Cyclization-activated Prodrugs


1
Centro de Investigação em Química da Universidade do Porto, Departamento de Química, Faculdade de Ciências, Universidade do Porto, Porto, Portugal
2
Centro de Estudos de Ciências Farmacêuticas, Faculdade de Farmácia, Universidade de Lisboa, Lisboa, Portugal
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(11), 2484-2506; https://doi.org/10.3390/12112484
Submission received: 6 October 2007
/
Revised: 8 November 2007
/
Accepted: 9 November 2007
/
Published: 12 November 2007
(This article belongs to the Special Issue Prodrugs)
Abstract
:Many drugs suffer from an extensive first-pass metabolism leading to drug inactivation and/or production of toxic metabolites, which makes them attractive targets for prodrug design. The classical prodrug approach, which involves enzyme-sensitive covalent linkage between the parent drug and a carrier moiety, is a well established strategy to overcome bioavailability/toxicity issues. However, the development of prodrugs that can regenerate the parent drug through non-enzymatic pathways has emerged as an alternative approach in which prodrug activation is not influenced by inter- and intraindividual variability that affects enzymatic activity. Cyclization-activated prodrugs have been capturing the attention of medicinal chemists since the middle-1980s, and reached maturity in prodrug design in the late 1990s. Many different strategies have been exploited in recent years concerning the development of intramoleculary-activated prodrugs spanning from analgesics to anti-HIV therapeutic agents. Intramolecular pathways have also a key role in two-step prodrug activation, where an initial enzymatic cleavage step is followed by a cyclization-elimination reaction that releases the active drug. This work is a brief overview of research on cyclization-activated prodrugs from the last two decades.
1. Introduction
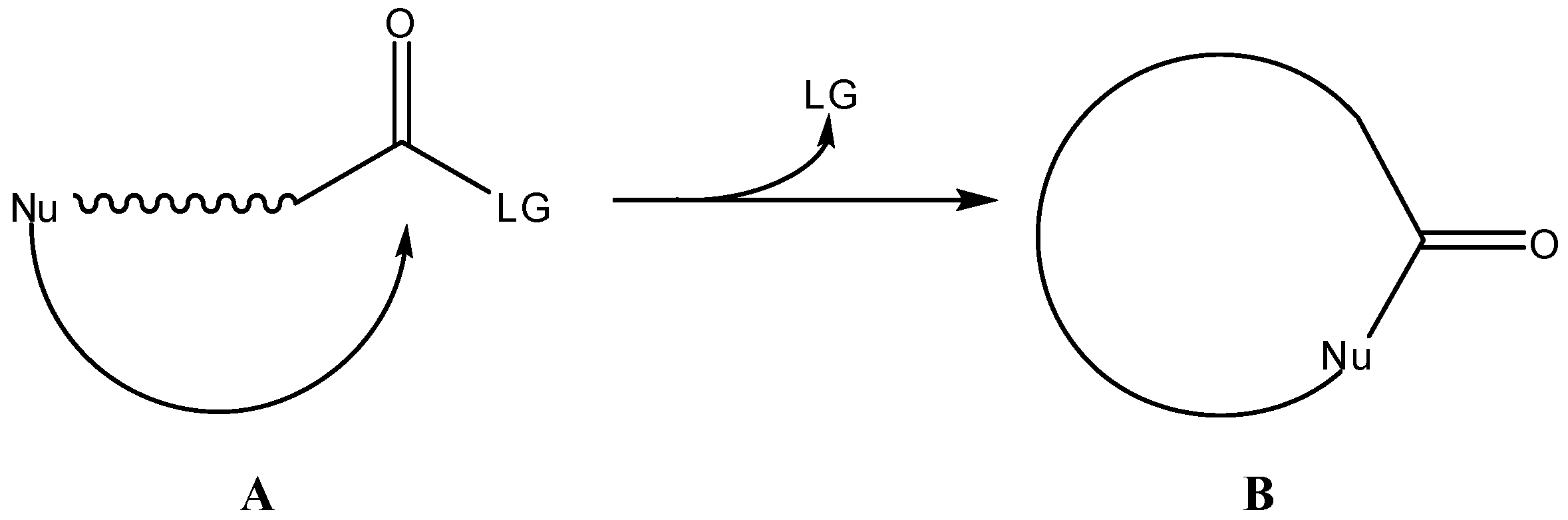
Many therapeutically active agents experience low bioavailability after oral administration due to poor absorption or susceptibility to first pass metabolism [1]. The prodrug approach has been widely used to improve delivery of drugs to their site of action by modulation of physico-chemical properties that affect absorption or by targeting to specific enzymes or membrane transporters [2,3,4,5]. Most of the prodrugs that are now in clinical use require enzymatic catalysis in order to be converted into the parent drug. This is particularly true for those prodrugs designed to liberate the parent drug in the blood stream following gastro-intestinal absorption: they are typically ester derivatives of drugs containing carboxyl or hydroxyl groups, which are readily converted into the parent drug by esterase-catalyzed hydrolysis [5]. However, applying enzymatic activation for other functional groups may result in high chemical reactivity that precludes either liquid or solid formulation of the prodrug (e.g. some phenol esters) or low chemical reactivity, resulting in reduced regeneration of the parent drug (e.g. amide derivatives of amine-containing drugs). Thus, the development of prodrugs that can regenerate the parent drug through non-enzymatic pathways has emerged as an alternative approach in which prodrug activation is not influenced by inter- and intra-individual variability that affects the enzymatic activity. In particular, cyclization-activated prodrugs have been capturing the attention of medicinal chemists since the middle-1980s, and reached maturity in prodrug design in the late 1990s. Activation of prodrugs via a cyclization pathway allows a fine tuning of the rate of drug release through the appropriate choice of the functional groups involved in ring closure and stereoelectronic constraints in the course of the cyclization step. Many different strategies have been exploited in recent years concerning the development of intramoleculary-activated prodrugs. These can be summarized as follows: (i) the cyclization reaction is used to release the active drug as the cyclization product (the drug is the cyclic species formed, B in Scheme 1), (ii) the cyclization involves the elimination of the parent drug (the drug is a leaving group, LG in Scheme 1, in the process of cyclization of the carrier moiety) and (iii) the cyclization is preceded by an enzymatic reaction that generates the internal nucleophile (also called two-step activation). Intramolecular activation or cyclization-elimination strategies for drug release from prodrugs has been almost exclusively based on the attack of nitrogen- (amino, amido) or oxygen- (hydroxyl, carboxylate) nucleophiles over a carbonyl moiety, as depicted in Scheme 1. The present work is a brief overview of research on cyclization-activated prodrugs over the past two decades. Reviews addressing this particular issue have previously been published by Shan et al. [6], Testa & Mayer [7] and by Wang et al. [8], in the late 1990s, and more recently by Vinšová and Imramovský [9]. Another interesting review on anticancer prodrugs selectively activated by elimination and cyclization pathways was published by Papot et al. [10] in 2002. Also, a commentary by Testa on prodrug research [11] is a key reference for those working on, or interested in, this field. Finally, a most recent and extensive description of prodrug development, including a chapter on cyclization-elimination strategies for prodrug activation, can be found in [12].
Scheme 1.
Intramolecular cyclization-elimination reaction for activation of a general prodrug A.
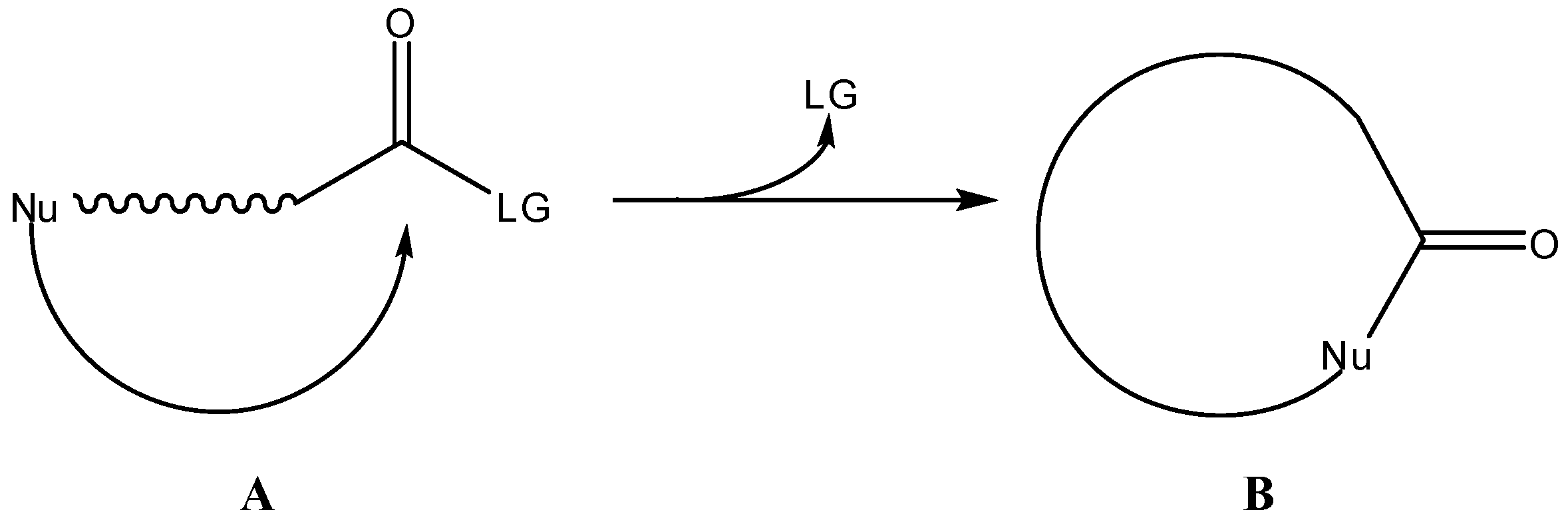
2. Active Drug as the Cyclic Product of Intramolecular Activation
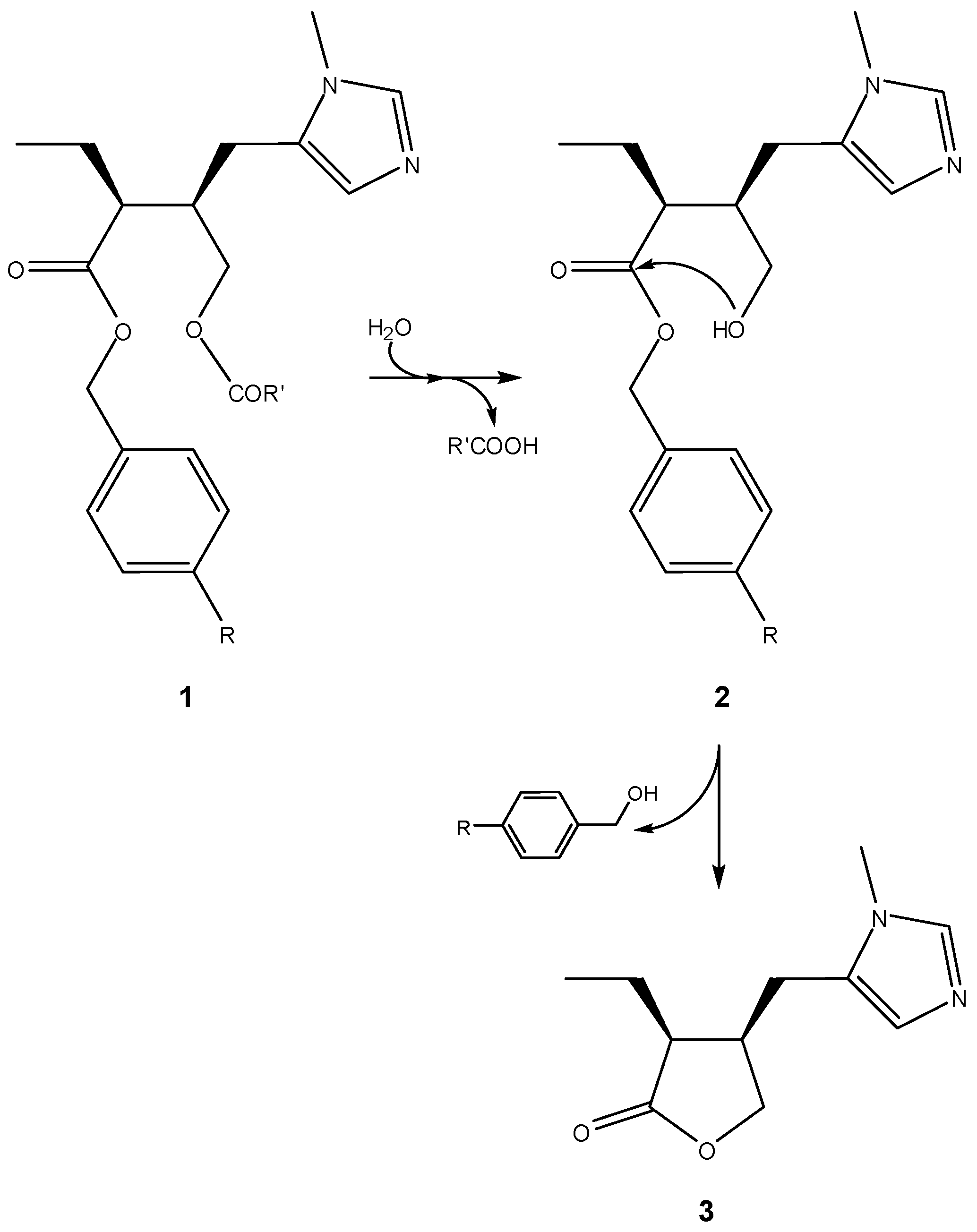
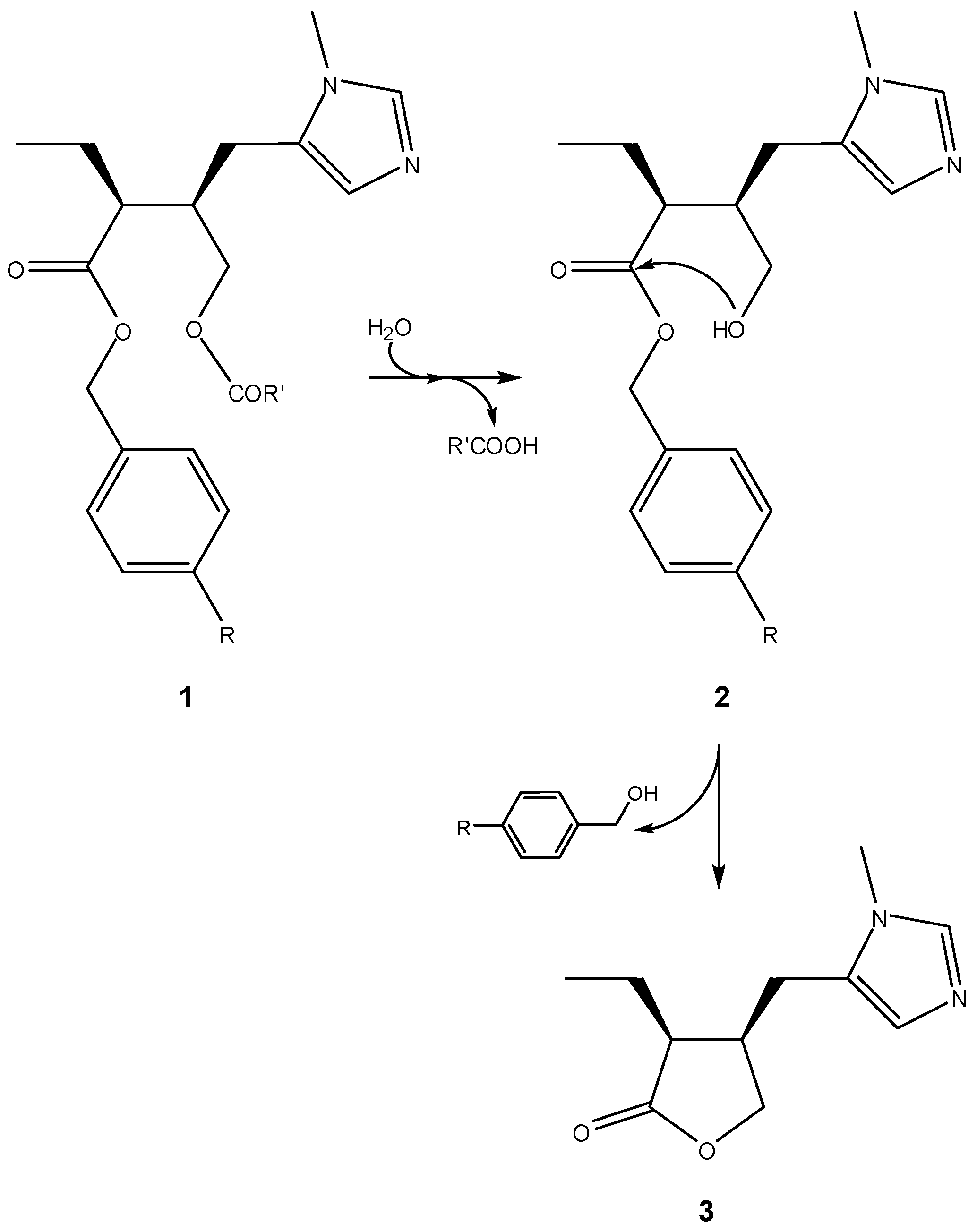
A classical example of this approach can be traced back to 1985, when Bundgaard and co-workers proposed pilocarpine prodrugs based on pilocarpic acid double esters 1 [13]. These were shown to work as prodrugs of pilocarpine both in vitro and in vivo and, in aqueous solution, to undergo a quantitative and apparently specific-base-catalyzed lactonization to pilocarpine (3). This process was based on an initial ester hydrolysis step that leaves a hydroxyl nucleophile free to attack the benzyl ester moiety (2), thus promoting the final cyclization-elimination reaction (Scheme 2) [13].
Scheme 2.
Activation of pilocarpic acid double esters 1 to release pilocarpine (3) [13].
Scheme 2.
Activation of pilocarpic acid double esters 1 to release pilocarpine (3) [13].
Another case where the intramolecular cyclization product is the active drug involves the α-amino acid prodrugs of camphothecins proposed by Song et al. [14]. These authors based their proposal on previous findings about the high lactone stability in human blood [15]. Thus, linear carboxylate precursors could be used as pro-moieties of the active lactone that should be formed in vivo through nucleophilic attack of the hydroxyl to the carboxylate group. In fact, the ester prodrugs undergo quantitative conversion to their pharmacologically active lactones via a non-enzymatic cyclization mechanism that is favored over direct hydrolysis at pH 7.4 [14]. The authors further proposed that the observed pH dependence of the non-enzymatic pathway for activation of the prodrugs suggests that these may be useful for tumor-targeting via liposomes, as they can be stabilized in an acidic environment in the core of liposomes and readily converted into the active lactone following their intramural release [14].
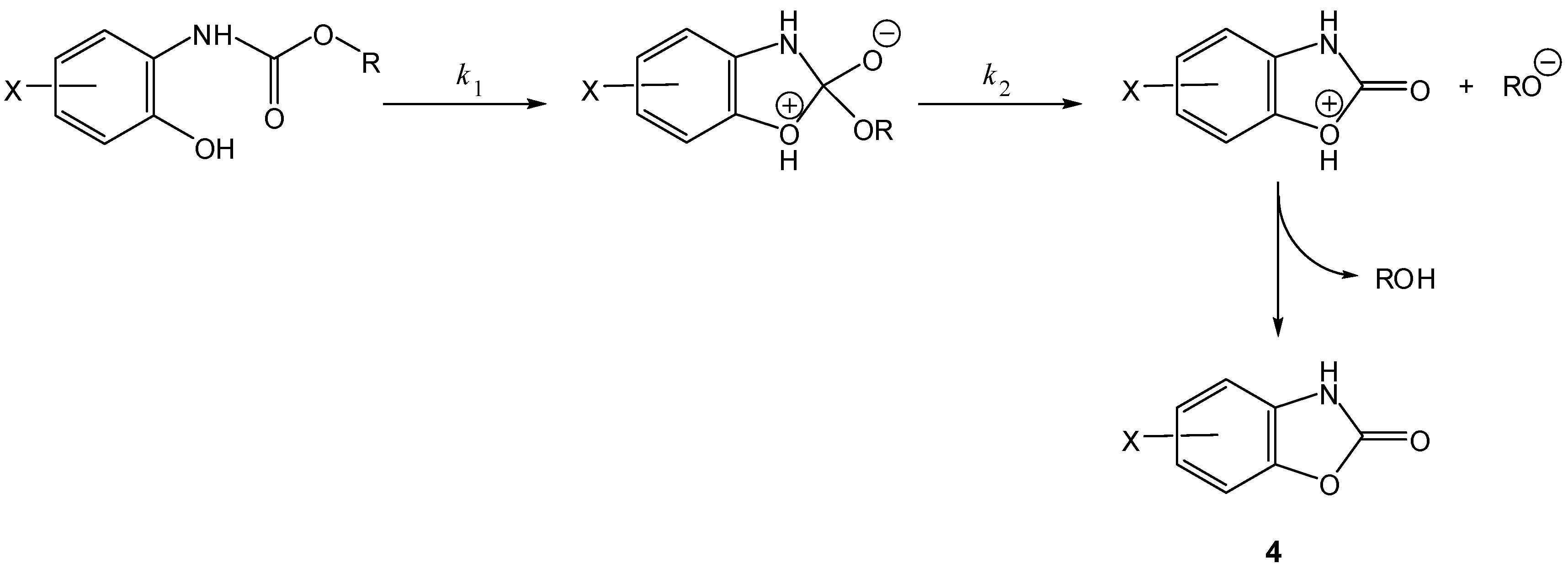
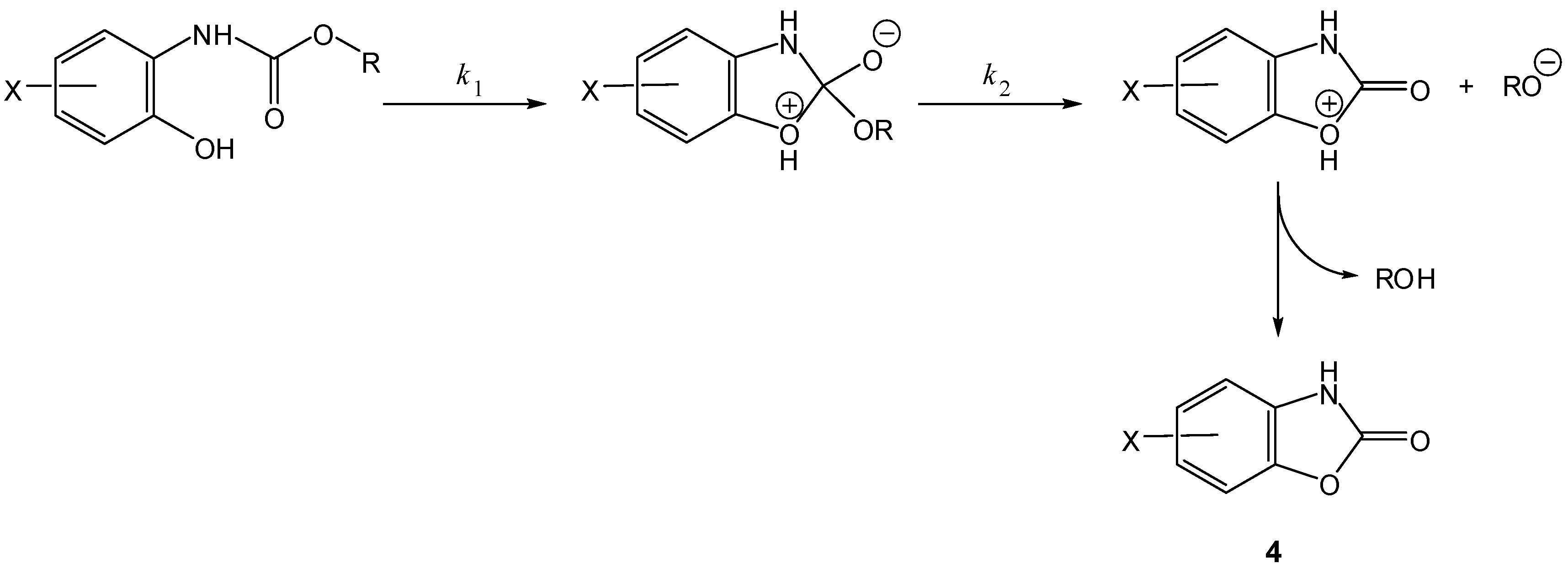
Scheme 3.
Intramolecular activation of prodrugs for active benzoxazolones (4) [16].
Scheme 3.
Intramolecular activation of prodrugs for active benzoxazolones (4) [16].
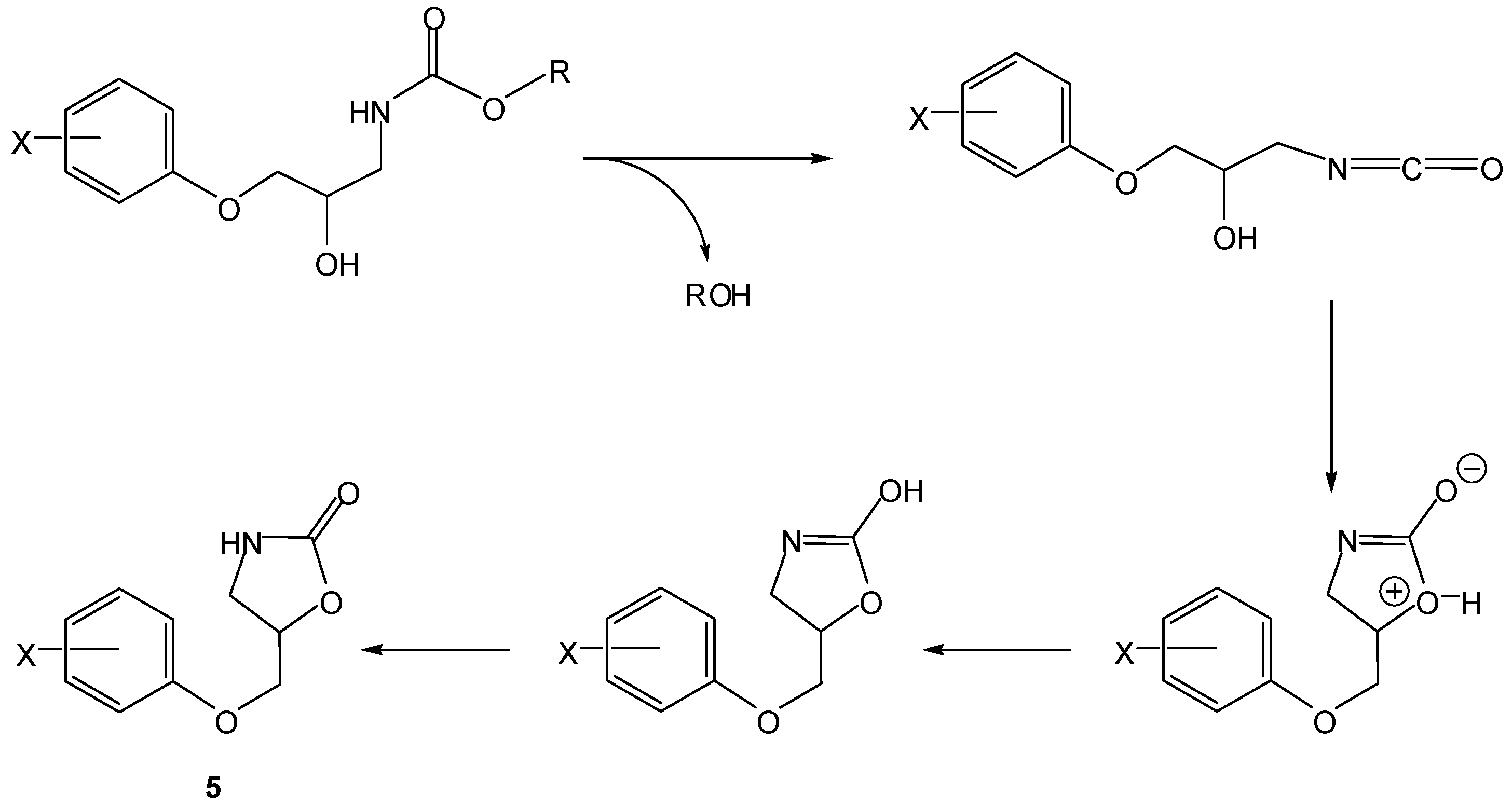
Scheme 4.
Intramolecular activation of prodrugs for active oxazolidinones (5) [16].
Scheme 4.
Intramolecular activation of prodrugs for active oxazolidinones (5) [16].
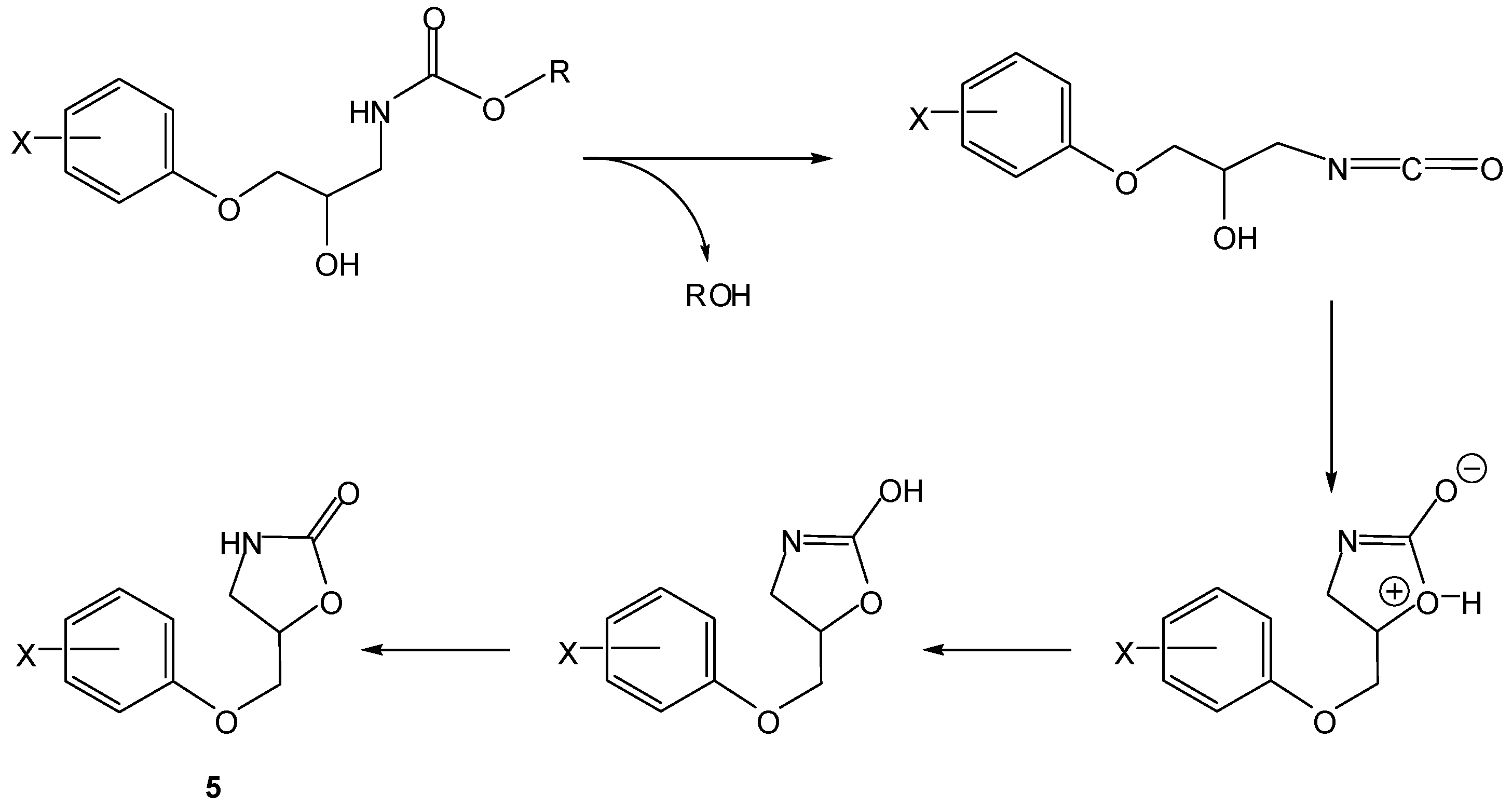
In the 1990s, Vigroux and co-workers demonstrated, both in aqueous buffer and in plasma, the efficiency of novel drug delivery systems for active benzoxazolones (4, Scheme 3) and oxazolidinones (5, Scheme 4). In the first case, one of the benzoxazolone precursors prepared was the 4-acetamido-phenyl ester chlorzacetamol, which is a mutual prodrug of chlorzoxazone and paracetamol (=ROH). Similarly, the second approach included two mutual prodrugs of paracetamol and active oxazolidinones (metaxalone and mephenoxalone) that were obtained using the appropriate amines [16]. All the carbamate prodrugs thus prepared were found to release the parent drugs in aqueous buffer (pH 6-11) and plasma (pH 7.4) through intramolecular reactions due to a hydroxyl nucleophile. Benzoxazolone release occurred by a cyclization mechanism involving a change in the rate-limiting step from formation of a cyclic tetrahedral intermediate (Scheme 3, k1) to departure of the leaving group ROH (Scheme 3, k2) when the leaving group ability decreased. However, oxazolidinones were released from their mutual prodrugs by means of a rate-limiting elimination-addition reaction (Scheme 4) [16].
3. Active Drug as the Leaving Group in the Cyclization-Elimination Reaction
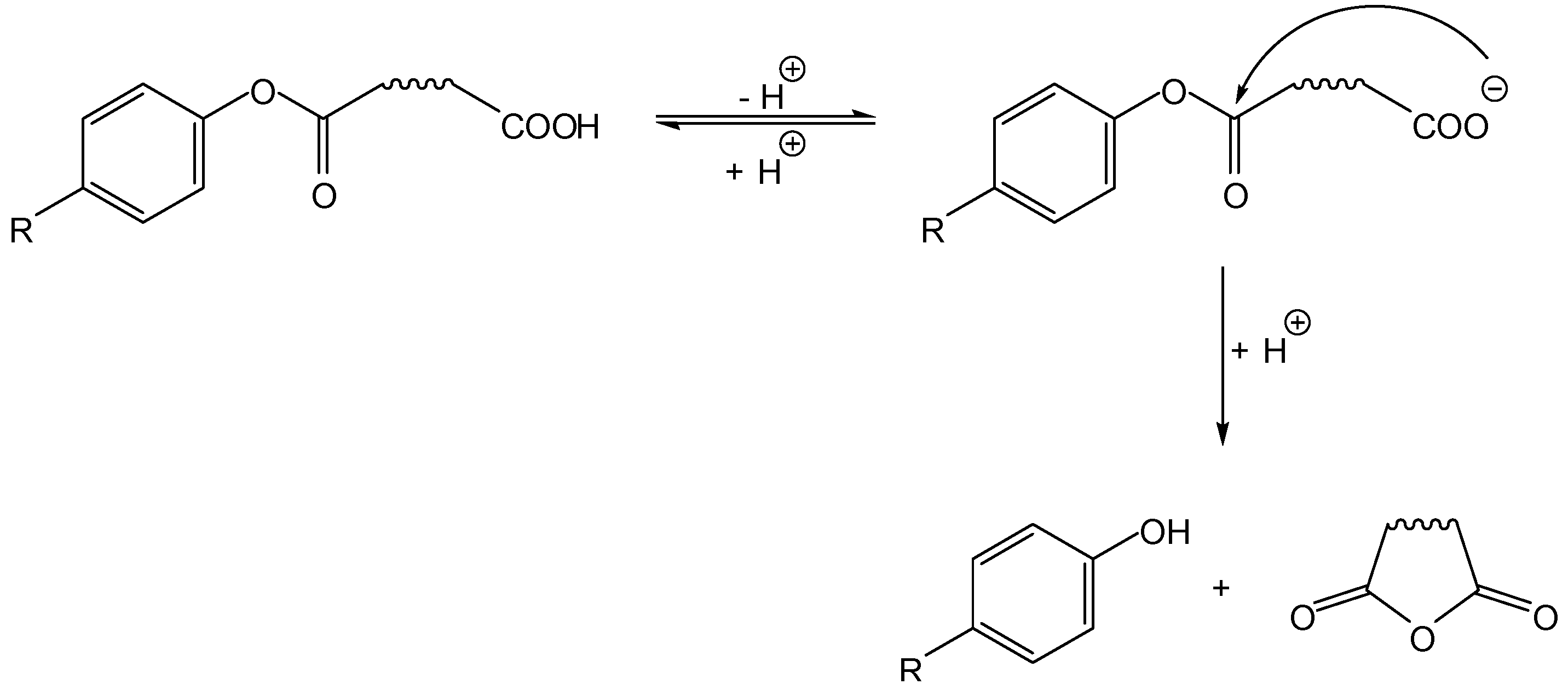
Most intramolecularly activated prodrugs proposed in the literature fall in this category, i.e., the active drug is the leaving group (LG in Scheme 1) in the cyclization-elimination process. Of these, the vast majority involves a nitrogen nucleophile (acidic amide or basic amine), so only such cases will be dealt with in the following sections. Notwithstanding, oxygen-based nucleophiles have a role, and a brief reference to a classical example seems worthwhile: Begtrup and co-workers proposed, in 1995, that besides hydroxyl nucleophiles (illustrated on Scheme 2, Scheme 3, Scheme 4 and Scheme 5), carboxylate groups could also be used for identical purposes. Thus, these authors have developed hemiesters of aliphatic dicarboxylic acids as cyclization-activated prodrugs of phenols that were released after attack of the carboxylate on the ester bond (Scheme 5) [17].
Scheme 5.
Carboxylate nucleophiles in the intramolecular activation of phenol prodrugs [17].
Scheme 5.
Carboxylate nucleophiles in the intramolecular activation of phenol prodrugs [17].
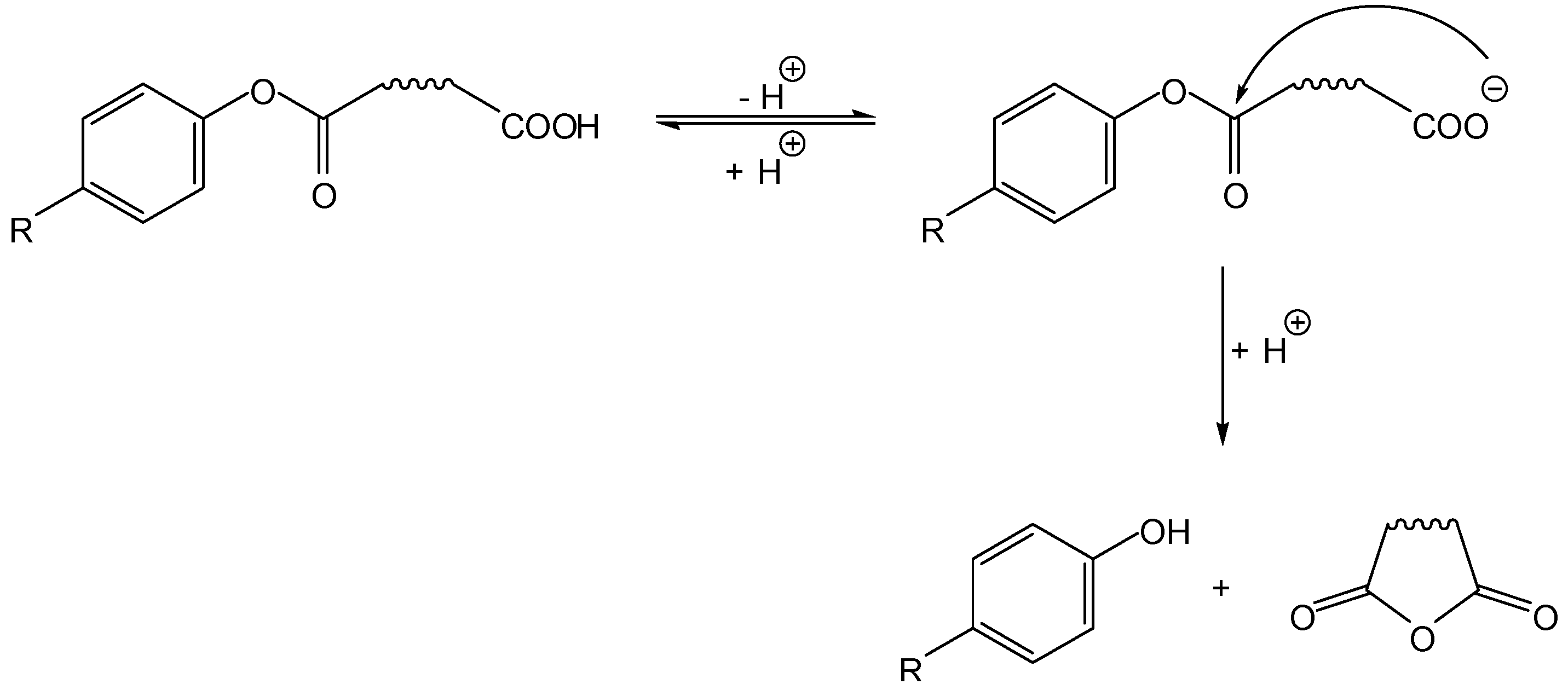
Besides the pKa of the phenolic group, other factors influenced the reactivity of the hemiesters under physiological conditions (t1/2 ranging from 1 to 350 min, at pH 7.4 and 37 °C). Hence, the size of the anhydride formed (i.e., the length of the aliphatic chain represented by the wavy line in Scheme 5) was relevant, as succinate esters reacted 133- and 151-times faster than their glutarate counterparts, which is explained by the greater proximity between the reacting groups in the former. Also, an increasing number of methyl substituents hanging on the aliphatic chain increased the rate of intramolecular cyclization, a phenomenon that has been previously attributed to a decrease in unprofitable (unreactive) rotamer distribution in the ground state [18].
3.1. Cyclization-elimination involving an amido group
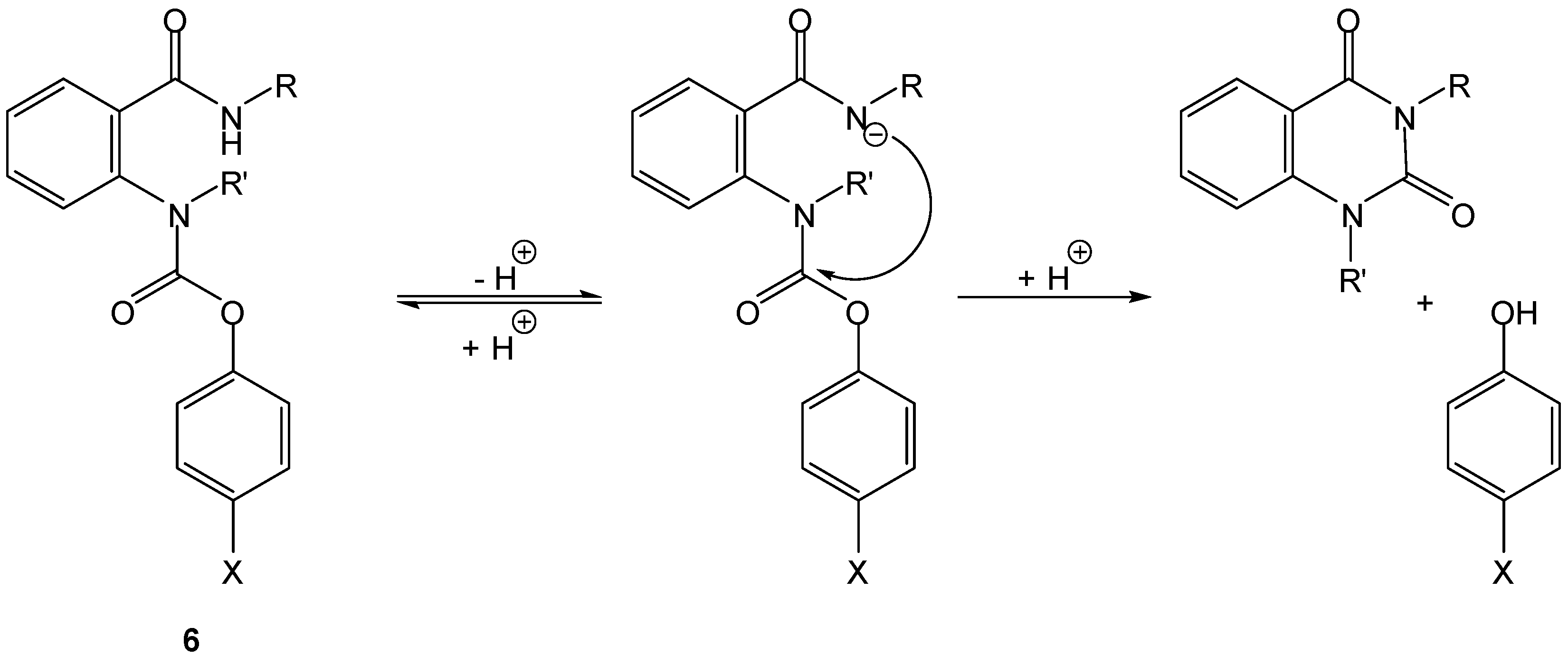
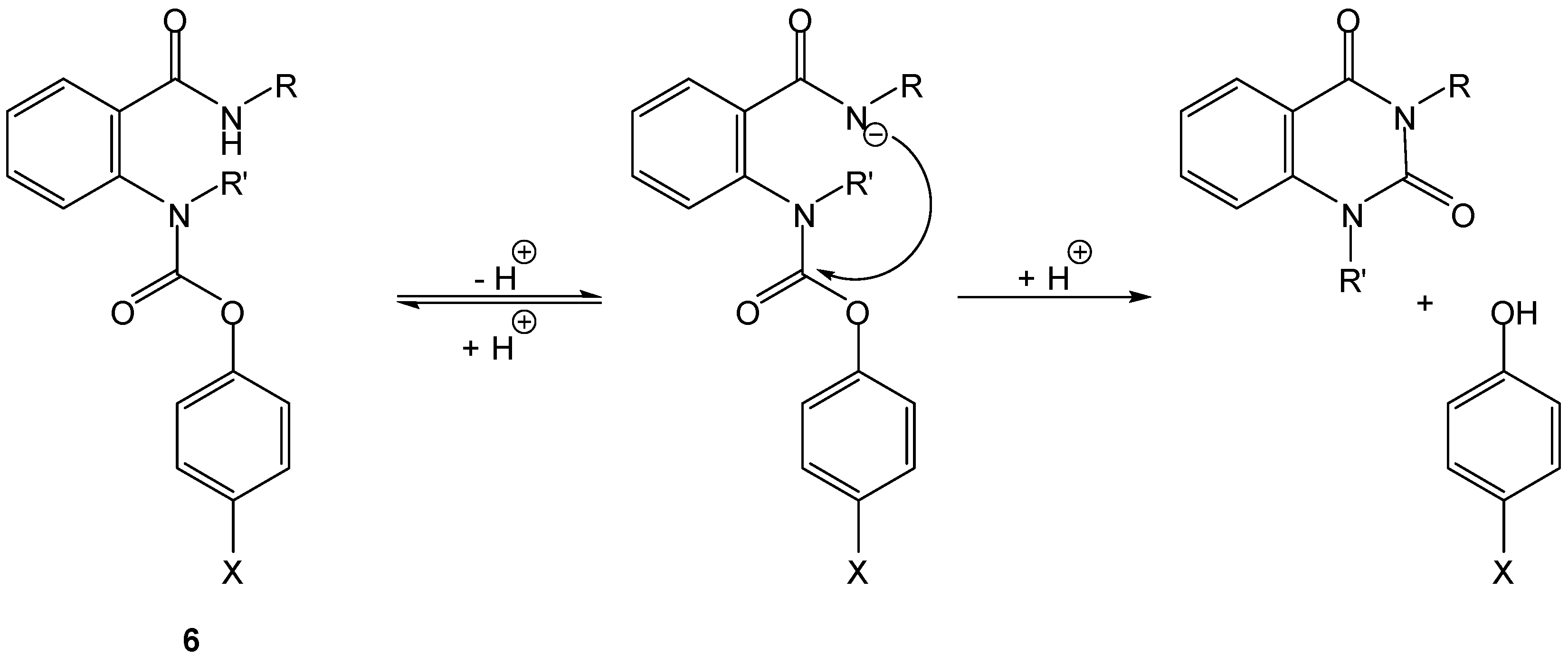
Acidic amides can supply a nucleophilic nitrogen to promote intramolecular prodrug activation. Thomsen and Bundgaard were pioneers in this approach, through preparation and study of cyclization-activated phenyl carbamate prodrugs of phenols, 6 [19]. Compounds 6, as in the case of Begtrup’s hemiesters of aliphatic dicarboxylic acids, were designed as models to test the protection of phenolic drugs from first-pass metabolism. Though highly stable at pH 1 to 6, these compounds underwent an apparent specific base-catalyzed cyclization at higher pH values (Scheme 6) leading to phenol release. The rate of cyclization was not affected by enzymes and was modulated by the pKa of the phenol and by nature and size of the carboxamido substituent (t1/2 ranging from 10 to 60 min, at pH 7.4 and 37 °C) [19].
Scheme 6.
Intramolecular activation of phenyl carbamate prodrugs (6) of phenols [19].
Scheme 6.
Intramolecular activation of phenyl carbamate prodrugs (6) of phenols [19].
More recent reports by Sohma and co-workers describe the synthesis and characterization of water-soluble prodrugs of HIV-1 protease inhibitors [20,21]. Thus, water-soluble prodrugs of KNI-727 (7a-b, 8a-c, Figure 1) were designed to release the parent drug through intramolecular activation promoted by an acidic amide nucleophile [20].
Figure 1.
Intramolecular activation of water-soluble prodrugs of KNI-727 [21].
Figure 1.
Intramolecular activation of water-soluble prodrugs of KNI-727 [21].
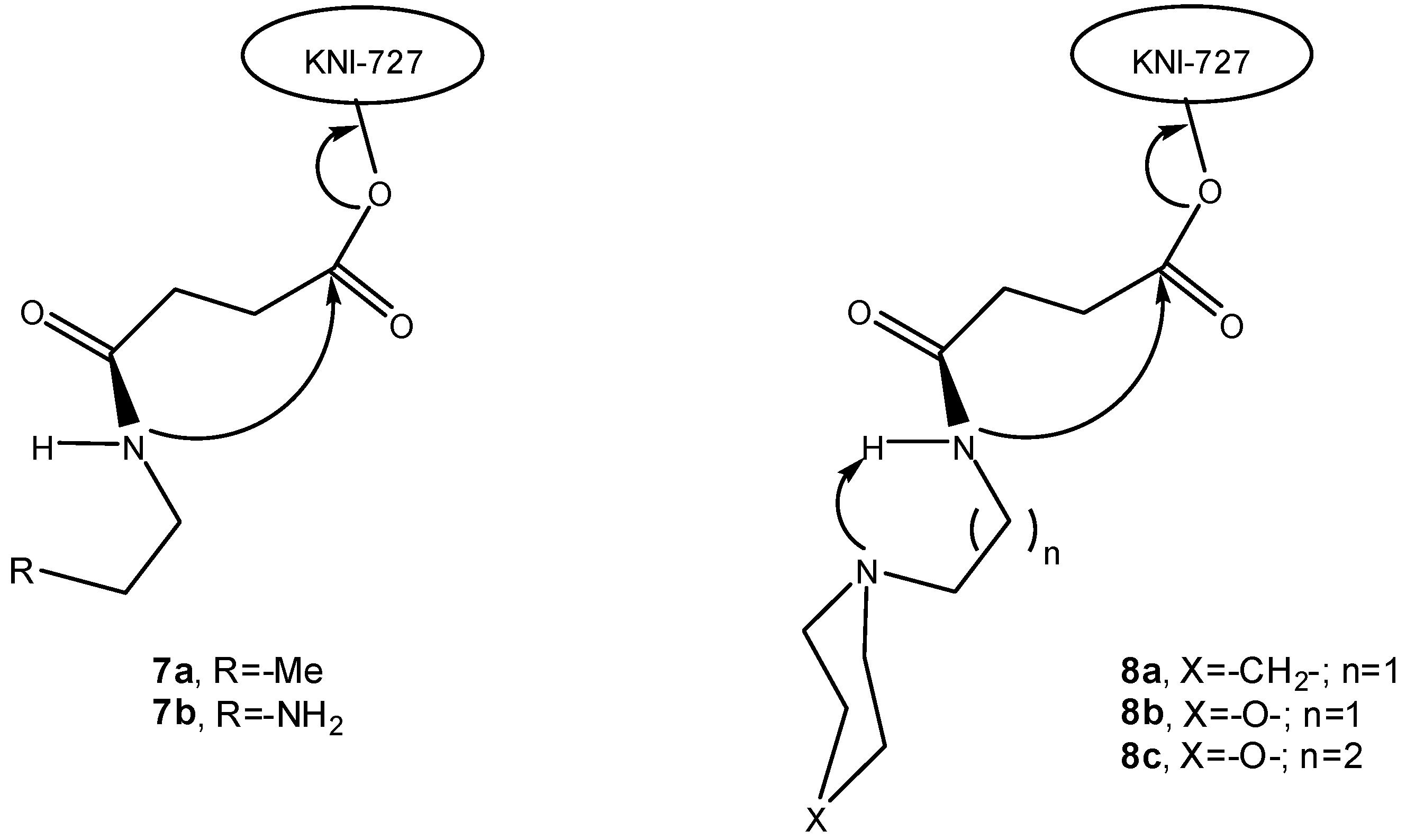
These water-soluble prodrugs were found to release the parent drug exclusively through the non-enzymatic process under physiological conditions, by cleavage at the spacer-drug bond via an intramolecular cyclization-elimination reaction involving imide formation. This work yielded a series of water-soluble KNI-727 prodrugs and diversified drug-release rates in vitro (half-lives ranging from 4 min to 34.3 h) that were developed by modifications focused at the chemical structure of the solubilizing/carrier moiety [21]. Thus, the introduction of sterically hindered cyclic tertiary amines with different electronic states (8a-c, Figure 1) accelerated the conversion, probably because the bulky cyclic structures forced the tertiary amine to be confined to a conformation suitable for proton abstraction on the neighbouring amide, rather than affording steric hindrance [21]. Prodrug 8b presented a 10-fold increase in t1/2 as compared to compound 8a, which can be explained by the electron-inductive effect exerted by the oxygen of the morpholine structure. Moreover, the propyl-morpholine derivative (8c) displayed a further increased t1/2, due to the additional methylene inserted between the tertiary amine and the amide proton [21]. The same research group also investigated a series of linkers connecting the hydroxyl group of KNI-727 to AZT. Under mild alkaline conditions, these mutual prodrugs were seen to spontaneously release KNI-727 also via an intramolecular cyclization through imide formation [22].
3.2. Cyclization-elimination involving an amino group
As previously mentioned, cyclization-elimination is the most well studied pathway in cyclization-activated prodrugs, where the leaving group is the active drug. This is particularly true when the carriers contain a terminal nucleophilic amino group so that drug release would occur through amino-mediated intramolecular cyclative cleavage of the drug-carrier bond, i.e., intramolecular aminolysis. Peptides have also been used/proposed as carriers to provide the basic amino group for such purpose [11,12,23,24].
3.2.1. General basic amine carriers
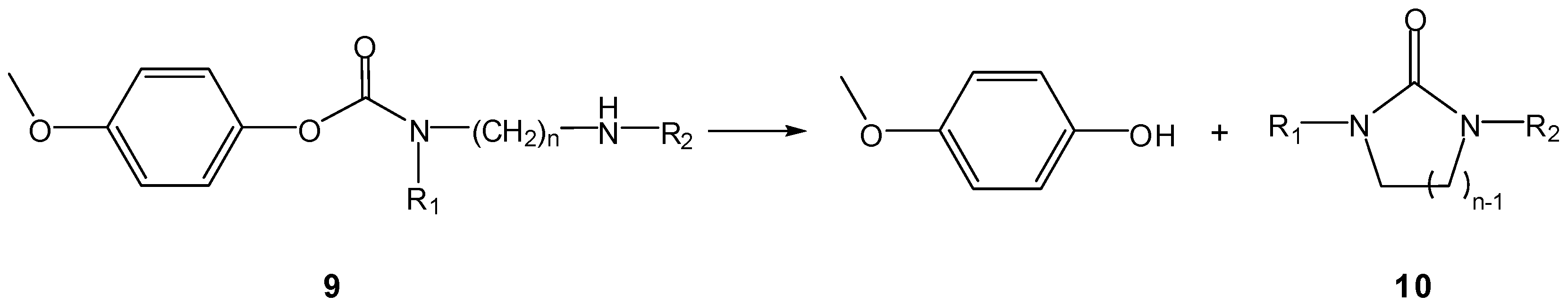
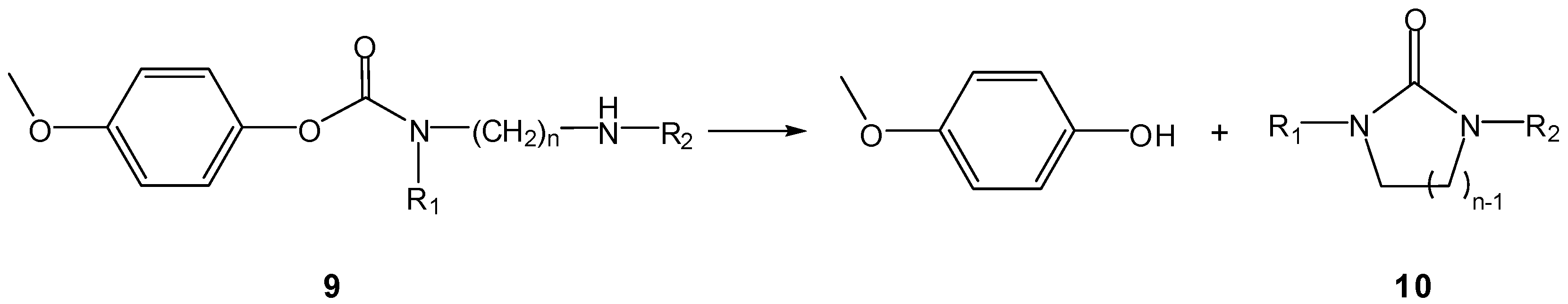
A classical example is that of aminocarbamate prodrugs 9 of 4-hydroxyanisole, prepared by Saari et al., who found them to release the parent compound by intramolecular cyclization of the carrier moieties to imidazolidin-2-ones 10 (Scheme 7) and not to be susceptible to murine plasma esterases [25]. All the compounds tested were stable at low pH but were reactive in neutral and alkaline medium, releasing 4-hydroxyanisole at rates that were structure dependent (t1/2 ranging from 36 to 942 min, at pH 7.4 and 37 °C). As previously noticed by Begtrup and co-workers [17], the reactivity of compounds 9 decreased with increasing n, i.e., formation of 10 was favoured for n=2 as compared to n=3 (Scheme 7), as the reacting groups are farther apart in the latter case. This implies that, for n>3, intramolecular activation is unlikely to increase reaction rates. In what concerns the introduction of N-methyl (R1, R2, R3) or N-ethyl substituents (R1, R2), di- and tri-methyl derivatives were more reactive than their unsubstituted, mono-methyl and di-ethyl counterparts [25].
Scheme 7.
Intramolecular decomposition of 4-hydroxyanisole carbamates 9 (n=2,3) [25].
Scheme 7.
Intramolecular decomposition of 4-hydroxyanisole carbamates 9 (n=2,3) [25].
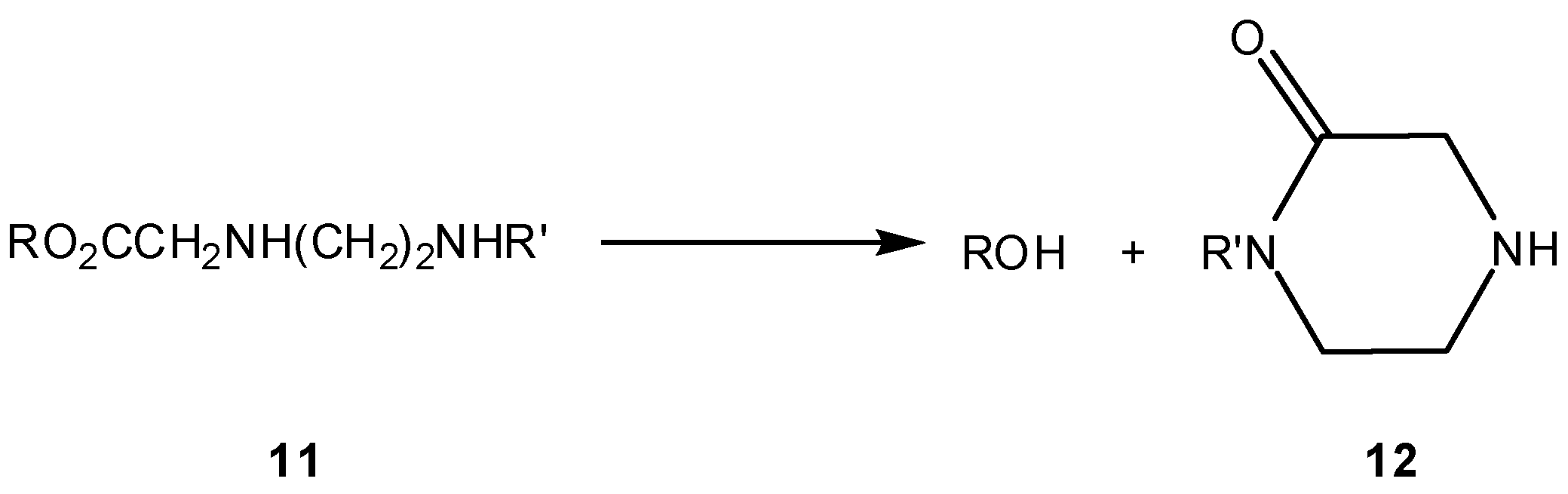
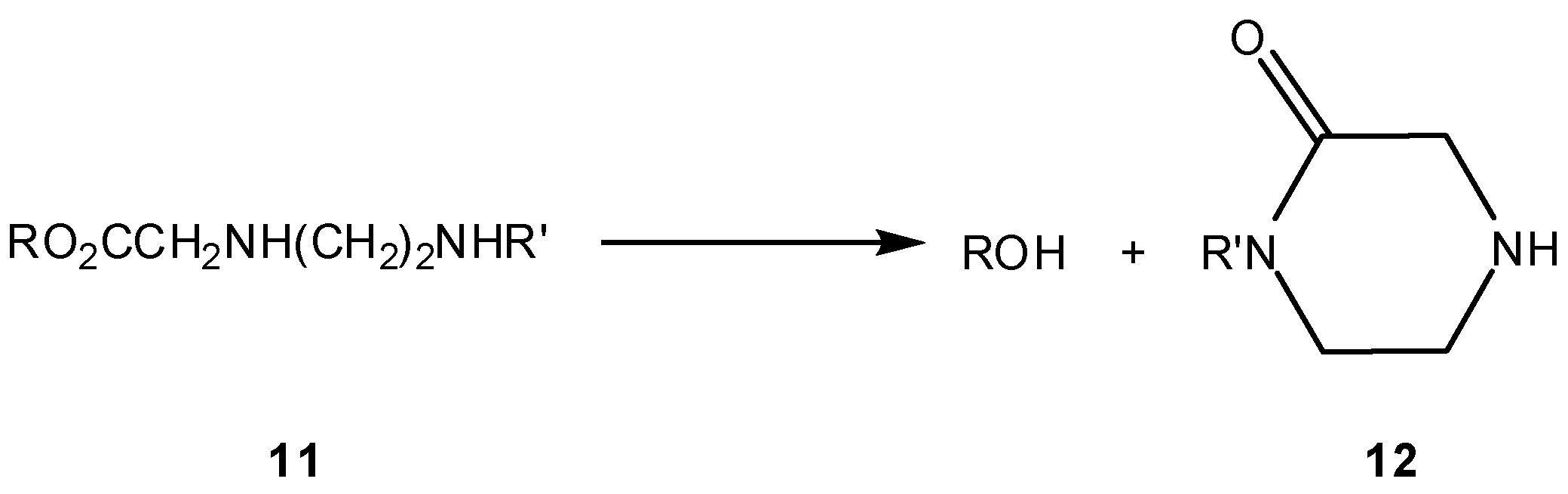
The same authors developed in parallel three ester prodrugs 11 of 5-bromo-2’-deoxyuridine (ROH in Scheme 8), that were also found to be stable at pH 2.5 and to release the alcohol at a slower rate at pH 6.8 than at pH 7.4 [26]. At this pH, all three esters released the parent alcohol at virtually the same rate (t1/2=23, 26.7 and 29.7 min), with slight differences being correlated with steric constraints imposed by R (Scheme 8) in the course of the cyclization reaction. These findings demonstrated the need for a non-protonated amine function to activate the prodrug, being consistent with alcohol release through a cyclization reaction. Confirmation of this assumption was enabled by quantitative isolation of the piperazinone cyclization product (12) after prodrug incubation at 37 °C and pH 7.4 [26]. Last, but not least, compounds 11 displayed similar or even higher stability in human plasma, but reacted much faster in rat plasma. This was explained by the generally low esterase activity of human plasma, as compared to rodent plasma, and also by a possible stabilizing effect due to binding of the nucleoside ester to the human plasma proteins [26].
Scheme 8.
Release of a drug (ROH) via piperazinone (12) formation [26].
Scheme 8.
Release of a drug (ROH) via piperazinone (12) formation [26].
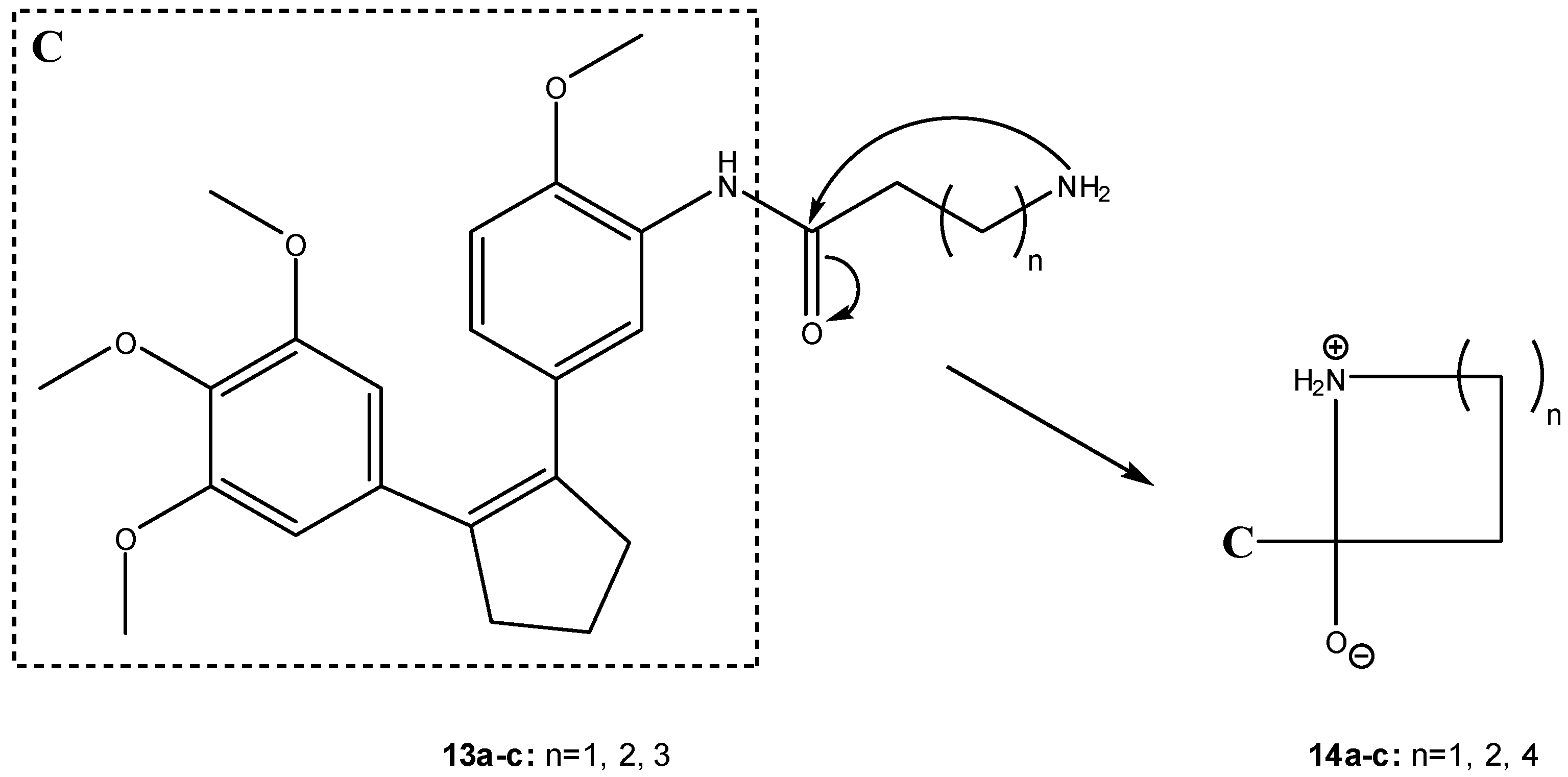
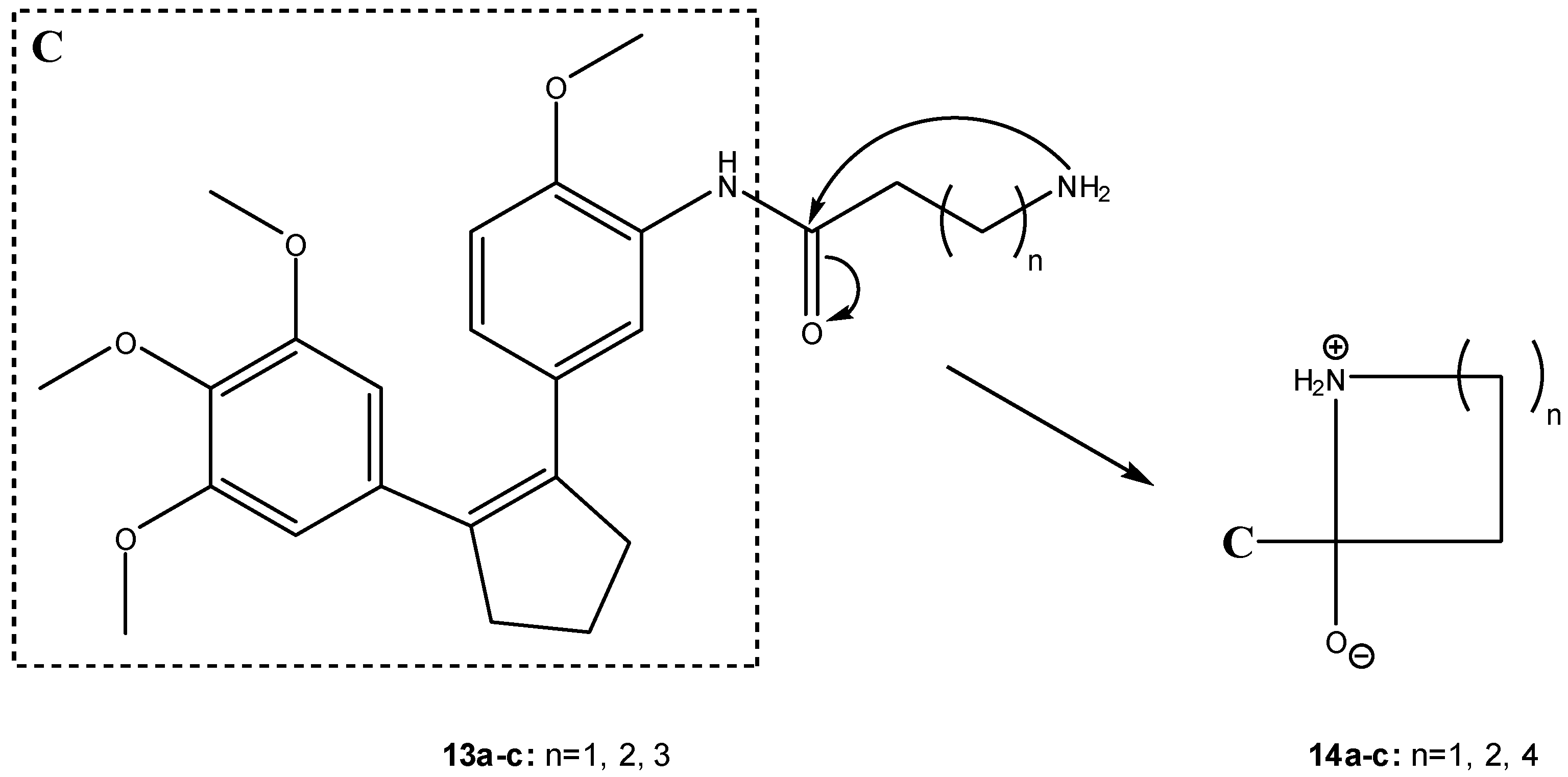
More recently, Nam et al. have developed water-soluble prodrugs 13a-c of the antitumor agent 3-[(3-amino-4-methoxy)phenyl]-2-(3,4,5-trimethoxyphenyl)cyclopent-2-ene-1-one (C, Scheme 9) based on β-, γ- and δ-amino acid carriers [27]. These authors found that the length of the amino acid aliphatic chains could be used to modulate the anti-tumor activity of the compounds, suggesting that folding of the terminal amino branch might play an important role on the rate of prodrug activation. Thus, the formation of the more stable five-membered ring intermediate (14b, Scheme 9) was more favorable than those of the more constrained four- or the larger seven-membered rings in intermediates 14a and 14c, respectively (Scheme 9) [27].
Scheme 9.
Intermediates in the intramolecular aminolysis of prodrugs 13a-c [27].
Scheme 9.
Intermediates in the intramolecular aminolysis of prodrugs 13a-c [27].
3.2.2. Peptide carriers
a) Peptide cyclization and prodrug design
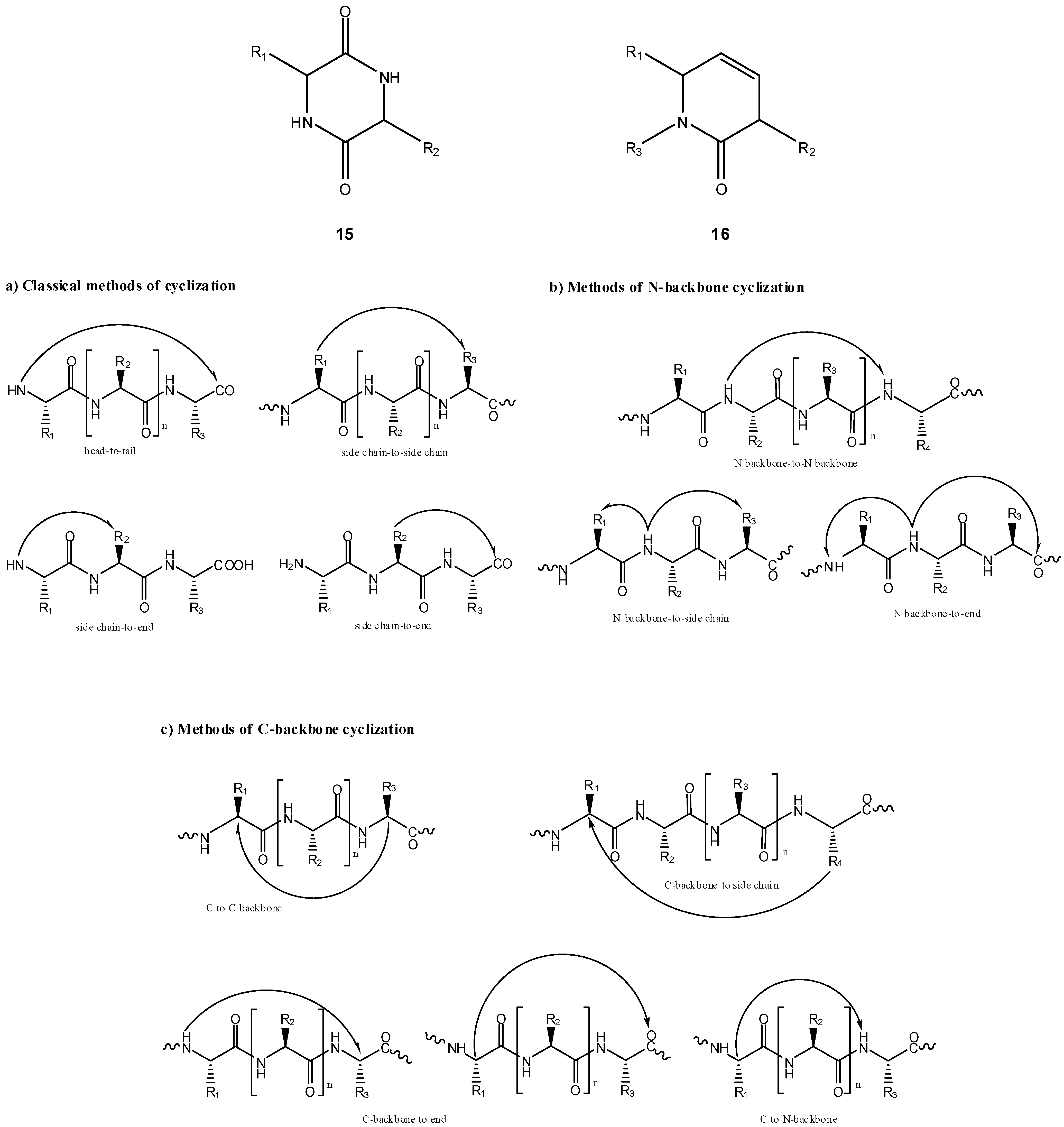
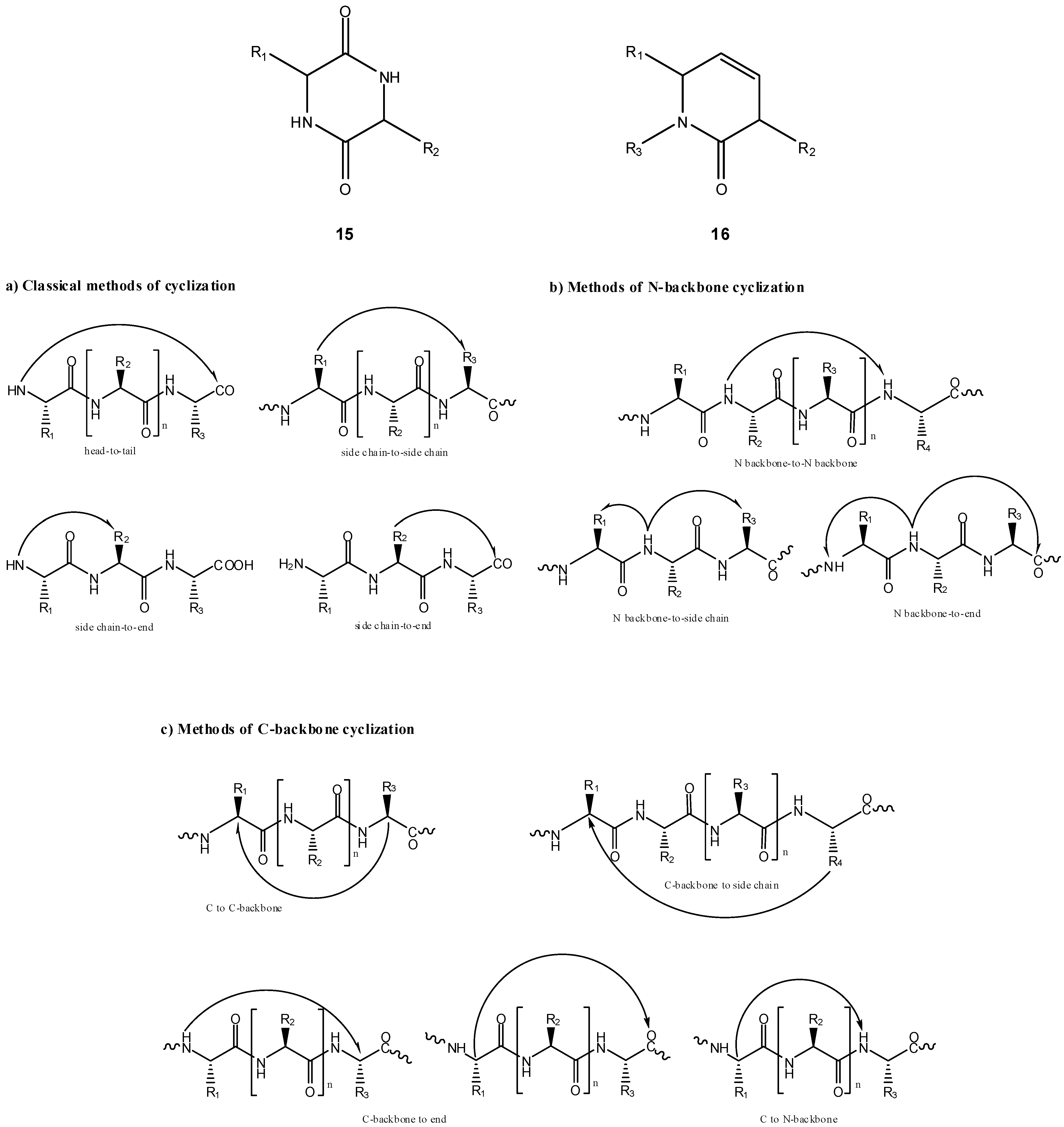
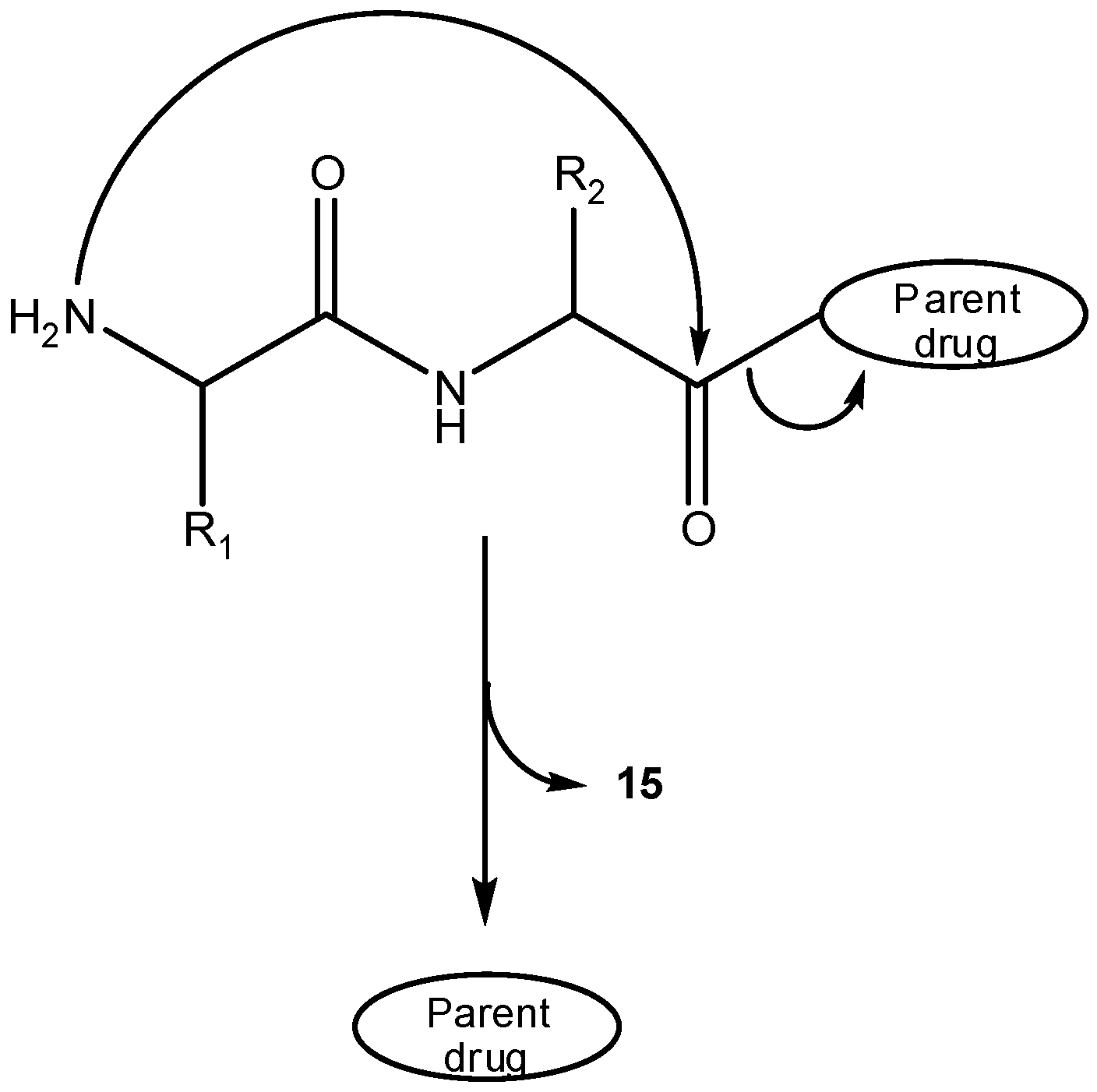
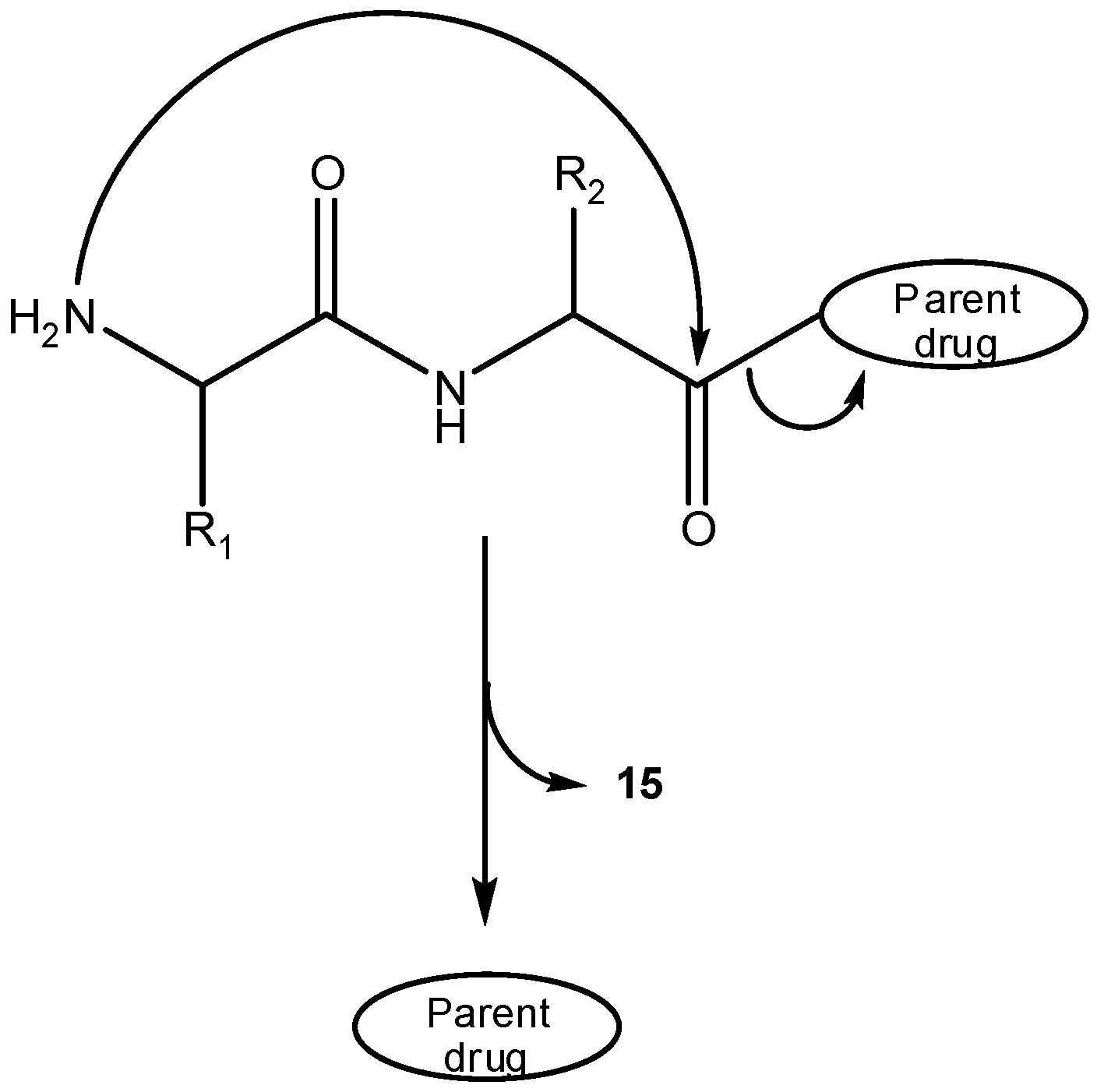
Intramolecular cyclization has been demonstrated to improve biological properties of bioactive peptides [28,29,30,31,32,33,34,35,36,37,38], in many cases allowing one to improve selectivity for a given receptor and/or metabolic stability [39,40,41]. Cyclic peptides can be divided into homodetic and heterodetic, the first being obtained by formation of an intramolecular peptide (amide) bond, whereas heterodetic refers to all cyclic peptides where the intramolecular bond newly formed is other than an amide (e.g., lactone, ether, thioether and, most commonly, disulphide bridges). Scheme 10 summarizes intramolecular cyclizations that peptides can undergo, which can be split into three categories: a) classical routes (head-to-tail, head-to-side chain, side chain-to-side chain and side chain-to-tail cyclizations); b) N-backbone cyclization; and, c) C-backbone cyclization [39,42]. The concept of backbone cyclization was introduced by Gilon et al. in 1991 to stabilize the peptide bioactive conformation without affecting its functional groups [39]. From Scheme 10, one can easily infer that peptide cyclization could be used in prodrug design by two different approaches: (i) linear peptides acting as cyclization-activated prodrugs of bioactive cyclic peptides; (ii) linear peptides acting as drug carriers in prodrugs that can be activated through cyclization of the peptide moiety. Approach (i) is not easy to accomplish if a strictly chemical drug release pathway is desired, as spontaneous cyclization of oligopeptides in solution does not easily occur without the intervention of specific enzymes; exception is made to some dipeptides that easily cyclize to piperazine-2,5-diones or diketopiperazines (DKP, 15) [43,44,45,46,47,48,49]. The DKP scaffold is widely found in compounds of biological interest and could serve as a drug template with appropriate arrayed pharmacophores. To this point, studies showed that the replacement of a DKP cis-amide bound with structurally similar (z)-alkene units could provide DKP mimetics (16) as novel templates for creating drug-like structures [50,51,52,53,54]. Therefore, adequate linear dipeptide derivatives could act as prodrugs for DKP-based drugs.
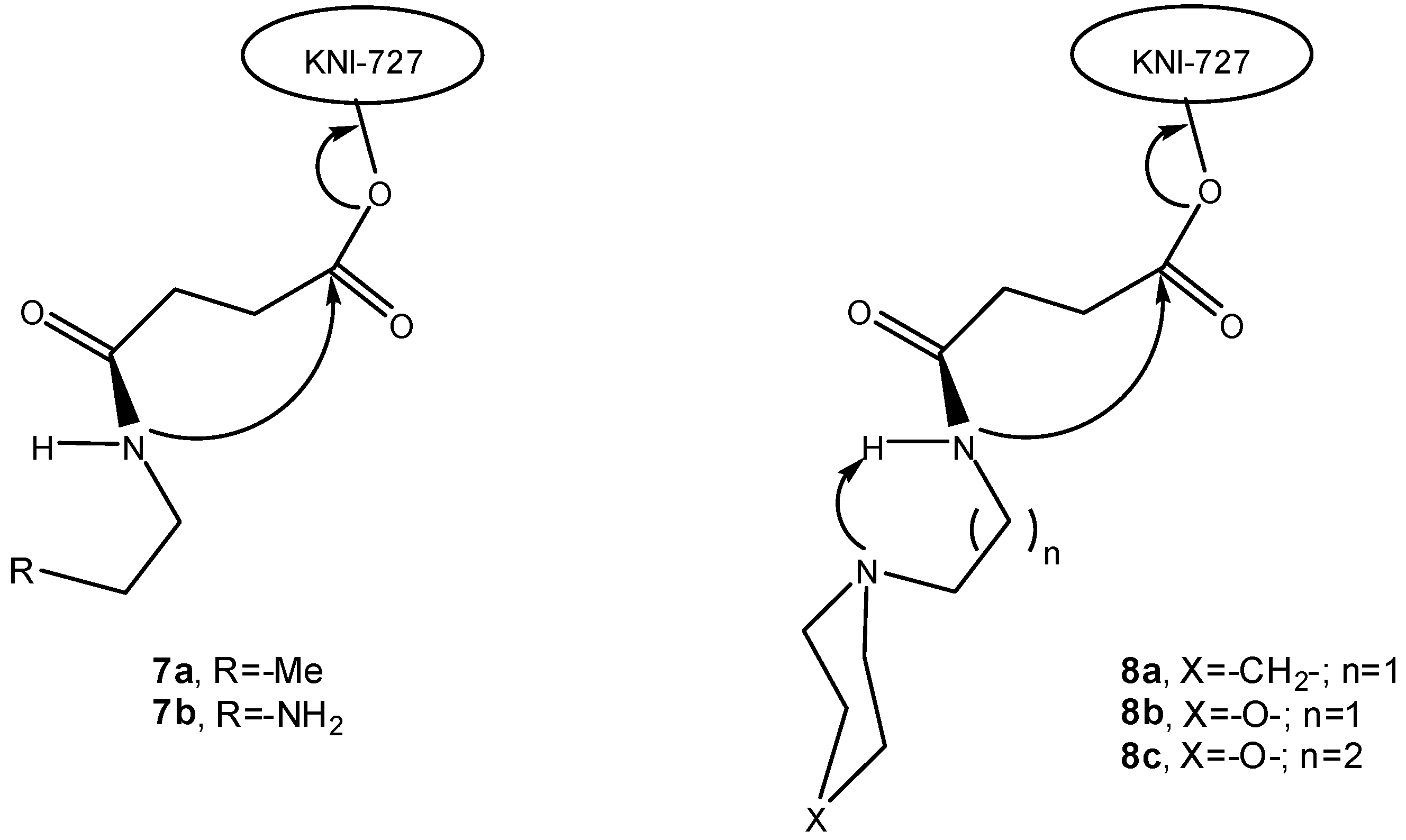{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
However, the major role of DKP in prodrug design falls in the domain of approach (ii): by linking adequate dipeptide carriers to a drug, a prodrug can be created which undergoes a strictly chemical cyclization-elimination process by intramolecular aminolysis of the dipeptide moiety to a DKP, with simultaneous departure of the free parent drug (Scheme 10) [55,56]. In this connection, peptide derivatives of some drugs have been prepared and evaluated as prodrug candidates where prodrug activation processes involved DKP formation. One example is that of peptide conjugates of the cytotoxic agent vinblastine, designed as potential prodrugs targeted at prostate cancer cells [57]. In vitro and in vivo evaluation showed the best derivative to be a conjugate bearing an octapeptide segment attached by an ester linkage to position 4 of vinblastine. This conjugate was found to undergo fast (t1/2=12 min) and specific cleavage by prostate enzymes of the Gln-Ser peptide bond and additional data from metabolism studies supported that the final spontaneous vinblastine release occurred from a dipeptidyl intermediate and was driven by DKP formation [57].
Scheme 10.
Prodrug intramolecular activation via DKP formation.
b) Prodrug activation via DKP formation
Over 30 years ago, formation of DKPs was reported as a major degradation pathway for simple alkyl esters [58,59,60,61] and dipeptide p-nitroanilides [62]. Dipeptides have been thus proposed as drug carriers to deliver the parent drug through enzyme-independent processes, namely, via DKP formation. Further, dipeptides are readily accessible carriers that can be easily modified to optimize the rate of release of the parent drug [63].
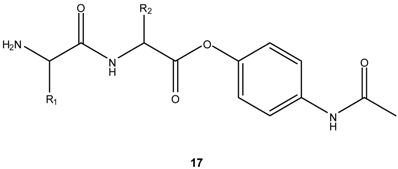
In view of the above, some authors have considered that dipeptides might play a crucial role as carriers for hydroxyl-containing drugs. Santos et al. have tested this hypothesis on a systematic study of the reactivity of dipeptide esters of paracetamol, 17, as this phenolic drug represents an excellent leaving group in the course of intramolecular aminolysis of the dipeptide esters [23] and is known to originate hepatotoxic metabolites [64]. Dipeptide esters 17 were found to be quantitatively hydrolyzed to the parent drug and corresponding DKPs at physiological pH and temperature [23]. Additional evidence supporting the intramolecular pathway for paracetamol release was that its rate depended on the structure of the dipeptide carrier. Thus, C-terminal bulky amino acids led to significantly higher half-lives and the presence of amino acids that can easily drive the peptide bond to adopt the cis configuration (e.g., proline, glycine, N-alkyl amino acids), i.e., more prone to cyclize to a DKP, [58,65] led to remarkably reactive substrates [23]. Paracetamol esterification with dipeptides also led to significant decrease or even elimination of the drug’s hepatotoxic effects on mice, hence reinforcing the great potential of dipeptide carriers in prodrug design [23]. However, when incubated in human plasma, the dipeptide esters released paracetamol at a much faster rate than in buffer, evidencing a significant role for enzymatic activation of these prodrugs [Santos et al., unpublished data].
From what has been described, it is clear that a major drawback of dipeptide-based prodrugs is their susceptibility to non-specific peptidases. However, this problem can be easily solved by incorporating at least one non-natural amino acid. For instance, enzymatically stable dipeptides (e.g. containing Aib or Sar) have been successfully employed in prodrugs of cytarabine [66] and cyclosporine A (CsA) [67]. Dipeptide esters of CsA thus obtained showed high thermodynamic stability, differential conversion rates under physiological conditions and strongly increased water solubility, offering a novel route for the design of CsA prodrugs [67].
Also to improve prodrug stability against protease/esterase action, unnatural amino acids were used to construct prodrugs of 5-fluorodeoxyuridine (FdU) [68]. Interestingly, these compounds were designed, on the one hand, to be resistant to certain enzymes and, on the other hand, to be susceptible to others. In fact, the antibacterial prodrugs of FdU developed by Wei and Pei are first activated in vivo by peptide deformylase, after which they suffer spontaneous intramolecular aminolysis of the resulting dipeptide carrier to form a DKP, with simultaneous release of FdU [68]. Consequently, they undergo two-step activation, constituting a fine example of the third major strategy for designing intramolecularly-activated prodrugs. Two-step prodrugs like these will be further explored in the next and last section of the present review.
4. Two-step Activation
Intramolecular activation of prodrugs has also been proposed as “the subsequent step” in drug release mechanisms involving a first enzymatic conversion step. In other words, double prodrugs have been proposed that would be enzymatically converted into a cyclization-activated prodrug. Actually, the example of pilocarpic acid double ester prodrugs of pilocarpine given in section 2 (Scheme 2) may fall in this category, as the nucleophilic hydroxyl can be released after enzymatic cleavage of the ester moiety [13]. Vinblastine peptide conjugates mentioned in 3.2.2.a) can also be included in this category, as the dipeptide intermediate undergoing intramolecular cyclization via DKP to release vinblastine is a product of enzymatic cleavage of a larger oligopeptide precursor [57].
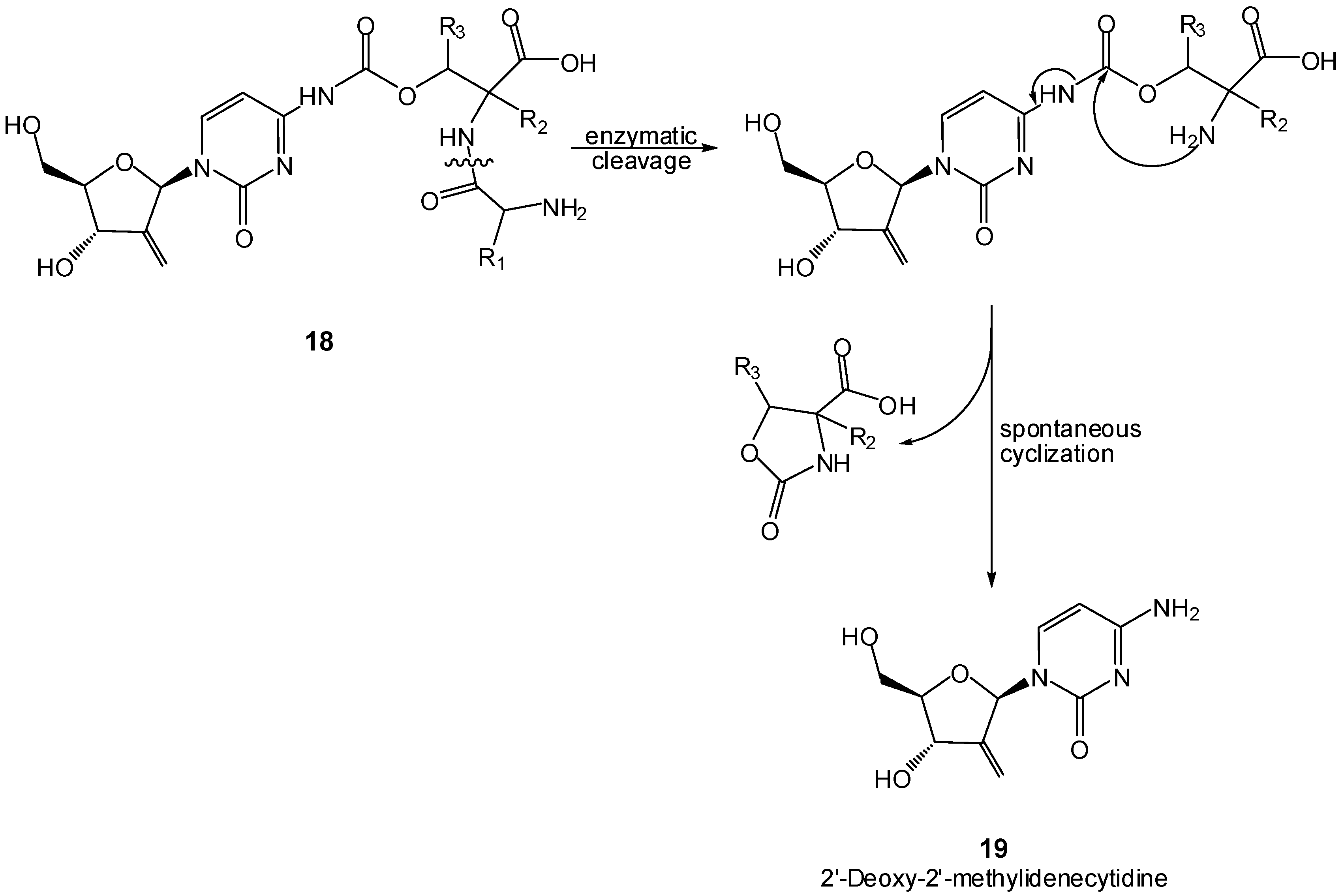
Cyclization of peptide esters mediated by the enzyme subtiligase has also been proposed over a decade ago for intramolecular activation of prodrugs [69]. Although it describes a strictly enzymatic pathway for drug release, this work constitutes another relevant example of the key role of peptide carriers in prodrug design. In this connection, cutting-edge work by Kohchi et al. proposes membrane dipeptidase (MDP)-mediated activation of prodrugs 18 of the anti-tumoral 2’-deoxy-2’-methylidene-cytidine (DMDC, 19) [70]. MDP specifically cleaves amide bonds in dipeptides and is over-expressed in tumors, which makes it an attractive target for anti-cancer therapy. Prodrugs 18 are activated by MDP in tumors by hydrolysis of the dipeptide bond followed by spontaneous cyclization of the promoiety, as depicted in Scheme 11 [70].
Scheme 11.
Enzymatic cleavage followed by intramolecular aminolysis of DMDC prodrugs [70].
Scheme 11.
Enzymatic cleavage followed by intramolecular aminolysis of DMDC prodrugs [70].
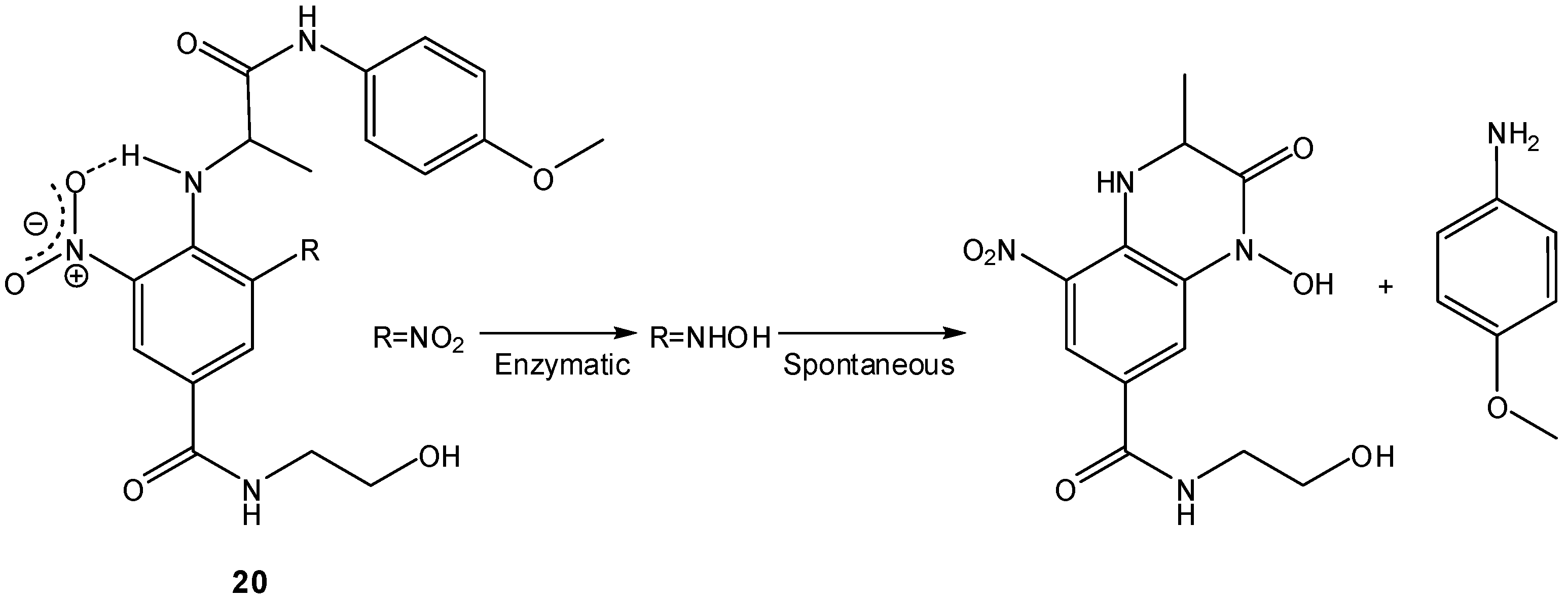
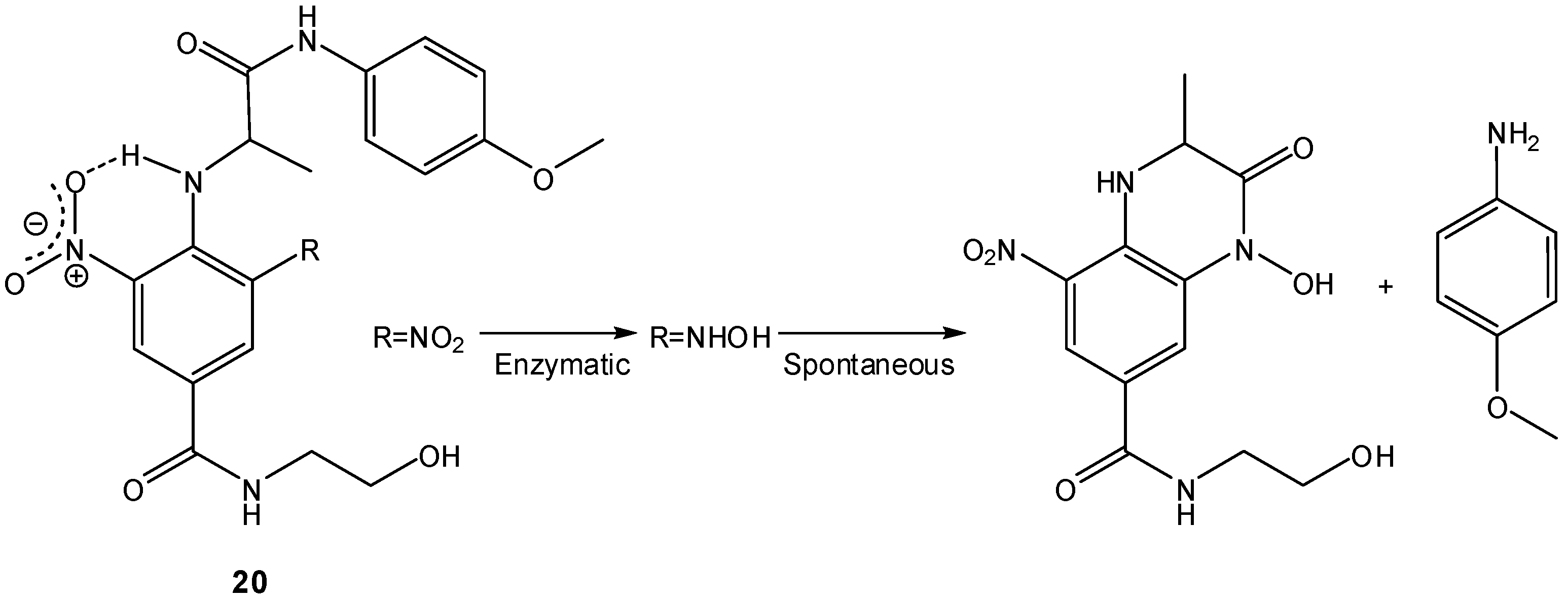
Other two-step prodrugs based on nitrogen nucleophiles have been developed, for instance, by Atwell et al., who proposed 2-nitroaryl amides as bio-reductive two-step prodrugs capable of releasing cytotoxic aminoaniline mustards by spontaneous cyclization of the corresponding 2-aminoaryl amides produced in a previous bio-reduction step [71]. These authors analyzed three factors that could influence the overall cyclization rate: the nucleophilicity of the amine, the geometry of the compound and the nature of the leaving group. Both the nucleophilicity of the amino group and the 4-substituent of the leaving aniline had little effect on the cyclization rate, whereas the geometry of the compound was a determinant factor, indicating that the rate of cyclization was greatly influenced by the pre-organization of the molecule [71]. A series of N-dinitrophenylamino acid amides 20 was also prepared as potential bio-reductive two-step prodrugs and the rates of their intramolecular cyclization after radio-promoted reduction were analyzed. The second (symmetric) nitro group was found to not only raise the reduction potential, but also, through H-bonded locking, correctly position the initial hydroxylamine for amide cyclization-extrusion (Scheme 12) [72]. The high rates of cyclization-extrusion on these highly electron-deficient hydroxylamines suggested that the process was greatly accelerated by the presence of the H-bonding “conformation lock” between the aniline proton and the adjacent o-nitro group [72], in agreement with previous findings on the effects of electronic variation in the attacking and leaving groups on the intramolecular aminolysis of amides [73]. A similar approach has been employed by Liu and Hu in the design of 5'-(2-Nitrophenylalkanoyl)-2'-deoxy-5-fluorouridines as bio-reductive prodrugs of FdU [74].
Scheme 12.
Bio-reduction and subsequent cyclization of N-dinitrophenylamino acid amides 20 [72].
Scheme 12.
Bio-reduction and subsequent cyclization of N-dinitrophenylamino acid amides 20 [72].
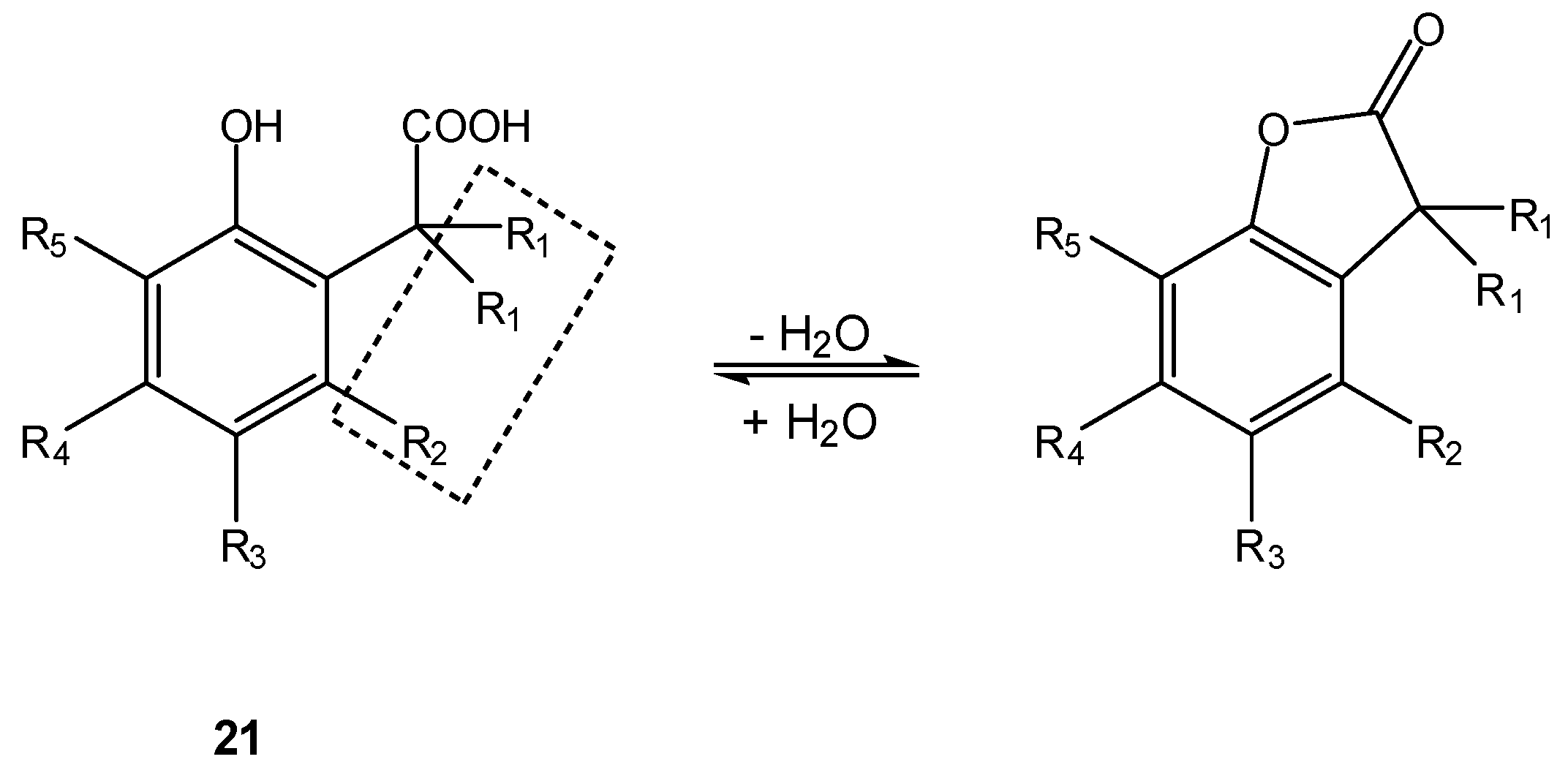
The most famous “conformation lock” in prodrug design is, however, the “trimethyl lock” (Scheme 13), whose presence in a molecular structure is known since the 1970s to greatly enhance the rate of lactonization of o-hydroxyhydrocinnamic acids (21) [75].
Scheme 13.
Lactonization of o-hydroxyhydrocinnamic acids 21; the “trimethyl lock” (R1=R2=Me) is depicted inside the dashed rectangle [75].
Scheme 13.
Lactonization of o-hydroxyhydrocinnamic acids 21; the “trimethyl lock” (R1=R2=Me) is depicted inside the dashed rectangle [75].
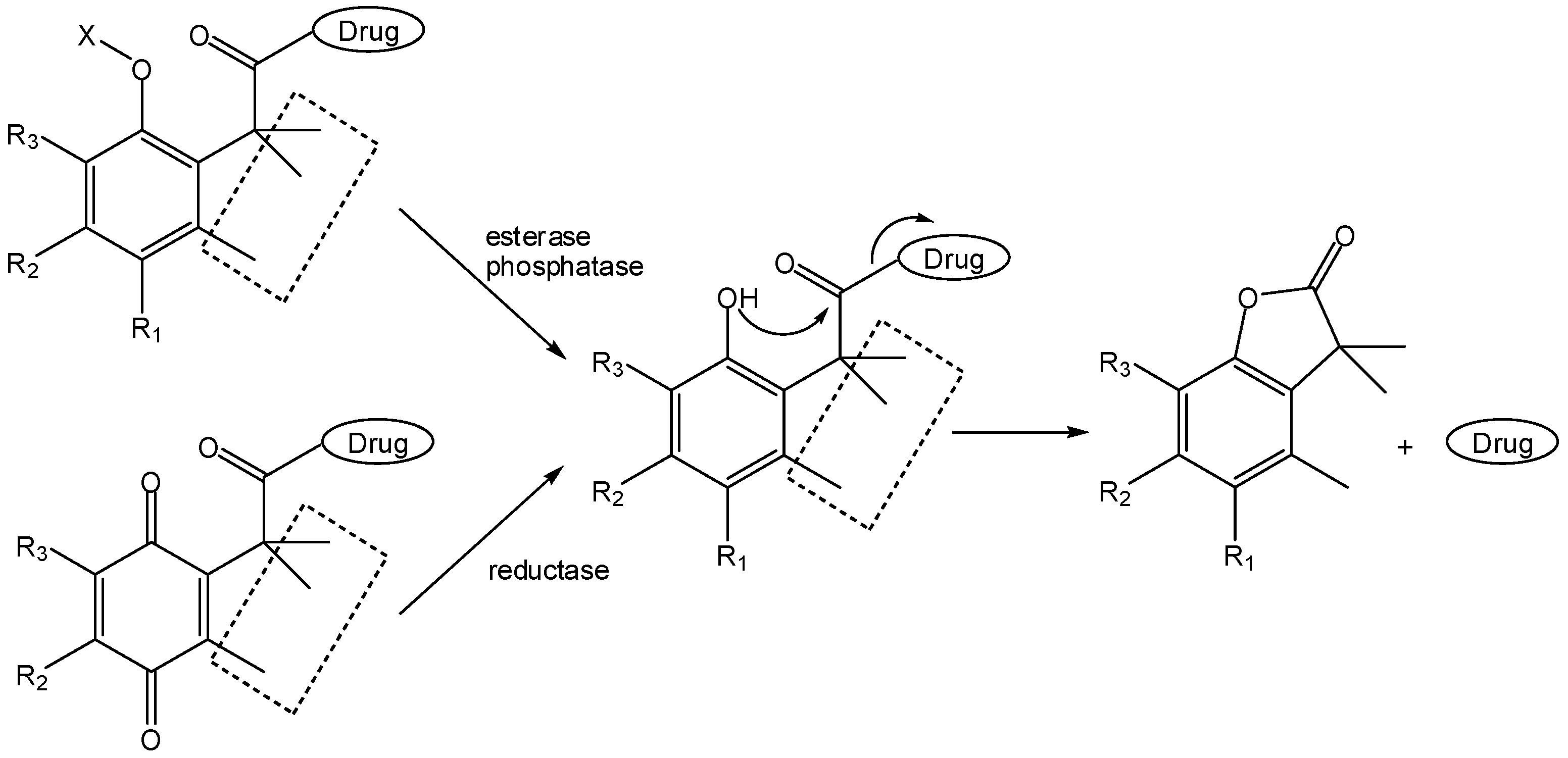
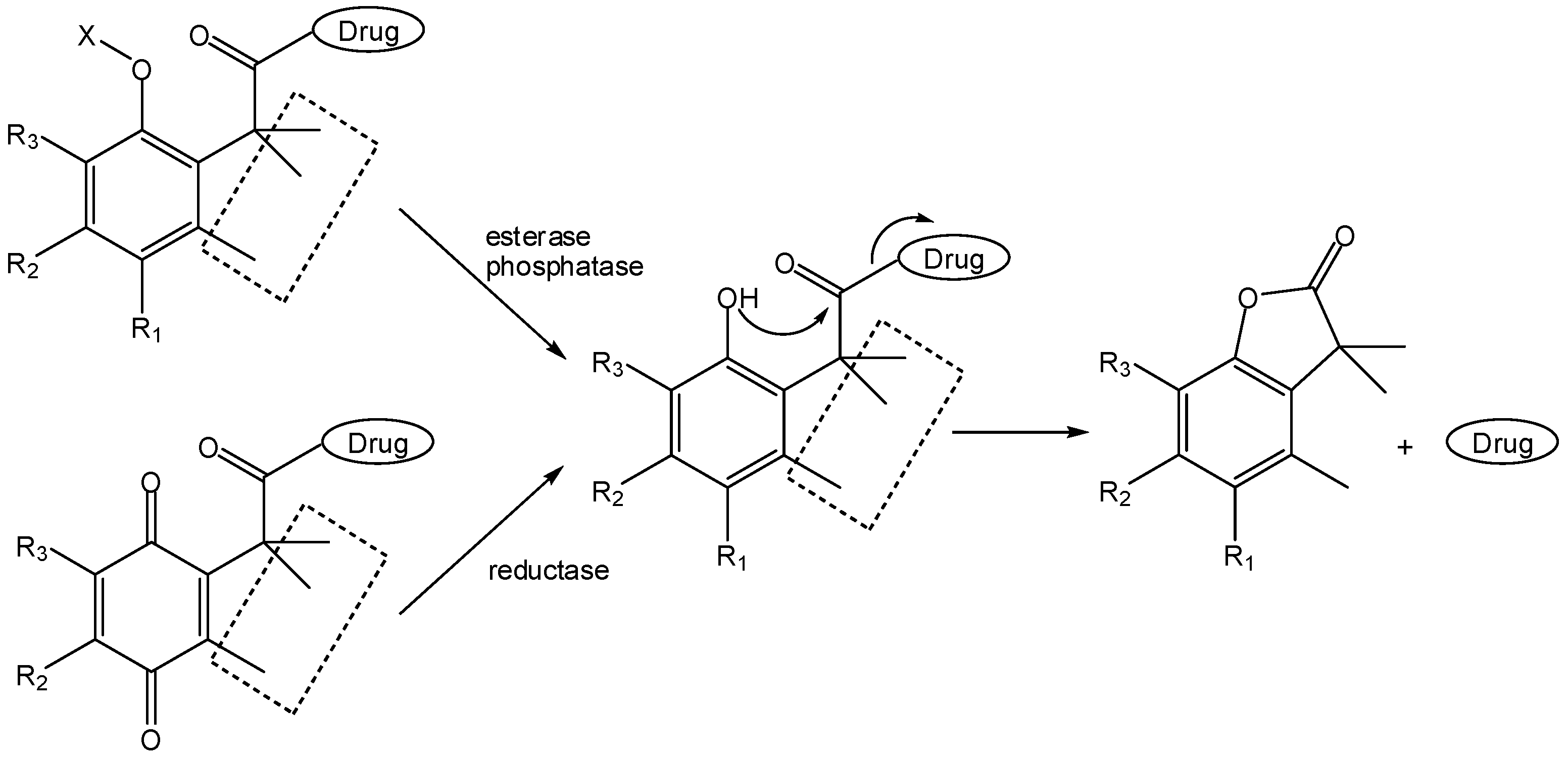
This has been exploited by medicinal chemists, especially by Borchardt and co-workers, to develop two-step activation prodrugs [6]. Thus, these authors carried out covalent attachment of model drugs to the carboxyl group of the hydrocinnamic acid moiety while masking the o-hydroxyl substituent as a precursor structure sensitive to either reductases [76,77,78], esterases [79,80,81] or phosphatases [82]. Consequently, the o-hydroxyl group could be released in a first enzymatically-promoted transformation, after which fast lactonization would lead to drug release (Scheme 14).
The “trimethyl lock” approach by Borchardt’s group yielded redox-sensitive pro-prodrugs of model amines that, after reduction of the hydroquinone, underwent fast lactonization with t1/2 ranging from 1.4 to 3.4 min [76], thus indicating that [bio-]reduction would be the rate-limiting step in the two-step activation of these double prodrugs [76,77,78]. Phosphatase-sensitive “trimethyl lock”-based two-step prodrugs of different amines and amino acids were also found to be good substrates for the human placental alkaline phosphatase (AP), although undistinguishable in terms of their activity for AP [82].
Scheme 14.
Application of the “trimethyl lock” concept by Borchardt and co-workers for the design of two-step prodrugs (X=RCOO or PO2O; R1=H, OH) [6,76,77,78,79,80,81,82].
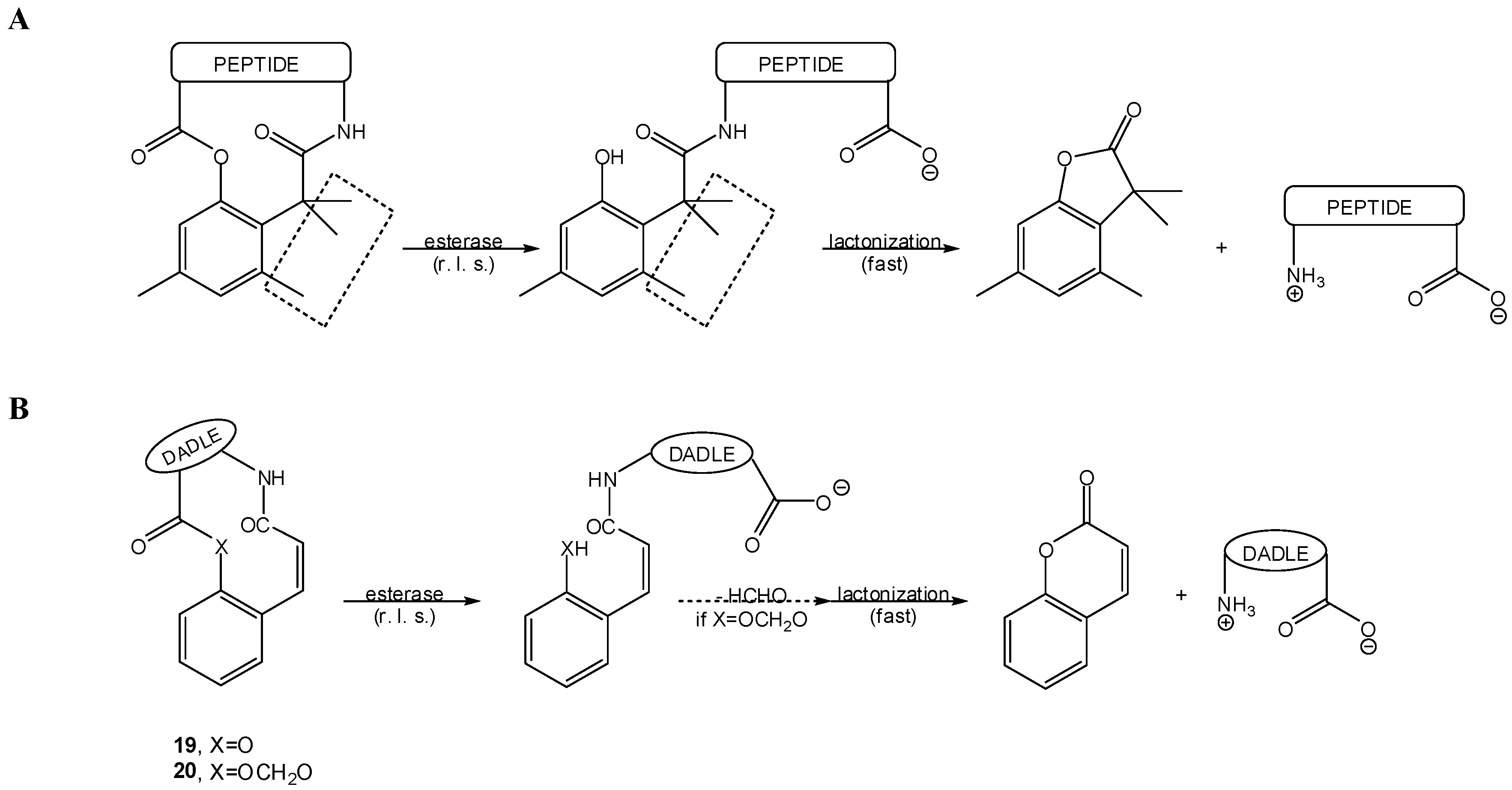
In what concerns similar esterase-sensitive pro-prodrugs of anisidine as model amine, half-lives determined at 37 °C in different media were as follows: 4030 min in phosphate buffer (pH 7.4), 11.9 min in the same buffer containing porcine liver esterase, 53.7 min in plasma and 475 min in plasma containing diisopropylfluorophosphate. Hence, these pro-prodrugs are enzymatically, rather than chemically, activated in biological media [79]. These observations prompted the same authors to design an esterase-sensitive cyclic two-step prodrug of a model hexapeptide [80,81]. This was also a “trimethyl lock”-based system that, in 90% human plasma, released the parent peptide (t1/2=504 min) by an apparent esterase-catalyzed hydrolysis of the phenol ester bond followed by fast lactonization (Scheme 15A). Moreover, this cyclic two-step prodrug presented a 70-fold increase relatively to the linear peptide in terms of permeation across Caco-2 cell monolayers [80,81].
Scheme 15.
Cyclic two-step prodrugs of peptides based on: (A) the “trimethyl lock” [80,81] and, (B) coumarinic acid derivatives [85,86,87,88,89,90,91].
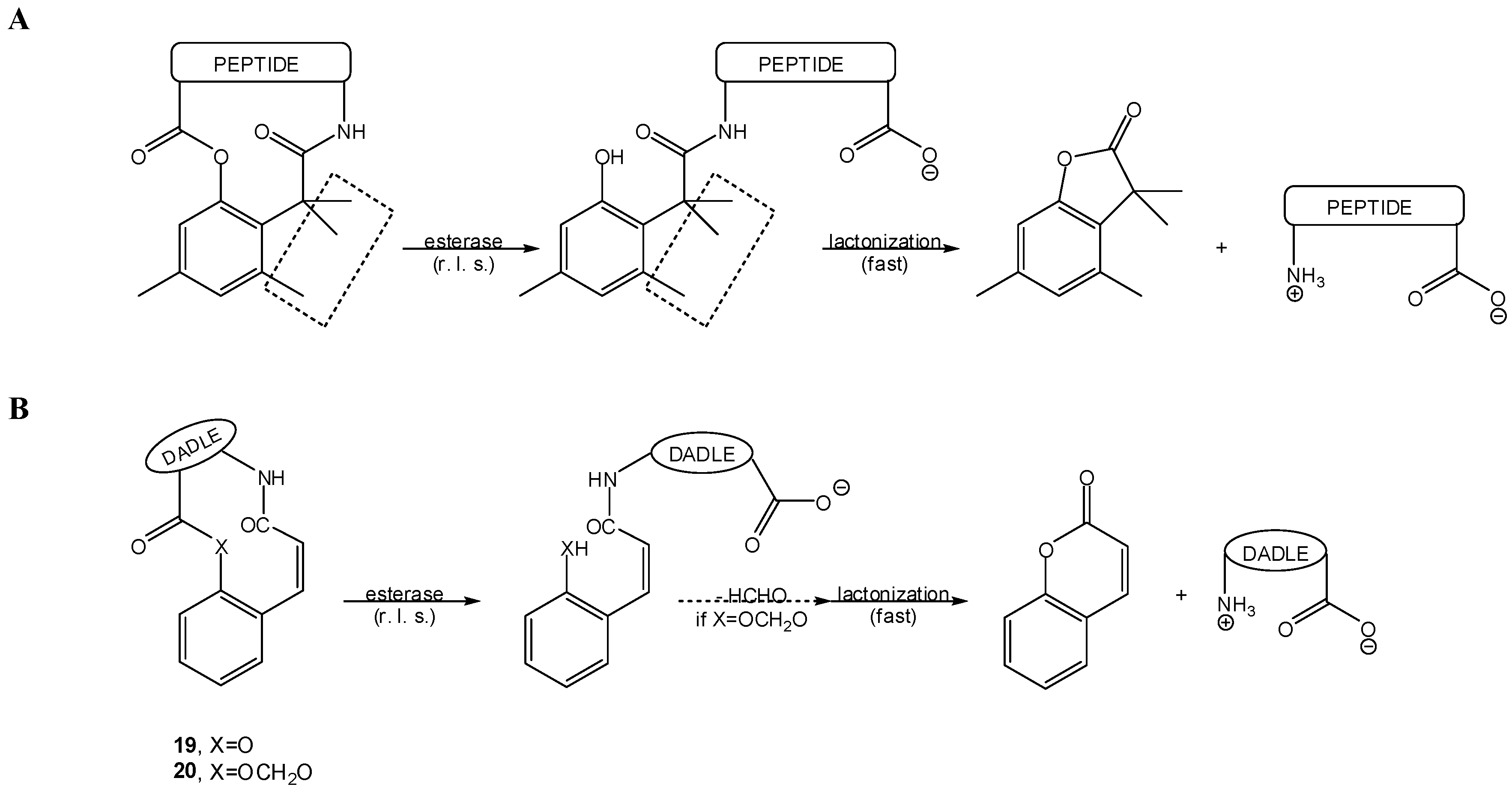
Another approach, quite similar to the “trimethyl lock” concept, for the design of two-step prodrugs has been based on coumarinic acid derivatives, as these equally undergo facile lactonization [83]. In this case, the “conformation lock” that favors lactonization is due to the cis double bond of the coumarinic acid aliphatic chain (Scheme 15B), and enzyme-sensitive two-step prodrugs can be conceived in a virtually superimposable way to that depicted above in Scheme 13 for “trimethyl lock”-based pro-prodrugs [6,83,84,85,86,87,88]. This approach yielded model two-step prodrugs of amines that were released upon incubation in the presence of porcine liver esterase, with t1/2 between 1.7 and 35 min [6,84]. The most relevant example of application of such coumarinic acid derivatives concerns cyclic pro-prodrugs of the opioid peptide DADLE (22), depicted in Scheme 15B [85,86]. This approach was also employed to oxymethyl-modified cyclic pro-prodrugs of DADLE (23, Scheme 15B), designed to promote better intrinsic cell permeation while preserving the desired sensitivity to esterases [87,88,89,90,91], and to RGD peptidomimetics [92].
Approaches to the design of two-step prodrugs based on concepts such as the “trimethyl lock”, apart from being quite elegant, were proven to promote significant increase in the oral bioavailability of known antivirals such as gancyclovir [93] and to be useful in the conception of PEG-daunorubicin prodrugs for the therapy of solid tumors [94]. More recently, the “trimethyl lock” concept has been applied by Raines and co-workers to the design of latent fluorophores as labels/indicators for biochemical and biological research [95,96]. Last, but not least, a 2007 report by Weerapreeyakul et al. describes “trimethyl lock” bioreductive prodrugs of fungal cytotoxic compounds aimed at cancer cells characterized by hypoxia and over-expression of reductases [97]. These two-step prodrugs were seen to be moderately to highly cytotoxic to cancer cells, while stable in the presence of esterases [97].
From what is above described, two-step activation can be regarded as an ingenious way to deal with the undeniable relevance of enzyme bioconversion of prodrugs, even of those originally designed to be activated by strictly chemical intramolecular pathways.
5. Concluding Remarks
Intramolecular activation of prodrugs began by being accidentally detected in the course of pharmaceutical research, but soon became a field of intense exploration by medicinal chemists. Cyclization-activated prodrugs are aimed at improving drug profiles while providing drug release pathways that are not exclusively dictated by the biological variability of enzyme activity.
Oligopeptides are promising carriers for cyclization-activated prodrugs, as they are generally non-toxic, non-immunogenic, specifically targeted at epithelial transporters such as hPEPT1 or hPEPT2 and provide chemical diversity through their side chains, so that drug release rates can be finely tuned. Further, their di- or tri-funcionality offers a wide span of chemical routes for both prodrug synthesis and intramolecular activation. Oligopeptides can be attached to a drug through their amino groups, hence offering the C-terminal carboxyl group as nucleophile to promote intramolecular activation. Conversely, if the drug is attached to the peptide’s carboxyl, the N-terminal amino group will become available to eventually engage in a cyclization-elimination for prodrug activation.
Despite the susceptibility of oligopeptides to suffer premature degradation by action of non-specific peptidases, the use of D- and other non-coded amino acids is a simple and effective way of circumventing this problem. More interestingly, the enzymatic susceptibility of oligopeptides can turn out to be an excellent tool for the development of two-step prodrugs. Hence, cyclization-activated prodrugs based on carefully designed peptide/peptidomimetic carriers are still a promising approach to improve drugs’ therapeutic indices. The same principle applies the other way around, i.e., when peptide drugs, often characterized by serious bioavailability limitations, can be released upon intramolecular cyclization of an adequate two-step pro-moiety. Elegant examples of such strategies for peptide delivery in vivo include those based on the “trimethyl lock” concept and alike.
Acknowledgments
NV thanks the Fundação para a Ciência e a Tecnologia (FCT, Portugal) for Ph.D. grant SFRH/BD/17754/2004. PG and RM thank FCT for financial support to CIQUP and CECF, respectively.
References and Notes
- Salama, N.N.; Fasano, A.; Thakar, M.; Eddington, N.D. The impact of ΔG on the oral bioavailability of low bioavailable therapeutic agents. J. Pharmacol. Exp. Ther. 2005, 312, 199–205. [Google Scholar]
- Lin, J.H.; Lu, A.Y.H. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS PharmSci. 2000, 2, E6. [Google Scholar]
- Sinkula, A.A.; Yalkowsky, S.H. Rationale for design of biologically reversible drug derivatives: prodrugs. J. Pharm. Sci. 1975, 64, 181–210. [Google Scholar]
- Yu, L.X.; Straughn, A.B.; Faustion, P.J.; Yang, Y.; Parekh, A.; Ciavarella, A.B.; Asafu-Adjaye, E.; Mehta, M.U.; Conner, D.P.; Lesko, L.J.; Hussain, A.S. The effect of food on the relative bioavailability of rapidly dissolving immediate-release solid oral products containing highly soluble drugs. Mol. Pharm. 2004, 1, 357–362. [Google Scholar]
- Shan, D.; Nicolaou, M.G.; Borchardt, R.T.; Wang, B. Prodrug strategies based on intramolecular cyclization reactions. J. Pharm. Sci. 1997, 86, 765–767. [Google Scholar]
- Testa, B.; Mayer, J.M. Design of intramolecularly activated prodrugs. Drug Metab. Rev. 1998, 30, 787–807. [Google Scholar]
- Wang, W.; Jiang, J.; Ballard, C.E.; Wang, B. Prodrug approaches to the improved delivery of peptide drugs. Curr. Pharm. Des. 1999, 5, 265–287. [Google Scholar]
- Vinšová, J.; Imramovský, A. Intramolekulární cycklizace využívané k uvolňování účinných látek z proléčiv. Chem. Listy 2005, 99, 21–29. [Google Scholar]
- Papot, S.; Tranoy, I.; Tillequin, F.; Florent, J.-C-; Gesson, J.-P. Design of selectively activated anticancer prodrugs: elimination and cyclization strategies. Curr. Med. Chem. – Anti-Cancer Agents 2002, 2, 155–185. [Google Scholar]
- Testa, B. Prodrug research: futile or fertile? Biochem. Pharmacol. 2004, 68, 2097–2106. [Google Scholar]
- Stella; Borchardt; Hageman; Oliyai; Maag; Tilley (Eds.) Prodrugs: challenges and rewards. Part 1 and 2; Springer-AAPS Press: New York, 2007; (the initial sections of Part 2 are particularly relevant in the present context).
- Bundgaard, H.; Falch, E.; Larsen, C.; Mesher, G.L.; Mikkelson, T. Pilocarpic acid esters as novel sequentially labile pilocarpine prodrugs for improved ocular delivery. J. Med. Chem. 1985, 28, 979–981. [Google Scholar]
- Song, L.; Bevins, R.; Anderson, B.D. Kinetics and mechanisms of activation of α-amino acid ester prodrugs of camptothecins. J. Med. Chem. 2006, 49, 4344–4355. [Google Scholar]
- Bom, D.; Curran, D.P.; Kruszewski, S.; Zimmer, S.G.; Strode, J.T.; Kohlhagen, G.; Du, W.; Chavan, A.J.; Fradey, K.A.; Bingcang, A.L.; Latus, L.J.; Pommier, Y.; Burke, T.G. The novel silatecan 7-tert-butyldimethylsilyl-10-hydroxycamptothecin displays high lipophilicity, improved human blood stability, and potent anticancer activity. J. Med. Chem. 2000, 43, 3970–3980. [Google Scholar]
- Vigroux, A.; Bergon, M.; Zedde, C. Cyclization-activated prodrugs: N-(substituted 2 hydroxyphenyl and 2-hydroxypropyl)carbamates based on ring-opened derivatives of active benzoxazolones and oxazolidinones as mutual prodrugs of acetaminophen. J. Med. Chem. 1995, 38, 3983–3994. [Google Scholar]
- Fredholt, K.; Mork, N.; Begtrup, M. Hemiesters of aliphatic dicarboxylic acids as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1995, 123, 209–216. [Google Scholar]
- Bruice, T.C.; Pandit, U.K. The effect of germinal substitution, ring size and rotamer distribution on the intramolecular nucleophilic catalysis of the hydrolysis of monophenyl esters of dibasic acids and the solvolysis of the intermediate anhydrides. J. Am. Chem. Soc. 1960, 82, 5858–5865. [Google Scholar]
- Thomsen, K.F.; Bundgaard, H. Cyclization-activated phenyl carbamate prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1993, 91, 39–49. [Google Scholar]
- Matsumoto, H.; Sohma, Y.; Kimura, T.; Hayashi, Y.; Kiso, Y. Controlled drug release: new water-soluble prodrugs of an HIV protease inhibitor. Bioorg. Med. Chem. Lett. 2001, 11, 605–609. [Google Scholar]
- Sohma, Y.; Hayashi, Y.; Ito, T.; Matsumoto, H.; Kimura, T.; Kiso, Y. Development of water-soluble prodrugs of HIV-1 protease inhibitor KNI-727: importance of the conversion time for higher gastrointestinal absorption of prodrugs based on spontaneous chemical cleavage. J. Med. Chem. 2003, 46, 4124–4135. [Google Scholar]
- Matsumoto, H.; Kimura, T.; Hamawaki, T.; Kumagai, A.; Goto, T.; Sano, K.; Hayashi, Y.; Kiso, Y. Design, synthesis, and biological evaluation of anti-HIV double-drugs: conjugates of HIV protease inhibitors with a reverse transcriptase inhibitor through spontaneously cleavable linkers. Bioorg. Med. Chem. 2001, 9, 1589–1600. [Google Scholar]
- Santos, C.; Mateus, M.L.; Santos, A.P.; Moreira, R.; Oliveira, E.; Gomes, P. Cyclization-activated prodrugs. Synthesis, reactivity and toxicity of dipeptide esters of paracetamol. Bioorg. Med. Chem. Lett. 2005, 15, 1595–1598. [Google Scholar]
- Gomes, P.; Gomes, J.R.B.; Rodrigues, M.; Moreira, R. Amino acids as selective sulfonamide acylating agents. Tetrahedron 2003, 59, 7473–7480. [Google Scholar]
- Saari, W.S.; Schwering, J.E.; Lyle, P.A.; Smith, S.J.; Engelhardt, E.L. Cyclization-activated prodrugs. Basic carbamates of 4-hydroxyanisole. J. Med. Chem. 1990, 33, 97–101. [Google Scholar]
- Saari, W.S.; Schwering, J.E.; Lyle, P.A.; Smith, S.J.; Engelhardt, E.L. Cyclization-activated prodrugs. Basic esters of 5-bromo-2’-deoxyuridine. J. Med. Chem. 1990, 33, 2590–2595. [Google Scholar] [CrossRef]
- Nam, N.H.; Kim, Y.; You, Y.J.; Hong, D.H.; Kim, H.M.; Ahn, B.Z. Water soluble prodrugs of the antitumor agent 3-[(3-amino-4-methoxy)phenyl]-2-(3,4,5-trimethoxyphenyl)cyclopent-2-ene-1-one. Bioorg. Med. Chem. 2003, 11, 1021–1029. [Google Scholar]
- Besser, D.; Müller, B.; Agricola, I.; Reissmann, S. Synthesis of differentially protected N-acylated reduced pseudodipeptides as building units for backbone cyclic peptides. J. Peptide Sci. 2000, 6, 130–138. [Google Scholar]
- Besser, D.; Müller, B.; Kleinwächter, P.; Greiner, G.; Seyfarth, L.; Steinmetzer, T.; Arad, O.; Reissmann, S. Synthesis and characterization of octapeptide somatostatin analogues with backbone cyclization: comparison of different strategies, biological activities and enzymatic stabilities. J. Prakt. Chem. 2000, 342, 537–545. [Google Scholar]
- Kaul, R.; Surprenant, S.; Lubell, W.D. Systematic study of the synthesis of macrocyclic dipeptide β-turn mimics possessing 8-, 9-, and 10- membered rings by ring-closing metathesis. J. Org. Chem. 2005, 70, 3838–3844. [Google Scholar]
- Poteau, R.; Trinquier, G. All-cis cyclic peptides. J. Am. Chem. Soc. 2005, 127, 13875–13889. [Google Scholar]
- Nielsen, T.E.; Quement, S.L.; Meldal, M. Solid-phase synthesis of bicyclic dipeptide mimetics by intramolecular cyclization of alcohols, thiols, amines, and amides with N-acyliminium intermediates. Org. Lett. 2005, 7, 3601–3604. [Google Scholar]
- Liederer, B.M.; Fuchs, T.; Velde, D.V.; Siahaan, T.J.; Borchardt, R.T. Effects of amino acid chirality and the chemical linker on the cell permeation characteristics of cyclic prodrugs of opioid peptides. J. Med. Chem. 2006, 49, 1261–1270. [Google Scholar]
- Keramida, M.; Tselios, T.; Mantzourani, E.; Papazisis, K.; Mavromoustakos, T.; Klaussen, C.; Agelis, G.; Deraos, S.; Friligou, I.; Habibi, H.; Matsoukas, J. Design, synthesis, and molecular modeling of a novel amide-linked cyclic GnRH analogue cyclo(4-9)[Lys4,d-Trp6,Glu9]GnRH: stimulation of gonadotropin gene expression. J. Med. Chem. 2006, 49, 105–110. [Google Scholar]
- Flora, D.; Mo, H.; Mayer, J.P.; Khan, M.A.; Yan, L.Z. Detection and control of aspartimide formation in the synthesis of cyclic peptides. Bioorg. Med. Chem. Lett. 2005, 15, 1065–1068. [Google Scholar]
- Che, Y.; Marshall, G.R. Engineering cyclic tetrapeptides containing chimeric amino acids as preferred reverse-turn scaffolds. J. Med. Chem. 2006, 49, 111–124. [Google Scholar]
- Arnott, G.; Clayden, J.; Hamilton, S.D. Azabicyclic amino acids by stereoselective dearomatizing cyclization of the enolates of N-nicotinoyl glycine derivatives. Org. Lett. 2006, 8, 5325–5328. [Google Scholar]
- Norgren, A.S.; Büttner, F.; Prabpai, S.; Kongsaeree, P.; Arvidsson, P.I. β2-aminoacids in the design of conformationally homogeneous cyclo-peptide scaffolds. J. Org. Chem. 2006, 71, 6814–6821. [Google Scholar]
- Gilon, C.; Halle, D.; Chorev, M.; Selinger, Z.; Byk, G. Backbone cyclization: a new conformational constraint on peptides. Biopolymers 1991, 31, 745–750. [Google Scholar]
- Al-Obeidi, F.; Castrucci, A.M.L.; Hadley, M.E.; Hruby, V.J. Potent and prolonged acting cyclic lactam analogues off α-melanotropin: design based on molecular dynamics. J. Med. Chem. 1989, 32, 2555–2561. [Google Scholar]
- Charpentier, B.; Dor, A.; Roy, P.; England, P.; Pham, H.; Durieux, C.; Roques, B.P. Synthesis and binding affinities of cyclic and related linear analogues of CCK8 selective for central receptors. J. Med. Chem. 1989, 32, 1184–1190. [Google Scholar]
- Reissmann, S.; Imhof, D. Development of conformationally restricted analogues of bradykinin and somatostain using constrained amino acids and different types of cyclization. Curr. Med. Chem. 2004, 11, 2823–2844. [Google Scholar]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Peptide Sci. 2003, 9, 471–501. [Google Scholar]
- Bray, A.M.; Maeji, N.J.; Valerio, R.M.; Campbell, R.A.; Geysen, H.M. Direct cleavage of peptides from a solid support into aqueous buffer. Applications in simultaneous multiple peptide synthesis. J. Org. Chem. 1991, 56, 6659–6666. [Google Scholar]
- Capasso, S.; Vergara, A.; Mozzarella, l. Mechanism of 2,5-dioxopiperazine formation. J. Am. Chem. Soc. 1998, 120, 1990–1995. [Google Scholar]
- Besada, P.; Mamedova, L.; Thomas, C.J.; Costanzi, S.; Jacobson, K.A. Design and synthesis of new bicyclic diketopiperazines as scaffolds for receptor probes of structurally diverse functionality. Org. Biomol. Chem. 2005, 3, 2016–2025. [Google Scholar]
- Moyroud, J.; Gelin, J.; Chêne, A.; Mortier, J. Synthèse d’analogues structuraux de Thaxtomines, phytotoxines responsables de la gale de la pomme de terre. Tetrahedron 1996, 52, 8525–8534. [Google Scholar]
- Szardenings, A.K.; Burkoth, T.S. A simple procedure for the solid phase synthesis of diketopiperazine and diketomorpholine derivatives. Tetrahedron 1997, 53, 6573–6593. [Google Scholar]
- Sollis, S.L. Short and novel stereospecific synthesis of trisubstituted 2,5-diketopiperazines. J. Org. Chem. 2005, 70, 4735–4740. [Google Scholar]
- Niida, A.; Tanigaki, H.; Inokuchi, E.; Sasaki, Y.; Oishi, S.; Ohno, H.; Tamamura, H.; Wang, Z.; Peiper, S.C.; Kitaura, K.; Otaka, A.; Fujii, N. Stereoselective synthesis of 3,6-disubstituted-3,6-dihydropyridun-2-ones as potential diketopiperazine mimetics using organocopper-mediated anti-Sn2’ reactions and their use in the preparation of low-molecule CXCR4 antagonists. J. Org. Chem. 2006, 71, 3942–3951. [Google Scholar]
- Kanoh, K.; Kohno, S.; Katada, J.; Takahashi, J.; Uno, I.; Hayashi, Y. Synthesis and biological activies of phenylahistin derivatives. Bioorg. Med. Chem. 1999, 7, 1451–1457. [Google Scholar]
- Donkor, I.O.; Sanders, M.L. Synthesis of a reported calpain inhibitor isolated from Streptomyces griseus. Bioorg. Med. Chem. Lett. 2001, 11, 2647–2649. [Google Scholar]
- Nam, N.H.; Ye, G.; Sun, G.; Parang, K. Conformationally constrained peptide analogues of pTyr-Glu-Glu-Ile as inhibitors of the Src SH2 domain binding. J. Med. Chem. 2004, 47, 3131–3141. [Google Scholar]
- Borthwick, A.D.; Davies, D.E.; Exall, A.M.; Livermore, D.G.; Sollis, S.L.; Nerozzi, F.; Allen, M.J.; Perren, M.; Shabbir, S.S.; Woollard, P.M.; Wyatt, P.G. 2,5-Diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists. 2. Synthesis, chirality, and pharmacokinetics. J. Med. Chem. 2005, 48, 6956–6969. [Google Scholar] [CrossRef]
- Gomes, P.; Gomes, J.R.B.; Rodrigues, M.; Moreira, R. Amino acids as selective sulfonamide acylating agents. Tetrahedron 2003, 59, 7473–7480. [Google Scholar]
- Santos, C.; Moreira, R.; Gomes, P. Cyclization-activated prodrugs: dipeptide esters of paracetamol. In Peptides 2004; M. Flegel, M., Fridkin, C., Gilon e, J., Slaninova, Eds.; Kenes International: Tel A’viv, 2005. [Google Scholar]
- Brady, S.F.; Pawluczyk, J.M.; Lumma, P.K.; Feng, D.M.; Wai, J.M.; Jones, R.; DeFeo-Jones, D.; Wong, B.K.; Miller-Stein, C.; lin, J.H.; Oliff, A.; Freidinger, R.M.; Garsky, V.M. Design and synsthesis of a pro-drug of vinblastine targeted at treatment of prostate cancer with enhanced efficacy and reduced systemic toxicity. J. Med. Chem. 2002, 45, 4706–4715. [Google Scholar]
- Purdie, J. E.; Benoiton, N. L. Piperazinedione formation from esters of dipeptides containing glycine, alanine, and sarcosine: the kinetics in aqueous solution. J. Chem. Soc. Perkin II 1973, 13, 1845–1852. [Google Scholar]
- Meresaar, U.; Ågren, A. Hydrolysis of protolytic esters VI. Alkaline hydrolysis and ring closure of glycylglycine ethyl ester. Acta Pharm. Suec. 1968, 5, 85–94. [Google Scholar]
- Jensen, E.; Bundgaard, H. Peptide esters as water-soluble prodrugs for hydroxyl containing agents: chemical stability and enzymatic hydrolysis of benzyl esters of glycine, diglycine and triglycine. Int. J. Pharm. 1991, 71, 117–125. [Google Scholar]
- Larsen, S.W.; Ankersen, M.; Larsen, C. Kinetics of degradation and oil solubility of ester prodrug of a model dipeptide (Gly-Phe). Eur. J. Pharm. Sci. 2004, 22, 399–408. [Google Scholar]
- Goolcharran, C.; Borchardt, R. T. Kinetics of diketopiperazine formation using model peptides. J. Pharm. Sci. 1998, 87, 283–288. [Google Scholar]
- Shan, D.; Nicolaou, M.G.; Borchardt, R.T.; Wang, B. Prodrug strategies based on intramolecular cyclization reactions. J. Pharm. Sci. 1997, 86, 765. [Google Scholar]
- Bertoloni, A.; Ferrari, A.; Ottani, A.; Guerzoni, S.; Tacchi, R.; Leone, S. Paracetamol: new vistas of an old drug. CNS Drug Rev. 2006, 12, 250–275. [Google Scholar]
- Lloyd-Williams, P.; Albericio, F.; Giralt, E. Chemical Approaches to the Synthesis of Peptides and Proteins.; CRC Press: Boca Raton, 1997; p. 60. [Google Scholar]
- Wipf, P.; Li, W.L.; Adeyeye, C.M.; Rusnak, J.M.; Lazo, J.S. Synthesis of chemoreversible prodrugs of ara-C with variable time-release profiles. Biological evaluation of their apoptotic activity. Bioorg. Med. Chem. 1996, 4, 1585–1596. [Google Scholar]
- Hamel, A.R.; Hubler, F.; Carrupt, A.; Wenger, R.M.; Mutter, M. Cyclosporin A prodrugs: design, synthesis and biophysical properties. J. Peptide Res. 2004, 63, 147–154. [Google Scholar]
- Wei, Y.; Pei, D. Activation of antibacterial prodrugs by peptide deformylase. Bioorg, Med. Chem. Lett. 2000, 10, 1073–1076. [Google Scholar] [CrossRef]
- Jackson, D.Y.; Burnier, J.P.; Wells, J.A. Enzymatic cyclization of linear peptide esters using subtiligase. J. Am. Chem. Soc. 1995, 117, 819–820. [Google Scholar]
- Kohchi, Y.; Hattori, K.; Oikawa, N.; Mizuguchi, E.; Isshiki, Y.; Aso, K.; Yoshinari, K.; Shirai, H.; Miwa, M.; Inagaki, Y.; Ura, M.; Ogawa, K.; Okabe, H.; Ishitsuka, H.; Shimma, N. Design and synthesis of novel prodrugs of 2’-deoxy-2’-methylidenecytidine activated by membrane dipeptidase overexpressed in tumor tissues. Bioorg. Med. Chem. Lett. 2007, 17, 2241–2245. [Google Scholar]
- Atwell, G.; Sykes, B.M.; O’Connor, C.J.; Denny, W.A. Relationships between structure and kinetics of cyclization of 2-aminoaryl amides: potential prodrugs of cyclization-activated aromatic mustards. J. Med. Chem. 1994, 37, 371–380. [Google Scholar]
- Sykes, B.M.; Atwell, G.J.; Hogg, A.; Wilson, W.R.; O’Connor, C.J.; Denny, W.A. N-substituted 2-(2,6-dinitrophenylamino)propanamides: novel prodrugs that release a primary amine via nitroreduction and intramolecular cyclization. J. Med. Chem. 1999, 4, 346–355. [Google Scholar]
- Kirk, K.L.; Cohen, L.A. Intramolecular aminolysis of amides. Effects of electronic variation in the attacking and leaving groups. J. Am. Chem. Soc. 1972, 94, 8142–8147. [Google Scholar]
- Liu, B.; Hu, L. 5'-(2-nitrophenylalkanoyl)-2'-deoxy-5-fluorouridines as prodrugs of FUDR for reductive activation. Bioorg. Med. Chem. 2003, 11, 3889–3899. [Google Scholar]
- Milstien, S.; Cohen, L.A. Stereopopulation control. I. Rate enhancement in the lactonizations of o-hydroxyhydrocinnamic acids. J. Am. Chem. Soc. 1972, 94, 9158–9165. [Google Scholar] [CrossRef]
- Amsberry, K.L.; Borchardt, R.T. Amine prodrugs which utilize hydroxy amide lactonization. I. A potential redox-sensitive amide prodrug. Pharm. Res. 1991, 8, 323–330. [Google Scholar] [CrossRef]
- Wolfe, J.L.; Vander Velde, D.G.; Borchardt, R.T. Facilitated intramolecular conjugate addition of N-(p- methoxypheny1)-3-(3’,6’-dioxo-2’,4’-dimethylcyclohexa-1’,4’-dienyl)-3,3-dimethylpro-pionamide. 1. Product characterization. J. Org. Chem. 1992, 57, 6138–6142. [Google Scholar]
- Nicolaou, M.G.; Wolfe, J.L.; Schowen, R.L.; Borchardt, R.T. Facilitated intramolecular conjugate addition of amides of 3-(3’,6’-dioxo-2’,4’-dimethyl-1’,4’-cyclohexadienyl)-3,3-dimethylpropionic acid. 2. Kinetics of degradation. J. Org. Chem. 1996, 61, 6633–6638. [Google Scholar]
- Amsberry, K.L.; Gerstenberger, E.; Borchardt, R.T. Amine prodrugs which utilize hydroxy amide lactonization. II. A potential esterase-sensitive amide prodrug. Pharm. Res. 1991, 8, 455–461. [Google Scholar]
- Wang, B.; Gangwar, S.; Pauletti, G.M.; Siahaan, T.J.; Borchardt, R.T. Synthesis of a novel esterase-sensitive cyclic prodrug system for peptides that utilizes a “trimethyl lock”-facilitated lactonization reaction. J. Org. Chem. 1997, 62, 1363–1367. [Google Scholar]
- Pauletti, G.M.; Gangwar, S.; Wang, B.; Borchardt, R.T. Esterase-sensitive cyclic prodrugs of peptides: evaluation of a phenylpropionic acid promoiety in a model hexapeptide. Pharm. Res. 1997, 14, 11–17. [Google Scholar]
- Nicolaou, M.G.; Yuan, C.-S.; Borchardt, R.T. Phosphate prodrugs for amines utilizing a fast intramolecular hydroxy amide lactonization. J. Org. Chem. 1996, 61, 8636–8641. [Google Scholar]
- Hershfield, R.; Schmir, G.L. Lactonization of coumarinic acids. Kinetic evidence for three species of the tetrahedral intermediate. J. Am. Chem. Soc. 1973, 95, 8032–8040. [Google Scholar]
- Wang, B.; Zhang, H.; Wang, W. Chemical feasibility studies of a potential coumarin-based prodrug system. Bioorg. Med. Chem. Lett. 1996, 6, 945–950. [Google Scholar]
- Wang, B.; Wang, W.; Zhang, H.; Shan, D.; Smith, T.D. Coumarin-based prodrugs 2. Synthesis and bioreversibility studies of an esterase-sensitive cyclic prodrug of dadle, an opioid peptide. Bioorg. Med. Chem. Lett. 1996, 6, 2823–2826. [Google Scholar]
- Ouyang, H.; Borchardt, R.T.; Siahaan, T.J.; Vander Velde, D.G. Synthesis and conformational analysis of a coumarinic acid-based cyclic prodrug of an opioid peptide with modified sensitivity to esterase-catalyzed bioconversion. J. Peptide Res. 2002, 59, 183–195. [Google Scholar]
- Tang, F.; Borchardt, R.T. Characterization of the efflux transporter(s) responsible for restricting intestinal mucosa permeation of an acyloxyalkoxy-based cyclic prodrug of the opioid peptide DADLE. Pharm. Res. 2002, 19, 780–786. [Google Scholar]
- Tang, F.; Borchardt, R.T. Characterization of the efflux transporter(s) responsible for restricting intestinal mucosa permeation of a coumarinic acid-based cyclic prodrug of the opioid peptide DADLE. Pharm. Res. 2002, 19, 787–793. [Google Scholar]
- Yang, J.Z.; Chen, W.; Borchardt, R.T. In vitro stability and in vivo pharmacokinetic studies of a model opioid peptide, H-Tyr-D-Ala-Gly-Phe-D-Leu-OH (DADLE), and its cyclic prodrugs. J. Pharmacol. Exper. Ther. 2002, 303, 840–848. [Google Scholar]
- Chen, W.; Yang, J.Z.; Andersen, R.; Nielsen, L.H.; Borchardt, R.T. Evaluation of the permeation characteristics of a model opioid peptide, H-Tyr-D-Ala-Gly-Phe-D-Leu-OH (DADLE), and its cyclic prodrugs across the blood-brain barrier using an in situ perfused rat brain model. J. Pharmacol. Exper. Ther. 2002, 303, 849–857. [Google Scholar]
- Liederer, B.M.; Borchardt, R.T. Stability of oxymethyl-modified coumarinic acid cyclic prodrugs of diastereomeric opioid peptides in biological media from various animal species including human. J. Pharm. Sci. 2005, 94, 2198–2206. [Google Scholar]
- Camenish, G.P.; Wang, W.; Wang, B.; Borchardt, R.T. A comparison of the bioconverstion rates and the Caco-2 cell permeation characteristics of coumarin-based cyclic prodrugs and methylester-based linear prodrugs of RGD peptidomimetics. Pharm. Res. 1998, 15, 1174–1181. [Google Scholar]
- Dillon, M.P.; Cai, H.; Maag, H. Application of the "trimethyl lock” to ganciclovir, a pro-prodrug with increased oral bioavailability. Bioorg. Med. Chem. Lett. 1996, 6, 1653–1656. [Google Scholar]
- Greenwald, R.B.; Choe, Y.H.; Conover, C.D.; Shum, K.; Wu, D.; Royzen, M. Drug delivery systems based on trimethyl lock lactonization: poly(ethyleneglycol) prodrugs of amino-containing compounds. J. Med. Chem. 2000, 43, 475–487. [Google Scholar]
- Chandran, S.S.; Dickson, K.A.; Raines, R.T. Latent fluorophore based on the trimethyl lock. J. Am. Chem. Soc. 2005, 127, 1652–1653. [Google Scholar]
- Lavis, L.D.; Chao, T.-Y.; Raines, R.T. Latent blue and red fluorophores based on trimethyl lock. ChemBioChem. 2006, 7, 1151–1154. [Google Scholar]
- Weerapreeyakul, N.; Anorach, R.; Khuansawad, T.; Yenjai, C.; Isaka, M. Synthesis of bioreductive esters from fungal compounds. Chem. Pharm. Bull. 2007, 55, 930–935. [Google Scholar]
- Sample Availability: Not available.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Gomes, P.; Vale, N.; Moreira, R. Cyclization-activated Prodrugs. Molecules 2007, 12, 2484-2506. https://doi.org/10.3390/12112484
AMA Style
Gomes P, Vale N, Moreira R. Cyclization-activated Prodrugs. Molecules. 2007; 12(11):2484-2506. https://doi.org/10.3390/12112484
Chicago/Turabian StyleGomes, Paula, Nuno Vale, and Rui Moreira. 2007. "Cyclization-activated Prodrugs" Molecules 12, no. 11: 2484-2506. https://doi.org/10.3390/12112484