characterized compounds were tested for their respective antioxidant activities using the inhibition of lipid peroxidation method, trapping of peroxyl radical assays and the ORAC-FL method and all determinations were performed on the F1 and F2 isomer pair. The capacity to trap peroxyl radicals determined by the chemiluminescence method and expressed in mmoles peroxyl radicals scavenged per gram of the compound was 0.18. The in vitro inhibition of lipid peroxidation, expressed as an IC50 value (i.e., amount of pure isomers that inhibit lipid peroxidation by 50%) of the mixture of phenyl disaccharide isomers was 12.3 nM. The values obtained from these two assays, which point out the antioxidative capacity of the compounds, are comparable to those of the reference (Trolox) and with the flavonoids tested from the Populus tremula acetone extract []. The antioxidant properties were within a similar range to that reported for some knotwood flavonoids [,].
The ORAC value expressed as μmol Trolox equivalents per μmol mixture of the two compounds was 11.7 ± 0.9 (R2=0.995), which was determined for an interval range of the compounds of 0.2 to 1.5 μM. This ORAC value reveals a very significant antioxidant capacity compared to other previously reported ORAC values for phenolic acids and flavonoids, [,,].
Conclusions
Two different fractions consisting of a novel equilibrium mixture of isomeric phenyl disaccharides was isolated from residual knotwood material. The mixture of E/Z forms has been identified using conventional techniques and these isomers readily interconvert during the tests or structure elucidation of the compounds. These structures are novel and rather unusual in the metabolic route for these species, as other glycosidic esters of p-coumaric acid known in literature. These compounds were tested for their antioxidant capacity by different methods in order to fully characterize this extract. The biological activity reported also appears to be consistent with a p-coumaric acid ester.
Experimental
General
Melting points were determined on a Büchi model B-540 melting point apparatus and are uncorrected. Optical rotations were recorded with a manual Zuzi 404 polarimeter. UV-VIS determinations were recorded in ethanol using CECIL, CE 2041, 2000 series Spectrophotometer. FTIR spectra were obtained using a Nicolet model 520P spectrophotometer with samples prepared as KBr pellets. HRMS analysis of the exact mass giving the elemental composition was determined with a Fisons ZabSpec high-resolution MS instrument. The samples were applied through direct insertion by a solid probe and the ionisation mode was EI at 40 eV, electron energy (resolution was 10.000). All NMR spectra were recorded on a Bruker AVANCE 500 spectrophotometer at a resonance frequency of 500.0 MHz for 1H and 125.0 MHz for 13C, with tetramethylsilane (TMS) as an internal reference and methanol-d4 as the solvent. 2D experiments (HSQC) were recorded using the Bruker standard pulse sequences.
Plant material and purification of compounds.
European aspen (Populus tremula) stemwood discs containing knots were sampled from several trees in southern Finland. Knotwood material processing, preparation of the acetone extract of Populus tremula knotwood and isolation of pure fractions of unknown compounds through preparative HPLC were performed as previously described [].
HPLC-DAD-MS and MS/MS analyses.
The dried fractions obtained from the acetone extract of Populus tremula knotwood by preparative-HPLC were dissolved in methanol and analysed on an analytical HPLC system consisting of an Agilent LC1100 series HPLC (Agilent Technologies, Inc.) equipped with a quaternary pump, thermostated autosampler, column oven (25 ºC) and a diode array detector, controlled by the Chemstation software. An Esquire 3000+ (Bruker Daltonics, GmbH, Germany) mass spectrometer was coupled to the HPLC instrument and equipped with an ESI source and an ion trap mass analyzer and controlled by the Esquire Control and Data Analysis software. The HPLC column was a Merck LiChrospher 100 RP-18, 5 μm, 250x4.0 mm (i.d). The mobile phase was: solvent A, 5 % HCOOH, and solvent B, MeOH. Two different solvent gradient programs were used. The first program, used for the analysis of the extract, consisted of three linear gradients: from 7% to 14% B in 2 min, from 14% to 17% B in 40 min and from 40% to 65% B to 70 min, followed by a isocratic step up to 73 min, returning after that in 7 minutes to the initial conditions. The second solvent program consisted of three linear gradients: from 7 to 14% B in 2 min, from 14 to 17% B to 40 min, and returning to the initial conditions from 40 to 42 min for column re-equilibration. The flow rate in both programs was 1 to 0.7 mL min-1 from 0 to 2 min, and than kept to 0.7 until 40 min, returning to the initial conditions after that. The separation was monitored at 280 and 314 nm, the characteristic wavelengths for polyphenolic (flavonoidic) type compounds in the aglyconic and glycosylated forms. The injection volume was 20 μL. The electrospray ionisation source was operated in negative mode to generate [M-H]- ions with the following conditions: desolvation temperature was set at 300ºC; dry gas (nitrogen) and nebulizer were set at 10 L min-1 and 60 psi respectively. Mass and MS/MS spectra of the purified compounds were acquired over 50-1000 amu and the capillary voltage was 4.0kV. The MS/MS fragmentation amplitude was 0.80 V, and the gas was helium.
(Z) and (E)-p-coumarates of 1-O-rutinose (F1 and F2): Molecular formula: C21O12H28; yellowish powder; mp 121-2°C; 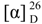 + 76.9º (EtOH, c 0.01), []. UV-vis λmax EtOH nm (log ε): 315 (4.45); IR, νmax cm-1: 3414 (-OH), 1712 (>C=O, unsaturated ester), 1631, 1604, 1515, 1450, 1380, 1260, 1204, 1170, 1067, 833; 1H-NMR: F2-E, δ 7.73 (1H, d, J=16Hz; H-3), 7.48 (2H, d, J=8Hz; H-5,-9), 6.81 (2H, d, J=8Hz; H-6,-8); 6.37 (1H, d, J=16Hz; H-2), 5.55 (1H, d, J=7.7Hz; H-1´), 4.72 (2H, multiplet s, H-1´´ in E/Z), 3.96-3.85 (4H, “multiplet” signals, H-6´ in E/Z), next overlapping signal for H-2´,-3´,-4´,-5´ and H-2”,-3”,-4”, 5” in E/Z isomers, 3.72- 3.63 (m), 3.55-3.54 (m), 3.49-3.31(m) total integration 16 H, and 1.26 ppm (6H, d, J=7.6 Hz, Me-Rha in E/Z); F1-Z, δ 7.71 (2H, d, J=8Hz; H-5,-9), 6.94 (1H, d, J=12Hz; H-3), 6.77 (2H, d, J=8Hz; H-6,-8), 5.84 (1H, d, J=12Hz; H-2), 5.52 (1H, d, J=8.2Hz; H-1´), 4.72 (2H, multiplet s, H-1´´ in E/Z), 3.96-3.85 (4H, “multiplet” signals, H-6´ in E/Z), next overlapping signal for H-2´,-3´,-4´,-5´ and H-2”,-3”,-4”, 5” in E/Z isomers, 3.72-3.63 (m), 3.55-3.54 (m), 3.49-3.31(m) total integration 16 H, and 1.26 ppm (6H, d, J=7.6 Hz, Me-Rha in E/Z), see Table 1; ESI/MS (negative mode) m/z 471.0 [M-H]- of F1 and F2; MS/MS, m/z (rel. int.) F1-(Z): 411 (21.4), 309.1 (5.2), 204.7 (3), 186.7 (12.7), 162.7 (100), 144.7 (11.2) and F2-(E): 411 (2.3), 204.7 (3), 186.8 (26), 162.7 (100), 144.7 (5). CG-MS TMSi, m/z obtained 904.3964 (calculated for C39H76O12Si6, 904.395249).
+ 76.9º (EtOH, c 0.01), []. UV-vis λmax EtOH nm (log ε): 315 (4.45); IR, νmax cm-1: 3414 (-OH), 1712 (>C=O, unsaturated ester), 1631, 1604, 1515, 1450, 1380, 1260, 1204, 1170, 1067, 833; 1H-NMR: F2-E, δ 7.73 (1H, d, J=16Hz; H-3), 7.48 (2H, d, J=8Hz; H-5,-9), 6.81 (2H, d, J=8Hz; H-6,-8); 6.37 (1H, d, J=16Hz; H-2), 5.55 (1H, d, J=7.7Hz; H-1´), 4.72 (2H, multiplet s, H-1´´ in E/Z), 3.96-3.85 (4H, “multiplet” signals, H-6´ in E/Z), next overlapping signal for H-2´,-3´,-4´,-5´ and H-2”,-3”,-4”, 5” in E/Z isomers, 3.72- 3.63 (m), 3.55-3.54 (m), 3.49-3.31(m) total integration 16 H, and 1.26 ppm (6H, d, J=7.6 Hz, Me-Rha in E/Z); F1-Z, δ 7.71 (2H, d, J=8Hz; H-5,-9), 6.94 (1H, d, J=12Hz; H-3), 6.77 (2H, d, J=8Hz; H-6,-8), 5.84 (1H, d, J=12Hz; H-2), 5.52 (1H, d, J=8.2Hz; H-1´), 4.72 (2H, multiplet s, H-1´´ in E/Z), 3.96-3.85 (4H, “multiplet” signals, H-6´ in E/Z), next overlapping signal for H-2´,-3´,-4´,-5´ and H-2”,-3”,-4”, 5” in E/Z isomers, 3.72-3.63 (m), 3.55-3.54 (m), 3.49-3.31(m) total integration 16 H, and 1.26 ppm (6H, d, J=7.6 Hz, Me-Rha in E/Z), see Table 1; ESI/MS (negative mode) m/z 471.0 [M-H]- of F1 and F2; MS/MS, m/z (rel. int.) F1-(Z): 411 (21.4), 309.1 (5.2), 204.7 (3), 186.7 (12.7), 162.7 (100), 144.7 (11.2) and F2-(E): 411 (2.3), 204.7 (3), 186.8 (26), 162.7 (100), 144.7 (5). CG-MS TMSi, m/z obtained 904.3964 (calculated for C39H76O12Si6, 904.395249).

Table 1.
NMR spectral data of the mixture of the two Z and E isomers of the p-coumarate of 1-O-rutinose
| δ in ppm*; 1H | Assignment | δ in ppm; 13C | Assignment | HSQC$ |
|---|---|---|---|---|
| 7.73 (d, 1H) | Ar-C
H=CH-CO2 Isomer (E) | 167.5; 166.4 | 2x C1; –CO2-Sugar | 2 x Q |
| 7.71 (d, 2H) | H5/H9 (Z) | |||
| 7.48 (d, 2H) | H5/H9 (E) | 161.5, 160.4 | 2xC7; p-coumaric | 2 x Q |
| 6.94 (d, 1H) | Ar-C
H=CH-CO2 Isomer (Z) | |||
| 6.81 (d, 2H);
6.77 (d, 2H) | H6/H8 (Z)/(E) | 148.0 | 2xC3; -CH=CH-CO2 | (E)-C3 |
| 147.1 | (Z)-C3 | |||
| 6.37 (d, 1H) | Ar-CH=C
H-CO2 Isomer (E) | 134.2 | C5/C9; in isomer (Z) of p-coumarin | (Z)-C5 |
| 5.84 (d, 1H) | Ar-CH=C
H-CO2 Isomer (Z) | 131.4 | C5/C9; in isomer (E) of p-coumarin | (E)-C5 |
| 5.55 (d, 1H);
5.52 (d, 1H) | H anomerics | 127.3; 127.0 | 2xC4; p-coumaroyl | 2 x Q |
| 4.72 (multiplet s, 2H) | H anomeric | 116.9; 115.9 | 2x C6/C8; p-coumaroyl | |
| 3.96 -3.85 (4H “multiplet” signals),
3.72-3.63 (m), 3.55-3.54 (m), 3.49-3.31(m), [total 16H]. | Glucosides | |||
| 115.4 | 2xC2 -CH=CH-CO2 | (Z)-C2 | ||
| 114.4 | (E)-C2 | |||
| 102.2 | C1´´ anomeric (Rha) | |||
| 95.7, 95.5 | 2 x C1´ anomeric (Glu) | |||
| 78.0, 77.9, 77.7, 74.0, 73.9, 72.3, 72.1, 71.1, 70.0, 69.9, 67.8, 67.7 | Glucosides
(Rha and Glu) | |||
| 1.26 (d, 6H) | Me-Rha | 18.0 | C6´´ (Rha) |
* In parentheses: multiplicity, integral of protons; $ HSQC correlation between 1H and 13C.
Antioxidative capacity determinations: Inhibition of lipid peroxidation assay.
The antioxidative capacity of the new compounds (in a mixture of two isomers) was measured as the capability to inhibit tert-butylhydroperoxide (t-BuOOH) induced lipid peroxidation in rat liver microsomes in vitro [].The lipid peroxidation was detected by luminol-enhanced chemiluminescence. The compound was added in a small volume to the incubation mixture (10 μL) and the lipid peroxidation potency was compared to that of the vehicle (solvent). The inhibition of lipid peroxidation assay was performed as following: buffer (50 mM sodium carbonate, pH 10.2, with 0.1 mM EDTA, 800 μL) was mixed with diluted rat liver microsomes (20 μL, final concentration 1.5 Kg protein/mL) in the luminometer cuvette. luminol (0.5 mg/mL, 10 μL) was added, followed by the test compounds (10 μL). The reaction was initiated by 0.9 mM t-BuOOH (0.05 mL) at 33oC. The chemiluminescence in two cuvettes was measured for 45 min at 1 min cycles.
Trapping of peroxyl radicals assay.
The capacity of the mixture of isomers to trap peroxyl radicals was assessed using a methodology based on chemiluminescence []. Peroxyl radicals were generated by thermal decomposition of 2,2’-azino-bis(2-amidinopropane) hydrochloride (ABAP) as follows: sodium phosphate buffer (0.45 mL, pH 7.4, containing 0.9% NaCl), 120 mM linoleic acid (0.02 mL) luminol (0.05 mL, 0.5 mg/mL) and test compound were mixed in the cuvettes. The assay was initiated by ABAP (50 μL, 83 mg/mL). The chemiluminescence was measured at 37oC, until a peak value was detected. The peroxyl radical trapping capacity is defined by the half-time peak point.
ORAC (oxygen radical absorbance capacity) assay.
In order to determine the capacity of the mixture of isomers to scavenge peroxyl radicals the ORAC method [] was used, with slight modification, and using fluorescein (FL) as the fluorescent probe (ORAC-FL). The pure compound was studied within the 0.2-1.5 μM concentration range, and prepared for the ORAC analysis, after appropriate dilution with 75 mM phosphate buffer solution at pH 7.4. The ORAC assay was carried out on a Fluostar Galaxy BMG Lab spectrofluorometric analyzer equipped with an incubator; the temperature of the incubator was set to 37 ºC. Fluorescence filters with an excitation wavelength of 485 nm and an emission wavelength of 520 nm were used. All the experiments were performed in 96 well black microplates. The reaction had the total final volume 200 μL, containing a mixture of FL (150 μL, 4nM), AAPH [2,2-azo-bis-(2-methylpropionamidine) dihydrochloride, 25 μL, 19 mM] and sample (25 μL). Several dilutions of Trolox (1-20 KM) were used in order to build the calibration curves. A freshly prepared AAPH solution was used for each experiment. Between two and three independent experiments by triplicate were performed. ORAC value was expressed as Trolox equivalents (μM Trolox/μM antioxidant, or μM Trolox/g antioxidant).
Acknowledgments
This work was supported by a research fellowship from Institute of Molecular and Cellular Biology (UMH, Elche, Spain) to M.N. Part of the results were obtained during a European Commission “Marie Curie Training Site’’ project (HPMT-CT-2001-00297) at the Åbo Akademi University Process Chemistry Centre, Finland. This investigation has also been partially supported by Grants AGL2004-06469/ALI and MAT-2005-1004 from MEC. J. Hemming, M. Reunanen, S. Pietarinen and M. Ahotupa are acknowledged for their help.
References
- Heuser, E; Broetz, A. The chemical nature of deciduos trees. Papierfabrikant 1925, 23, 69–88. [Google Scholar]
- Pearl, I.A. Water Extractives of American Populus Pulpwood Species Barks – a Review. Tappi 1969, 52, 428–431. [Google Scholar]
- Pearl, I.A.; Harrocks, J.A. Studies on Chemistry of Aspenwood. 10. Neutral Materials from Benzene Extractives of Populus Tremuloides. J. Org. Chem. 1961, 26, 1578–1583. [Google Scholar]
- Pietarinen, S.P.; Willför, S.M.; Sjöholm, R.E.; Holmbom, B.R. Bioactive phenolic substances in important tree species. Part 3: Knots and stemwood of Acacia crassicarpa and A-mangium. Holzforschung 2005, 59, 94–101. [Google Scholar]
- Neacsu, M.; Eklund, P.; Sjöholm, R.; Pietarinen, S.; Ahotupa, M.; Holmbom, B.; Willför, S.M. Antioxidant flavonoids from knotwood of Jack pine and European aspen. Holz Roh- Werks. 2007, 65, 1–6. [Google Scholar]
- Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Reunanen, M.H.; Eklund, P.C.; Sjöholm, R.E.; Eckerman, C.S.; Pohjamo, S.P.; Holmbom, B.R. Antioxidant activity of knotwood extractives and phenolic compounds of selected tree species. J. Agr. Food Chem. 2003, 51, 7600–7606. [Google Scholar]
- Holmbom, B.R.; Eckerman, C.; Eklund, P.; Hemming, J.; Nisula, L.; Reunanen, M.; Sjöholm, R.E.; Sundberg, A.; Sundberg, K.; Willför, S.M. Knots in trees: A new rich source of lignans. Phytochem. Rev. 2004, 2, 331–340. [Google Scholar]
- Baderschneider, B.; Winterhalter, P. Isolation and characterization of novel benzoates, cinnamates, flavonoids, and lignans from Riesling wine and screening for antioxidant activity. J. Agr. Food Chem. 2001, 49, 2788–2798. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteric Group Frequencies; John Wiley & Sons: Chichester, U.K, 2004. [Google Scholar]
- Yi, J.H.; Zhang, G.L.; Li, B.G.; Chen, Y.Z. Phenylpropanoid glycosides from Lamiophlomis rotata. Phytochemistry 1999, 51, 825–828. [Google Scholar] [CrossRef]
- Sun, X.F.; Sun, R.C.; Fowler, P.; Baird, M.S. Physicochemical characterization of liginin isolated with high yield purity from wheat straw. Int. J. Polym. Anal. Ch. 2004, 9, 317–337. [Google Scholar] [CrossRef]
- McNally, D.J.; Wurms, K.V.; Labbe, C.; Quideau, S.; Belanger, R.R. Complex C-glycosyl flavonoid phytoalexins from Cucumis sativus. Journal of Natural Products 2003, 66, 1280–1283. [Google Scholar] [CrossRef]
- Mun, S.P.; Ku, C.S. Characterization of low molecular weight polyphenols from pine (Pinus radiata) bark. Food Sci. Biotech. 2006, 15, 424–430. [Google Scholar]
- Cogne, A.L.; Queiroz, E.F.; Wolfender, J.L.; Marston, A.; Mavi, S.; Hostettmann, K. On-line identification of unstable catalpol derivatives from Jamesbrittenia fodina by LC-MS and LC-NMR. Phytochem. Anal. 2003, 14, 67–73. [Google Scholar] [CrossRef]
- Bergman, M.; Varshavsky, L.; Gottlieb, H.E.; Grossman, S. The antioxidant activity of aqueous spinach extract: chemical identification of active fractions. Phytochemistry 2001, 58, 143–152. [Google Scholar] [CrossRef]
- Bedir, E.; Tatli, I.I.; Khan, R.A.; Zhao, J.P.; Takamatsu, S.; Walker, L.A.; Goldman, P.; Khan, I.A. Biologically active secondary metabolites from Ginkgo biloba. J. Agr. Food Chem. 2002, 50, 3150–3155. [Google Scholar]
- Norbaek, R.; Nielsen, J.K.; Kondo, T. Flavonoids from flowers of two Crocus chrysanthus-biflorus cultivars: "Eye-catcher" and "Spring Pearl" (Iridaceae). Phytochemistry 1999, 51, 1139–1146. [Google Scholar] [CrossRef]
- Davalos, A.; Gomez-Cordoves, C.; Bartolome, B. Extending applicability of the oxygen radical absorbance capacity (ORAC-fluorescein) assay. J. Agr. Food Chem. 2004, 52, 48–54. [Google Scholar] [CrossRef]
- Ou, B.X.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agr. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Villano, D.; Fernandez-Pachon, M.S.; Troncoso, A.M.; Garcia-Parrilla, M.C. Comparison of antioxidant activity of wine phenolic compounds and metabolites in vitro. Anal. Chim. Acta 2005, 538, 391–398. [Google Scholar] [CrossRef]
- Li, Y.M.; Jiang, S.H.; Gao, W.Y.; Zhu, D.Y. Phenylpropanoid glycosides from Scrophularia ningpoensis. Phytochemistry 2000, 54, 923–925. [Google Scholar] [CrossRef]
- Ahotupa, M.; Mantyla, E.; Kangas, L. Antioxidant properties of the triphenylethylene antiestrogen drug toremifene. Naunyn-Schmiedebergs Arch. Pharmacol. 1997, 356, 297–302. [Google Scholar]
- Sample availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.