An Efficient Synthesis of a Spirocyclic Oxindole Analogue

{kind=link}
{kind=link}
Abstract
:Introduction
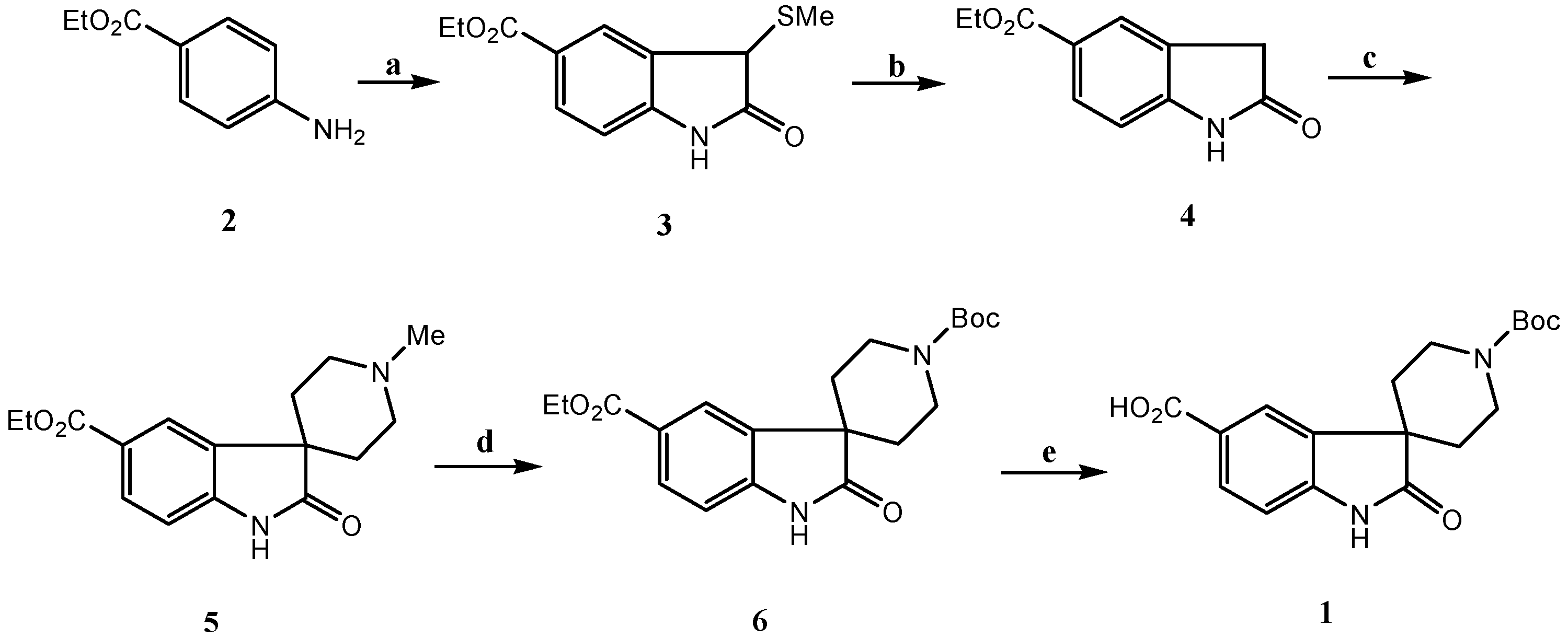
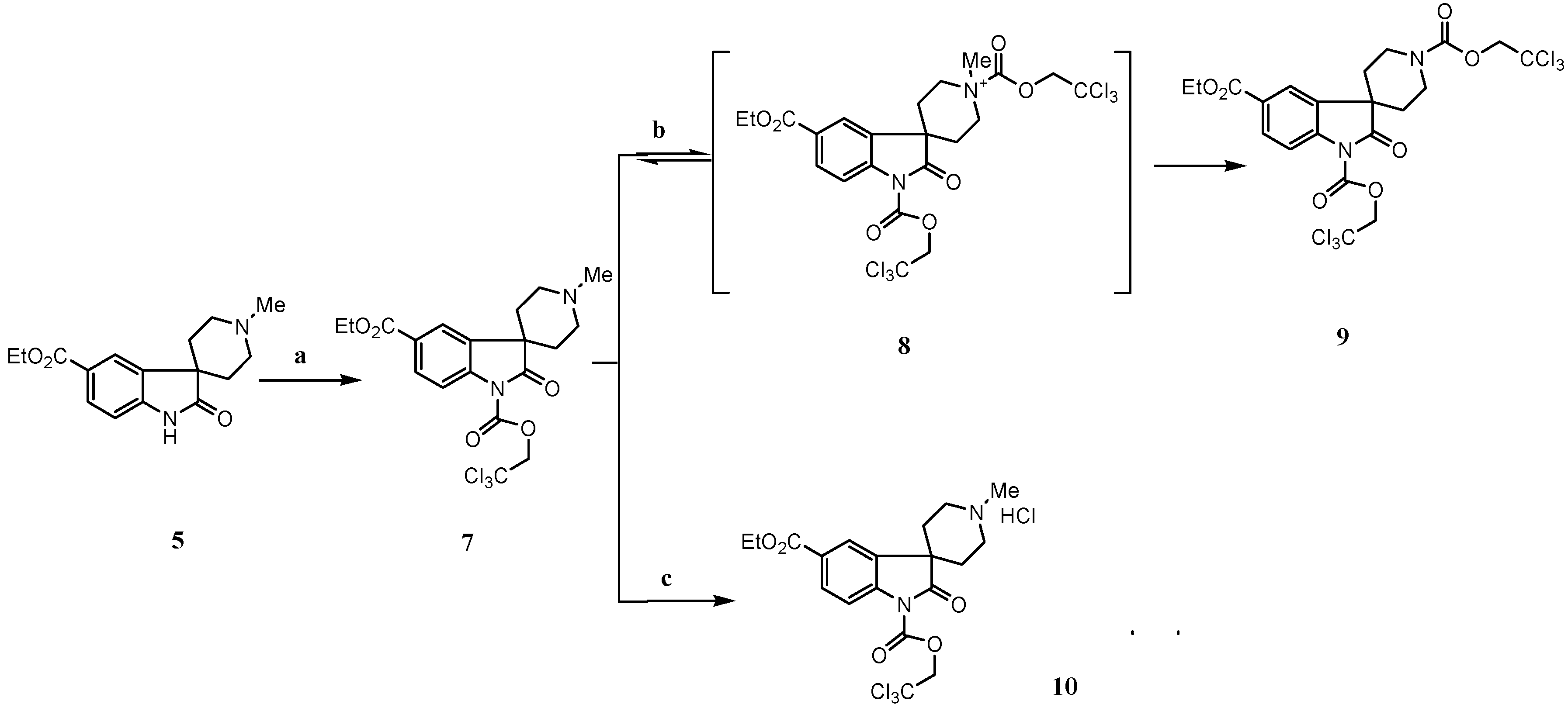
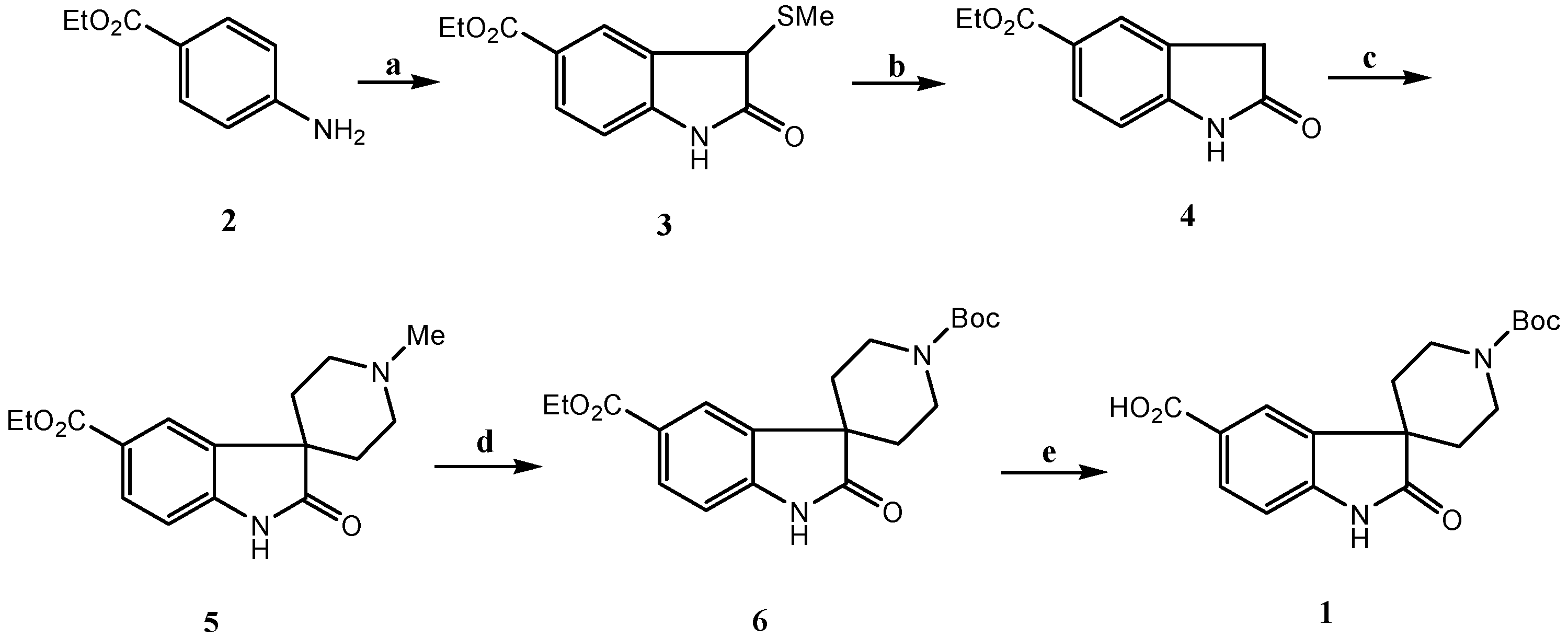
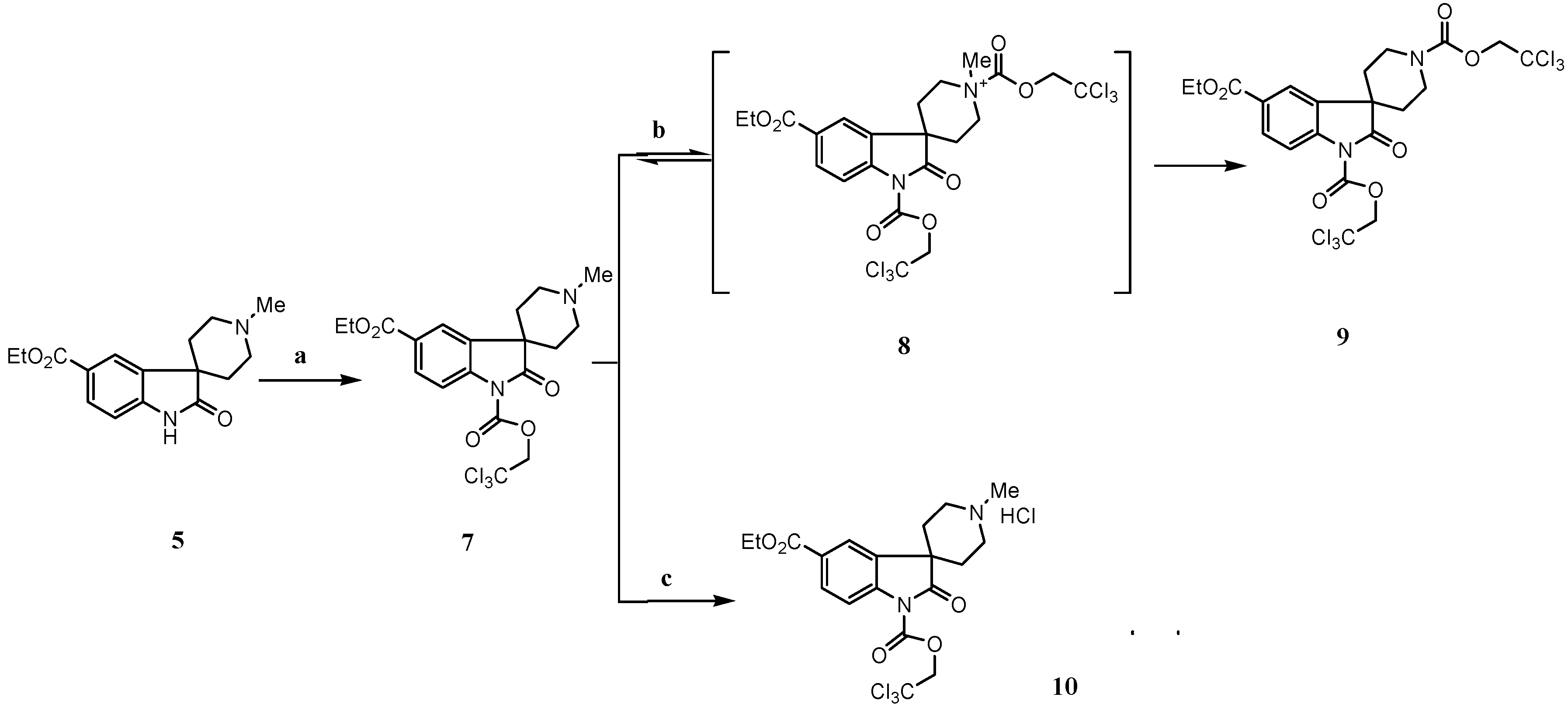
Results and Discussion
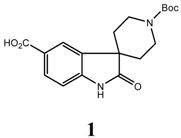
Conclusions
Experimental Section
General
Ethyl 3-(methylthio)-2-oxindoline-5-carboxylate (3).
Ethyl 2-oxindoline-5-carboxylate (4)
Ethyl 1’-methyl-2-oxospiro[indoline-3,4’-piperidine]-5-carboxylate (5)
1’-tert-Butyl 5-ethyl-2-oxospiro [indoline-3,4’-piperidine]-1’5-dicarboxylate (6)
1’-(tert-Butoxycarbonyl)-2-oxospiro[indoline-3,4’-piperidine]-5-carboxylic acid (1)
References and Notes
- Patchett, A. A.; Nargund, R. P.; Tata, J. R.; Chen, M.-H.; Barakat, K. J.; Johnston, D. B. R.; Cheng, K.; Chan, W. W.-S.; Butler, B.; Hickey, G. J.; Jacks, T.; Schleim, K.; Pong, S.-S.; Chuang, L.-Y. P.; Chen, H. Y.; Frazier, E.; Leung, K. H.; Chiu, S.-H. L.; Smith, R. G. Design and Biological Activities of L-163,191 (MK-0677): A Potent, Orally Active Growth Hormone Secretagogue. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7001–7005. [Google Scholar] Barakat, K. J.; Cheng, K.; Chan, W. W.-S.; Butler, B. S.; Jacks, T. M.; Schleim, K. D.; Hora, D. F., Jr.; Hickey, G. J.; Smith, R. G.; Patchett, A. A.; Nargund, R. P. Synthesis and biological activities of phenyl piperazine- based peptidomimetic growth hormone secretagogues. Bioorg. Med. Chem. Lett. 1998, 8, 1431–1436. [Google Scholar]
- Eliott, J. M.; Cascieri, M. A.; Chicchi, G.; Davies, S.; Kellecher, F. J.; Kurtz, M.; Ladduwahetty, T.; Lewis, R. T.; Macleod, A. M.; Merchant, K. J.; Sadowski, S.; Stevenson, G. I. Serine derived NK1 antagonists 1: The effect of modifications to the serine substituents. Bioorg. Med. Chem. Lett. 1998, 8, 1845–1850. [Google Scholar]
- Evans, B. E.; Leighton, J. L.; Rittle, K. E.; Gilbert, K. F.; Lundell, G. F.; Gould, N. P.; Hobbs, D. W.; Dipardo, R. M.; Veber, D. F.; Pettibone, D. J.; Clineschmidt, B. V.; Anderson, P. S.; Freidinger, R. M. Orally active, nonpeptide oxytocin antagonists. J. Med. Chem. 1992, 35, 3919–3927. [Google Scholar]
- Efange, S. M. N.; Kamath, A. P.; Khare, A. B.; Kung, M.-P.; Mach, R. H.; Parsons, S. M. N-Hydroxyalkyl Derivatives of 3β-Phenyltropane and 1-Methylspiro[1H-indoline-3,4'-piperidine]: Vesamicol Analogues with Affinity for Monoamine Transporters. J Med. Chem. 1997, 40, 3905–3914. [Google Scholar]
- Goehring, R. R. Synthesis of a Spiro Oxindole Analogue as a Putative Replacement for Pro2-pro3-gly4-phe5 in Bradkinin Antagonists. Org. Prep. Proced. Int. 1995, 27, 691–694. [Google Scholar]
- Edmondson, S.; Danishefsky, S. J.; Sepp-Lorenzino, L.; Rosen, N. Total Synthesis of Spirotryprostatin A, Leading to the Discovery of Some Biologically Promising Analogues. J. Am. Chem. Soc. 1999, 121, 2147–2155. [Google Scholar]
- For a review of oxindole synthesis, see Sundberg, R. J. “The Chemistry of Indoles”; Academic Press: New York, N. Y., 1970. [Google Scholar]
- Sumpter, W. C. The chemistry of Oxindole. Chem. Rev. 1945, 37, 443–479. [Google Scholar] Beckett, A. H.; Daisley, R. W.; Walker, J. Substituted oxindole—I : The preparation and spectral characteristics of some simple oxindole derivatives. Tetrahedron 1968, 24, 6093–6109. [Google Scholar] Walker, J.; Daisley, R. W.; Beckett, A. H. Substituted oxindoles. III. Synthesis and pharmacology of some substituted oxindoles. J. Med. Chem. 1970, 13, 983–985. [Google Scholar]
- Gassman, P. G.; van Bergen, T. J. Oxindoles. New, general method of synthesis. J. Am. Chem. Soc. 1974, 96, 5508–5512. [Google Scholar]
- Freund, R.; Mederski, W. W. K. R. A Convenient Synthetic Route to Spiro[indole-3,4′-piperidin]-2-ones. Helv. Chim. Acta 2000, 83, 1247–1255. [Google Scholar] ; (b) ref. [5]; Jackson, A. H.; Smith, A. E. Electrophilic substitution in indoles—II : The formation of 3,3-spirocyclic indole derivatives from tryptamines and their rearrangement to β-carbolines. Tetrahedron 1968, 24, 403–413. [Google Scholar]
- For examples of C-3 alkylation of the oxindole dianion, see: Kende, A. S.; Hodges, J. C. Regioselective C-3 alkylations of oxindole dianion. Synth. Commun. 1982, 12, 1–10. [Google Scholar] DeMarinis, R. M.; Gallagher, G.; Hall, R. F.; Franz, R. G.; Webster, C.; Huffman, W. F.; Schwarta, M. S.; Kaiser, C.; Ross, S. T.; Wilson, J. W.; Hieble, P. Syntheses and in vitro evaluation of 4-(2-aminoethyl)-2(3H)-indolones and related compounds as peripheral prejunctional dopamine receptor agonists. J. Med. Chem. 1986, 29, 939–947. [Google Scholar] Carroll, W. A.; Grieco, P. A. Biomimetic total synthesis of pseudotabersonine: a novel oxindole-based approach to construction of Aspidosperma alkaloids. J. Am. Chem. Soc. 1993, 115, 1164–1165. [Google Scholar] Karp, G. M. Preparation and Reactions of Indolin-2(3H)-ones. A Review. Org. Prep. Proc. Int. 1993, 25, 481–514. [Google Scholar]
- All reaction conditions are the same as the given procedure for compound 5. The N-methyl bis(2-chloroethyl)amine free base was generated in situ by neutralization of the dichloroethylmine hydrochloric salt with the base used.
- For the dealkylation of tertiary amine, see: Katritzky, A. R.; Meth-Cohn, O.; Rees, C. W. Comprehensive Organic Functional Group Transformations; Pergamon: Oxford, 1995; Volume 2, pp. 302–304. [Google Scholar] Cooley, J. H.; Evain, E. J. Amine Dealkylation with Acyl Chlorides. Synthesis 1989, 1–7. [Google Scholar] Olofson, R. A.; Martz, J. T.; Senet, J.-P.; Piteau, M.; Malfroot, T. A new reagent for the selective, high-yield N-dealkylation of tertiary amines: improved syntheses of naltrexone and nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. [Google Scholar] Howarth, N. M. Manipulation of substituents at nitrogen in tropanes, homotropanes, and dehydro- derivatives. Tetrahedron 1998, 54, 10899–10914. [Google Scholar] Olofson, R. A.; Schnur, R. C.; Bunes, L.; Pepe, J. P. Selective N-dealkylation of tertiary amines with vinyl chloroformate: An improved synthesis of naloxone. Tetrahedron Lett. 1977, 18, 1567–1570. [Google Scholar] Montzka, T. A.; Matiskella, J. D.; Partyka, R. A. 2,2,2-trichloroethyl chloroformate: A general reagent for demethylation of tertiary methylamines. Tetrahedron Lett. 1974, 15, 1325–1327. [Google Scholar]
- Sample Availability: Samples of compounds 1, 3, 4 and 5 are available from the authors.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Teng, D.; Zhang, H.; Mendonca, A. An Efficient Synthesis of a Spirocyclic Oxindole Analogue. Molecules 2006, 11, 700-706. https://doi.org/10.3390/11090700
Teng D, Zhang H, Mendonca A. An Efficient Synthesis of a Spirocyclic Oxindole Analogue. Molecules. 2006; 11(9):700-706. https://doi.org/10.3390/11090700
Chicago/Turabian StyleTeng, Dawei, Hongxing Zhang, and A. Mendonca. 2006. "An Efficient Synthesis of a Spirocyclic Oxindole Analogue" Molecules 11, no. 9: 700-706. https://doi.org/10.3390/11090700
APA StyleTeng, D., Zhang, H., & Mendonca, A. (2006). An Efficient Synthesis of a Spirocyclic Oxindole Analogue. Molecules, 11(9), 700-706. https://doi.org/10.3390/11090700