Introduction
Lanthanide complexation chemistry has generated considerable interest and has progressed rapidly over the past several years. In particular, gadolinium(III) complexes, derived from polyaminopoly-carboxylic acids, are habitually used as contrast agents (CAs) in Magnetic Resonance Imaging (MRI) [
1]. The most commonly Gd-complexes used for this propose are [dtpa(Gd)(H
2O)]
2- and [dota(Gd)(H
2O)]
-, where dtpa = diethylenetriaminepentaacetic and dota = 1,4,7,10-tetraazacyclo-dodecane-1,4,7,10-tetraacetic acids, respectively. Recently, several examples of chelating ligands containing heterocyclic rings have been reported. Concretely, pyridine and tetrazole systems have been studied as examples of nitrogen heterocyclic rings that participate in complexation with the metal center [
2]. As part of our studies of this topic, we have recently reported a novel series of bispyrazoles exhibiting improved relaxivity properties in an
in vitro assessment of contrast agent efficacy. Our results with their corresponding Gd(III)-complexes showed that the pyrazole ring N-2 participates in metal complexation, giving the corresponding double tetradentate complexes [
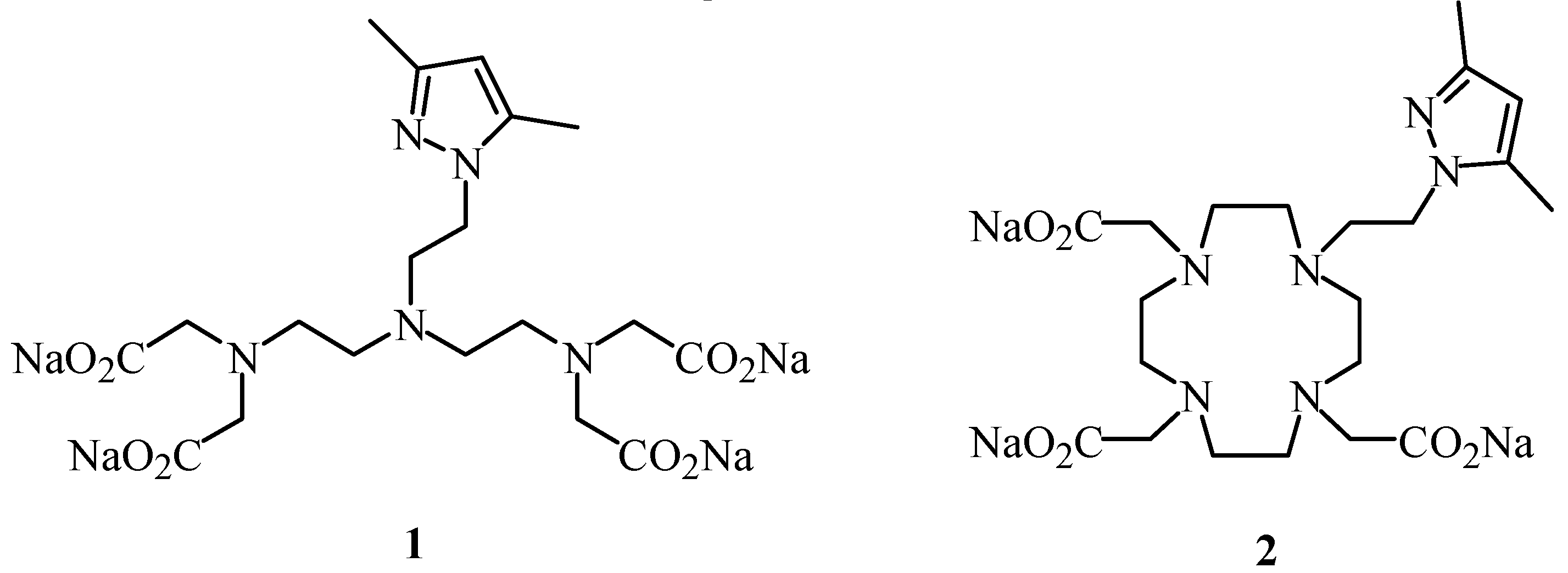
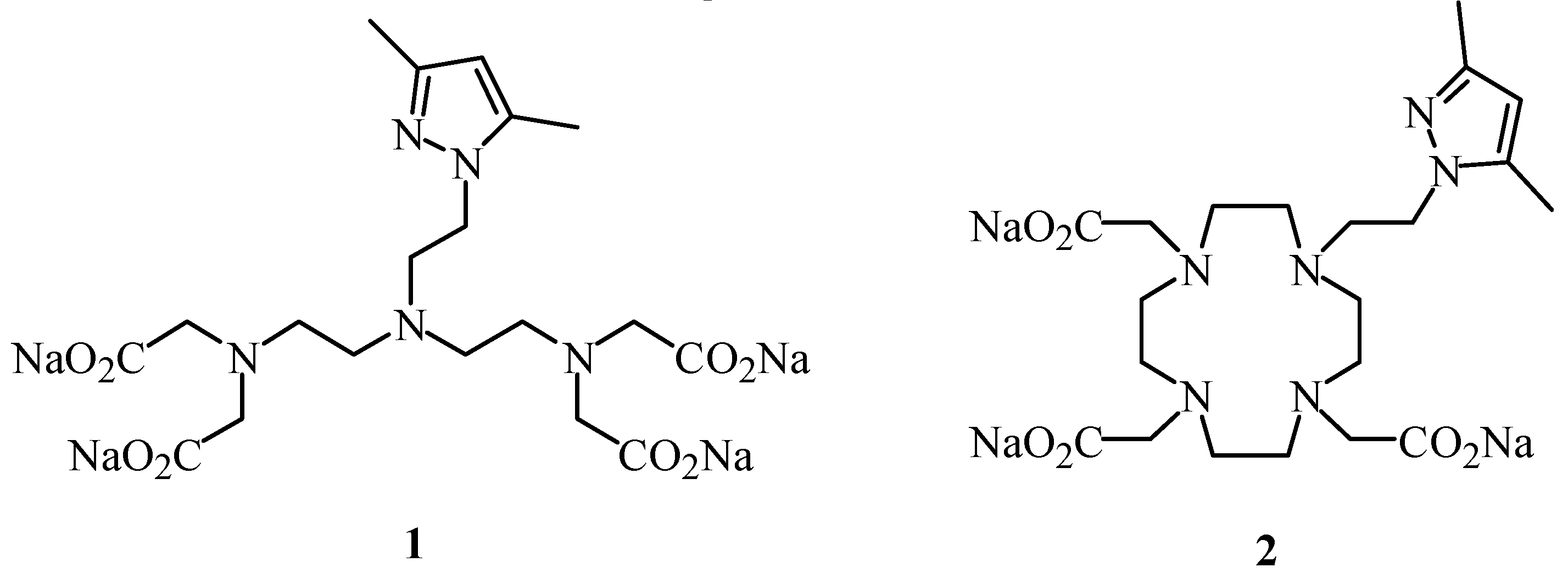
3]. In our efforts to find new contrast agents with higher relaxivity and improved thermodynamic stability, we now describe the synthesis of the chelating ligands
1 and
2, as examples of linear and macrocyclic polyaminepoly-carboxylic acids containing a pyrazolylethyl arm (
Figure 1).
We have analysed the effect of the 3,5-dimethylpyrazolylethyl moiety on the magnetic and complexation properties of the corresponding lanthanide complexes of 1 and 2, as compared with Gd(III)-dtpa and Gd(III)-dota. Furthermore, we have performed ab inito theoretical calculations that support our findings.
Results and Discussion
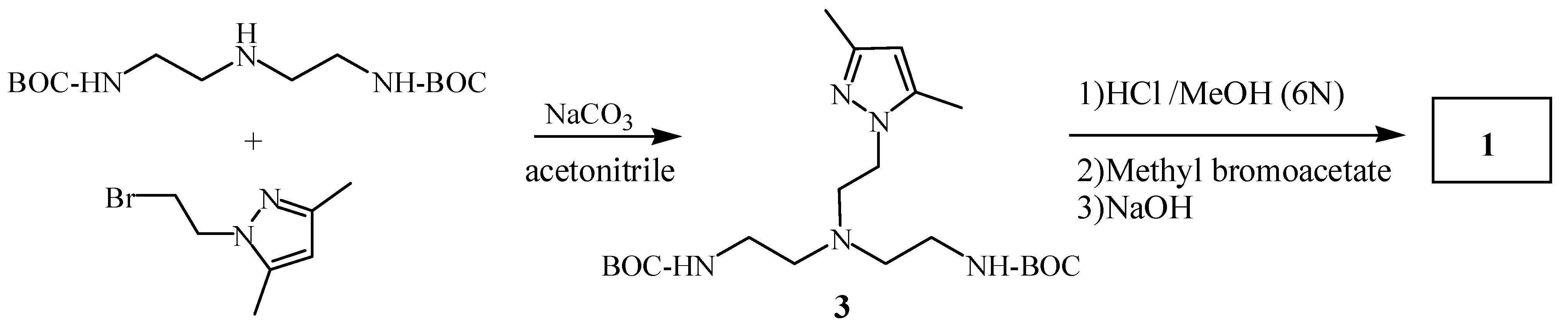
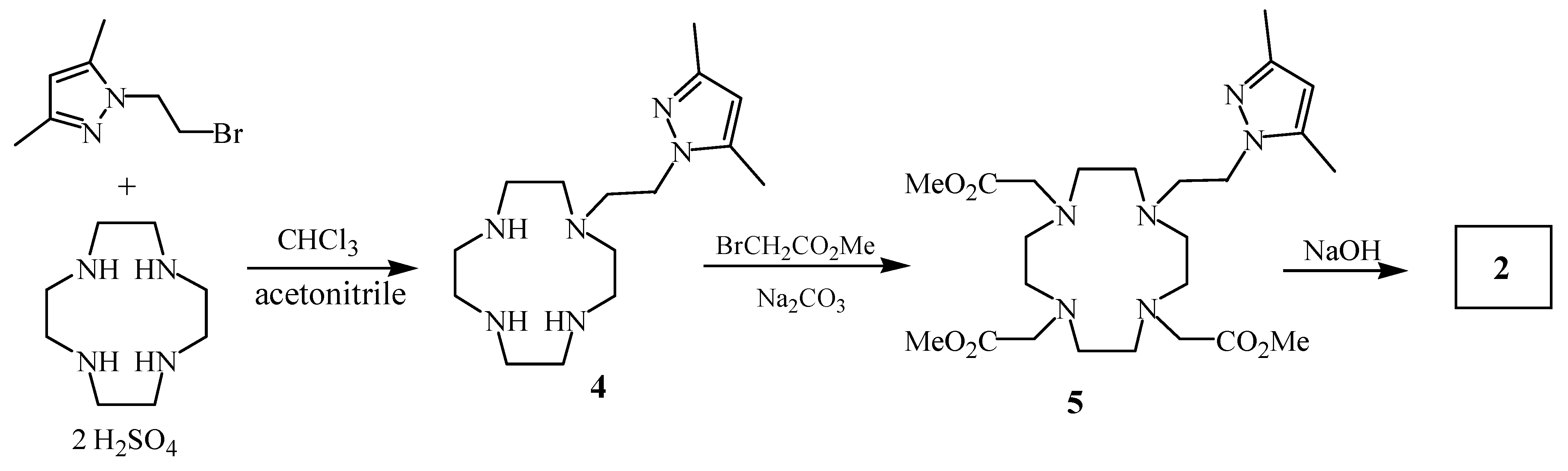
Ligand
1 was prepared starting from
N,
N’-Boc-diethylenetriamine [
4] and 2-bromoethyl-3,5-dimethylpyrazole [
5] yielding compound
3. Subsequently, the
tert-butoxycarbonyl groups of compound
3 were removed in an acidic medium and the resulting amine was alkylated with methyl bromoacetate. Finally, basic hydrolysis of the corresponding ester gave compound
1 in 83 % overall chemical yield (
Scheme 1).
Analogously, monoalkylation of 1,4,7,10-tetraazacyclododecane (cyclen) with 2-bromoethyl-3,5-dimethylpyrazole using the previously described reaction conditions [
6] yielded compound
4, which was refluxed with methyl bromoacetate in acetonitrile to give the ester
5. Ligand
2 was finally prepared by basic hydrolysis of the methyl aminoester
5 (
Scheme 2).
Gd(III)-complexes of chelating ligands 1 and 2 were synthesized by mixing equimolecular amounts of the corresponding ligands and GdCl3·6H2O in water at room temperature for several minutes. They were characterized by ESI-MS (negative ion mode) which indicated a 1:1 stoichiometry for the complexes.
The efficacy of a potential contrast agent can be evaluated by its proton relaxivity in aqueous solutions (r
1 and r
2) expressed in s
-1mM
-1.
Table 1 shows the longitudinal and transversal relaxivity values, r
1 and r
2, of
Gd(III)-1 and
Gd(III)-2, as determined at 60 MHz.
Gd(III)-1 exhibited maximum r
1(2) values, even higher than dtpa, while
Gd(III)-2 and dota presented similar relaxivity. These results probably result from a bigger complexation capacity and minor hydration number for macrocyclic complex,
Gd(III)-2.
Table 1.
Longitudinal and transversal relaxivities (r1 and r2) of aqueous solutions of Gd(III)-complexes of 1, 2 and Gd(III)-dtpa and Gd(III)-dota determined at 1.5 T.a
Table 1.
Longitudinal and transversal relaxivities (r1 and r2) of aqueous solutions of Gd(III)-complexes of 1, 2 and Gd(III)-dtpa and Gd(III)-dota determined at 1.5 T.a
| Gd-Complexb | r1(s-1mM-1) c | r2(s-1mM-1) c |
|---|
| Gd(III)-1 | 5.14 ± 0.03 | 5.40 ± 0.03 |
| Gd(III)-2 | 1.51 ± 0.003 | 1.73± 0.01 |
| Gd(III)-dtpa | 3.92 ± 0.01 | 4.39 ± 0.01 |
| Gd(III)-dota | 1.58 ± 0.01 | 1.82 ± 0.01 |
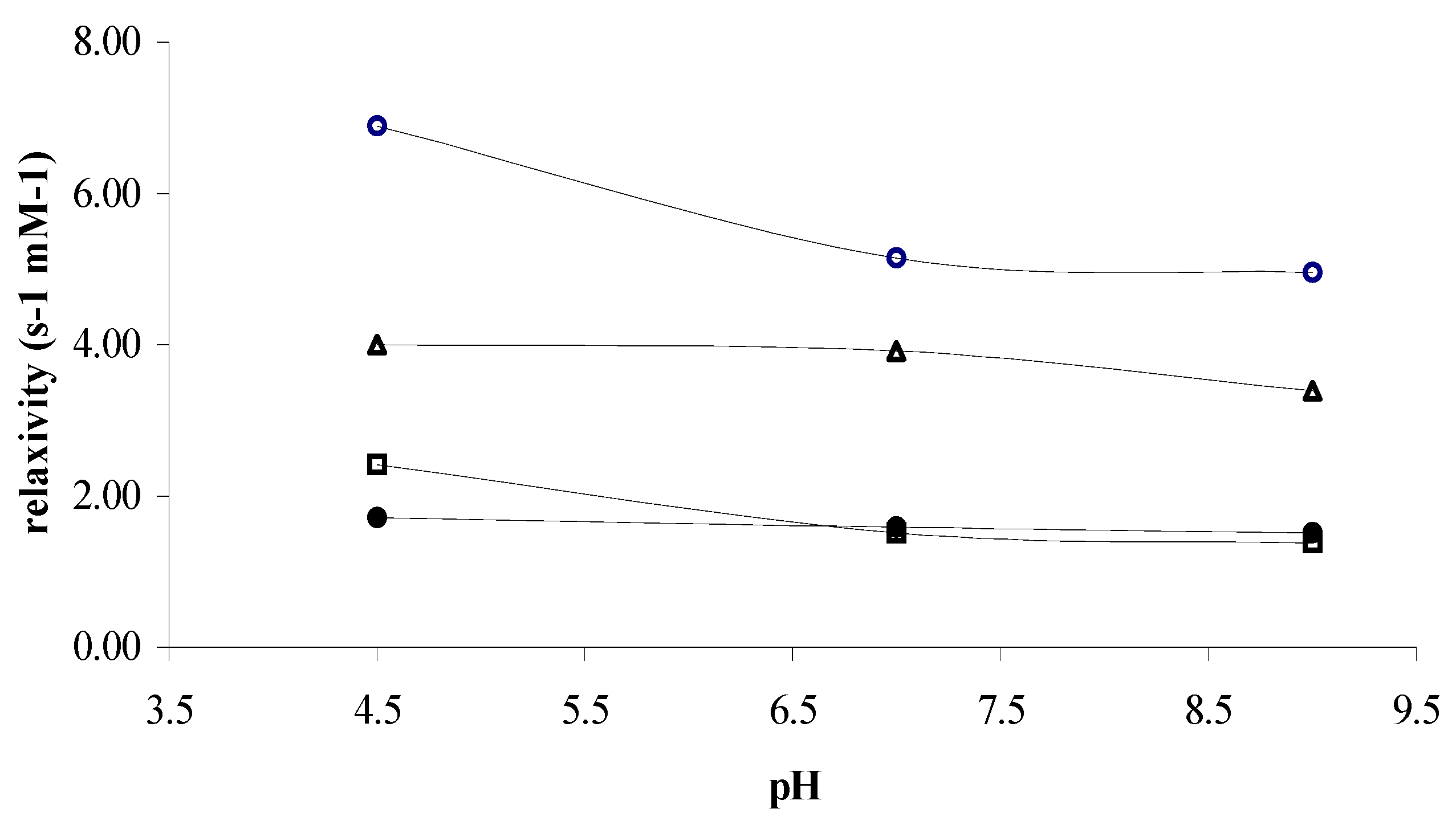
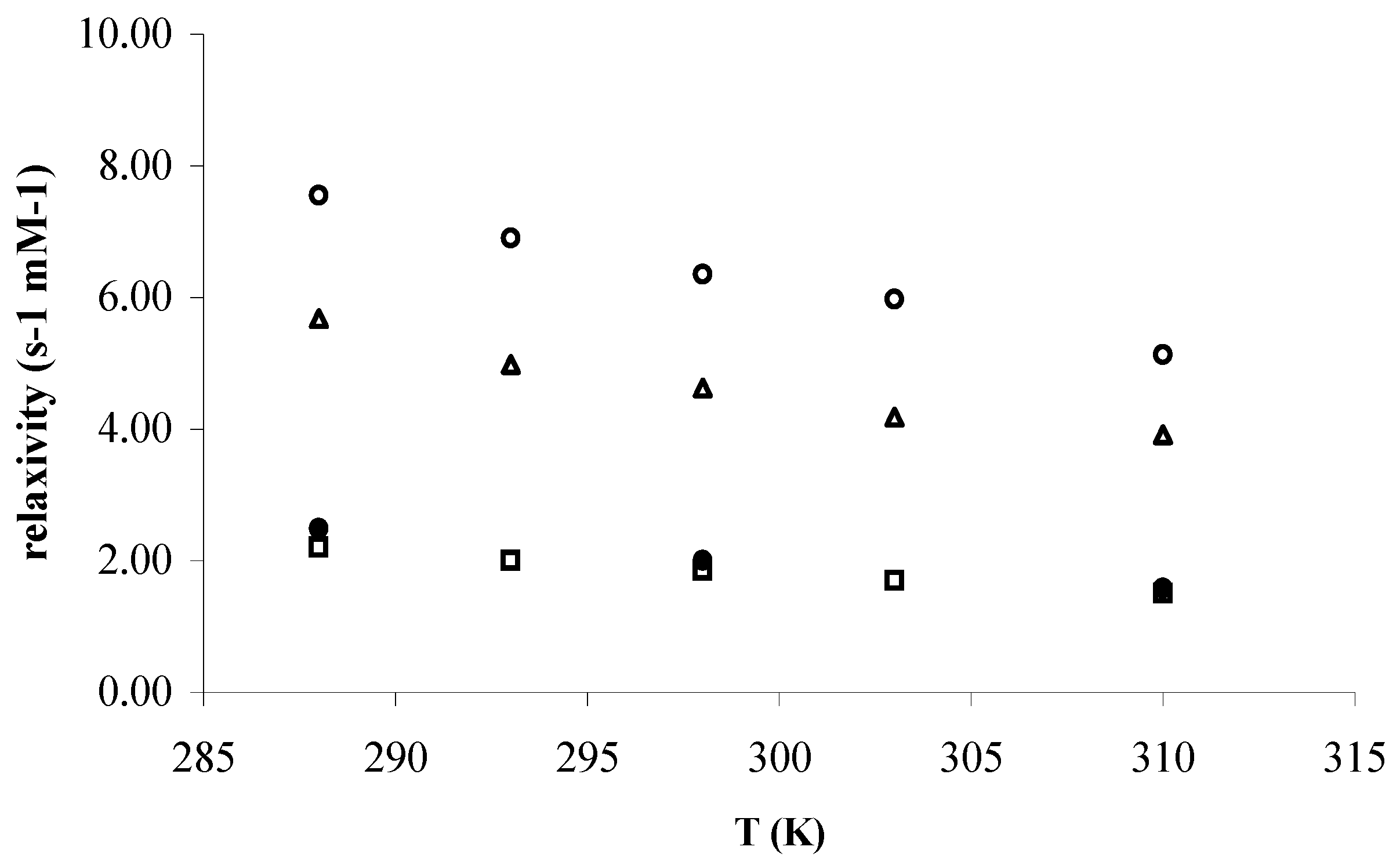
Figure 2 and
Figure 3 depict the temperature and pH dependences of r
1 for the mentioned complexes at 60 MHz, respectively. Considering the Solomon-Bloembergen–Morgan (SBM) Theory, the temperature dependence of r
1 is a qualitative assessment of the
τM, where r
1(2) is the longitudinal and transversal relaxivity, q, the hydration number (water molecules in the inner-sphere),
TM the relaxation time of water protons of the water bound to metal center and finally,
τM the residence time of water in inner-sphere (equation 1).
Considering our experimental results, when the temperature decreases a dramatic increase of the water exchange rate in the inner-sphere can be observed for
Gd(III)-1, whereas in the case of
Gd(III)-2 this acceleration is less pronounced (
Figure 2).
Figure 2.
Temperature dependence of the proton longitudinal relaxivity of the water solution 1 mM of ○) Gd(III)-1, △) Gd(III)-dtpa, □) Gd(III)-2, and ●) Gd(III)-dota at pH ~ 7 and 60 MHz.
Figure 2.
Temperature dependence of the proton longitudinal relaxivity of the water solution 1 mM of ○) Gd(III)-1, △) Gd(III)-dtpa, □) Gd(III)-2, and ●) Gd(III)-dota at pH ~ 7 and 60 MHz.
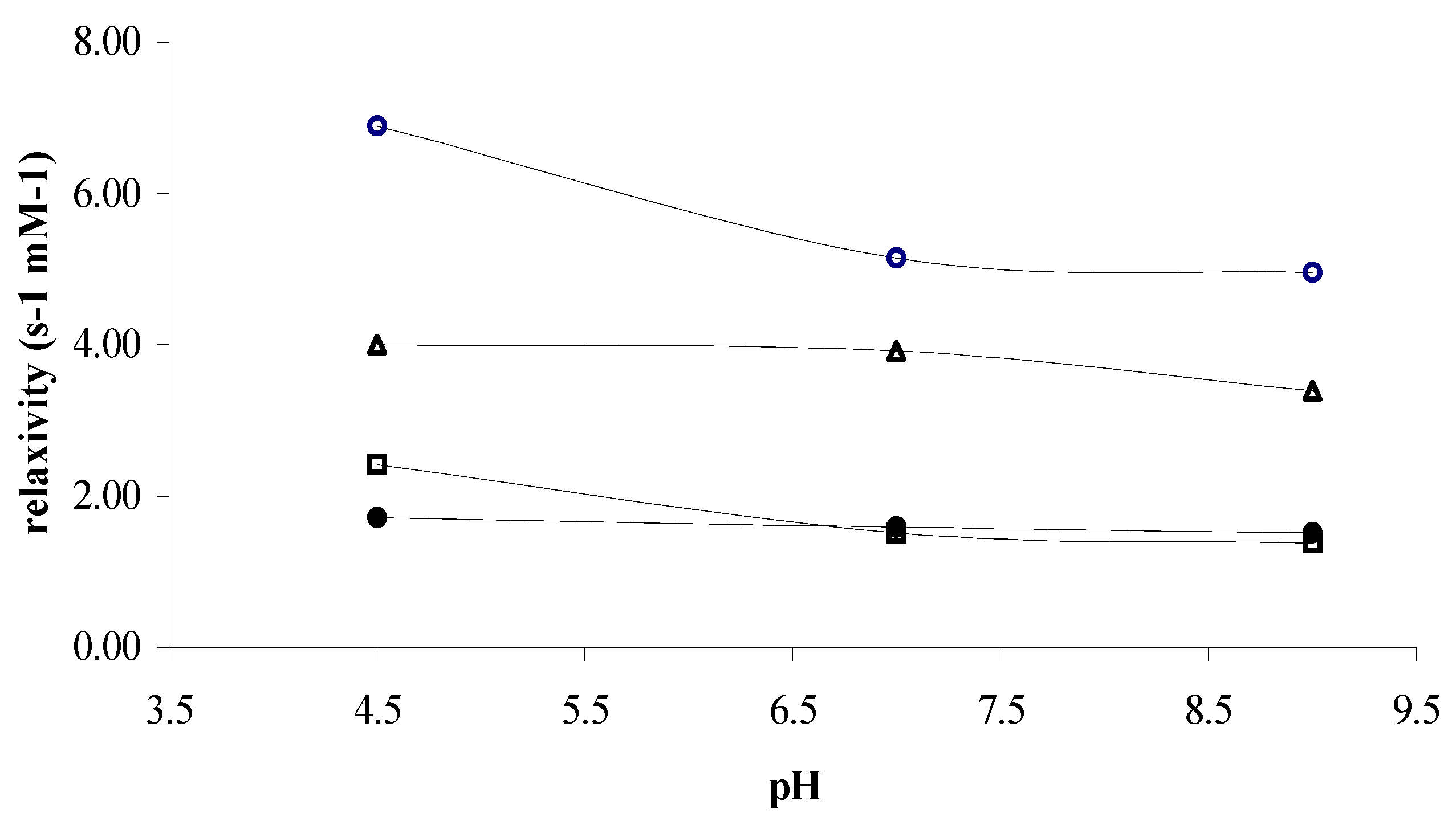
On the other hand, the relaxivity of
Gd(III)-dtpa and
Gd(III)-dota remains constant over a pH range between 4.5-9, exhibiting a constant hydration number under these conditions (
Figure 3). In our cases, an increase of r
1 was observed at acidic pH, with the highest value of relaxivity being observed for the Gd(III)-complex of
1. In contrast,
Gd(III)-2 showed a weak dissociation at acidic pH, probably due to the strong kinetic stability of this complex.
Figure 3.
pH dependence of the proton longitudinal relaxivity of the water solution 1 mM of ○) Gd(III)-1, △) Gd(III)-dtpa, □) Gd(III)-2, and ●) Gd(III)-dota, at 37 °C and 60 MHz.
Figure 3.
pH dependence of the proton longitudinal relaxivity of the water solution 1 mM of ○) Gd(III)-1, △) Gd(III)-dtpa, □) Gd(III)-2, and ●) Gd(III)-dota, at 37 °C and 60 MHz.
Furthermore, we have determined the hydration number, q, by
17O-NMR of the corresponding dysprosium complexes, following the method reported by Alpoim
et al. [
7], through a simple comparison of chemical shift induced by the aquodysprosium ion (d.i.s.) and the corresponding Dy-complexes, both of them as function of the complex concentration. Then, hydration numbers (q) of the corresponding complexes have been measured from the slope of the concerned line as compared with the slope for DyCl
3 (q = 8) [
8]. On the other hand, q values described for
Gd(III)-dtpa and
Gd(III)-dota are 1.3 [
8] and 1.0 [
9] water molecules in the coordination sphere. In this way, our results show that the slope of the lines for DyCl
3,
Gd(III)-1 and
Gd(III)-2, are 305.4 (r
2 0.98), 62.7 (r
2 0.95) and 39.1 (r
2 0.98) ppm M
-1, respectively. Consequently,
Gd(III)-1 and
Gd(III)-2 contain 1.6 and 1.0 water molecules in inner-sphere per dysprosium ion, respectively.
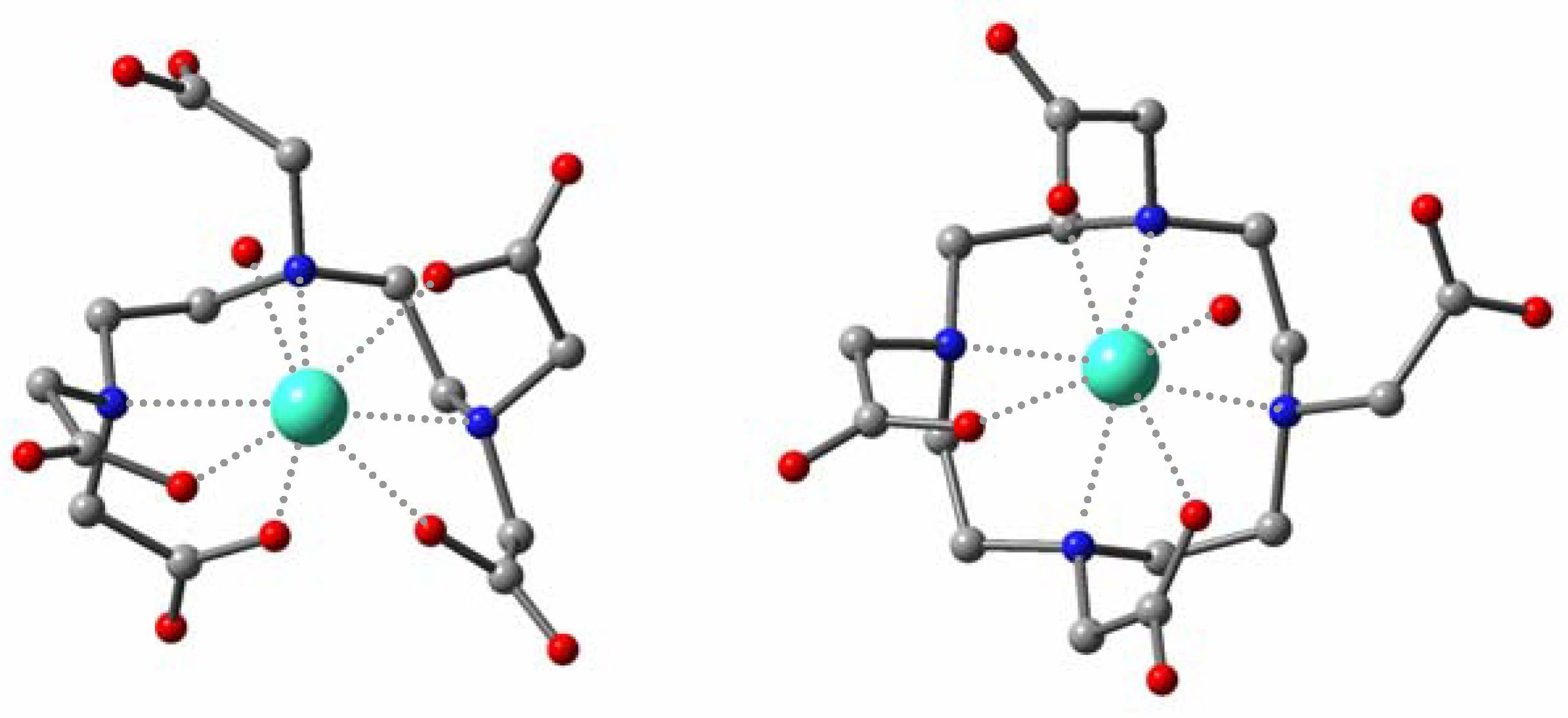
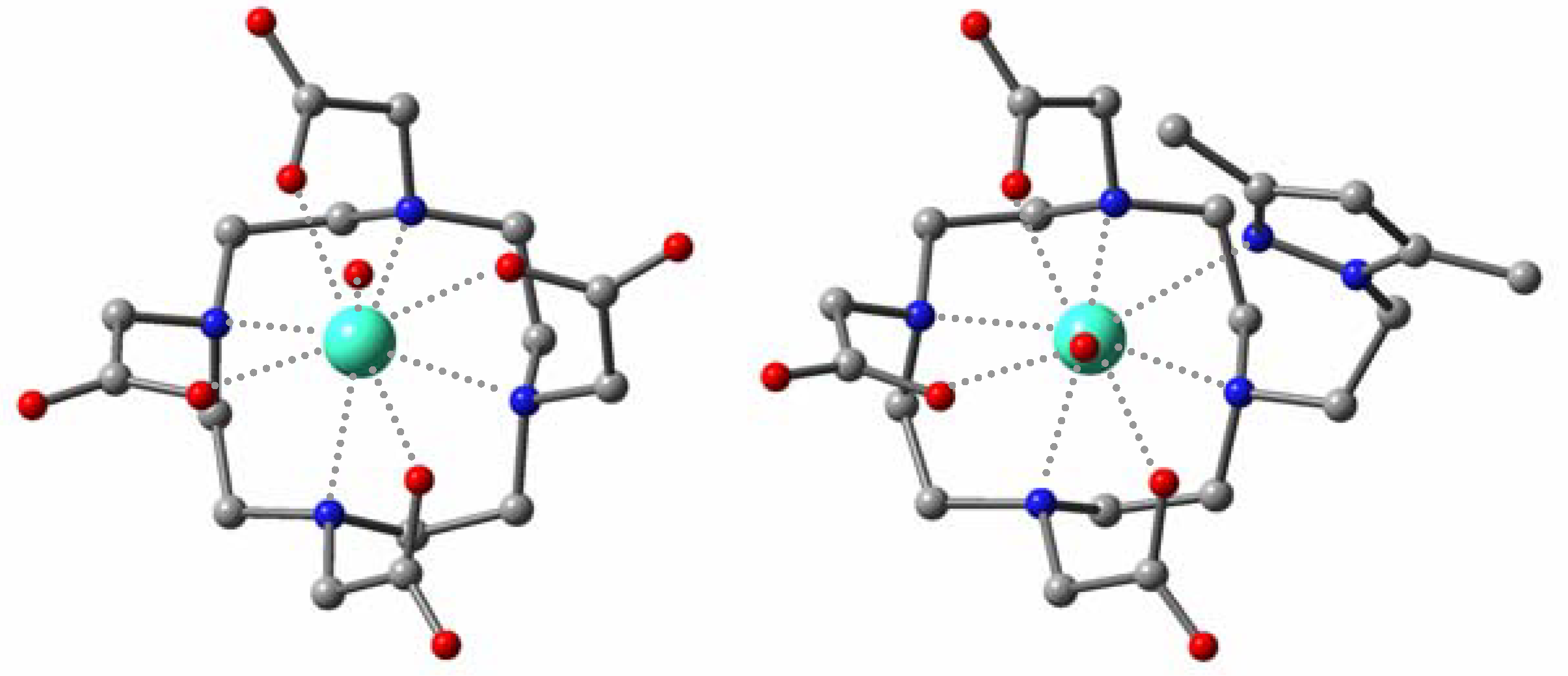
The optimized structures in aqueous phase at the
ab initio level for
Gd(III)-dtpa and
Gd(III)-dota systems revealed a good agreement with the available crystallographic data, so we adopted this methodology for the novel derivatives. The calculated structures show that the introduction of a pyrazolethyl arm results in a shortening of the Gd-O and Gd-N distances for the acyclic system (
Table 2). The methyl substituent induces steric compression around the water binding site, increasing the Gd-O
W distance and probably favouring its departure [
10]. For the macrocyclic species (
Figure 4), the metal center is displaced toward the plane formed by the carboxylate groups, giving rise to longer Gd-N and shorter Gd-O and Gd-O
W distances. Moreover, the azole ligand appears worse accommodated in the coordination sphere than in
Gd(III)-1 due to the higher rigidity of the cyclen framework.
Table 2.
Mean distances (Å) and standard deviations (in parenthesis) from theoretical calculations of [Gd-L(H2O)]n– complexes in aqueous phase.
Table 2.
Mean distances (Å) and standard deviations (in parenthesis) from theoretical calculations of [Gd-L(H2O)]n– complexes in aqueous phase.
| Complex | Gd-O | Gd-N | Gd-Naz | Gd-OW | Gd-PAa | Gd-PBb |
|---|
| Gd(III)-dtpa | 2.369 (0.035) | 2.689 (0.064) | –– | 2.616 | –– | –– |
| Gd(III)-1 | 2.356 (0.027) | 2.678 (0.075) | 2.758 | 2.698 | –– | –– |
| Gd(III)-dota | 2.351 (0.025) | 2.676 (0.010) | –– | 2.612 | 0.777 | 1.623 |
| Gd(III)-2 | 2.333 (0.017) | 2.686 (0.048) | 2.775 | 2.577 | 0.701 | 1.633 |
Figure 4.
Optimized geometries for the Gd(III)-dota (left) and Gd(III)-2 (right) complexes in aqueous phase.
Figure 4.
Optimized geometries for the Gd(III)-dota (left) and Gd(III)-2 (right) complexes in aqueous phase.
Regarding the thermodynamic stability, while
Gd(III)-dota is slightly more stable than the
Gd(III)-dtpa complex, which agrees with the experimental evidence,
Gd(III)-2 seems slightly less stable than
Gd(III)-1. The complexes bearing a pyrazolethyl ligand moiety are >10 kcal/mol less stable than the parent systems, for both acyclic and macrocyclic structures (
Table 3), as one might expect from the replacement of a charged carboxylate moiety by an azole ligand.
Table 3.
Energy difference, ΔETOTAL = ECOMPLEX – ELIGAND - EGd(III), and ΔEREL, energy relative to the pattern complex (in kcal/mol, DFT level).
Table 3.
Energy difference, ΔETOTAL = ECOMPLEX – ELIGAND - EGd(III), and ΔEREL, energy relative to the pattern complex (in kcal/mol, DFT level).
| Complex | ΔETOTAL | ΔEREL |
|---|
| Gd(III)-dtpa | –146.55 | 0.00 |
| Gd(III)-1 | –135.74 | +10.80 |
| Gd(III)-dota | –147.80 | 0.00 |
| Gd(III)-2 | –135.28 | +12.52 |
The kinetic stability of the Gd(III)-complexes plays a critical role in determining the toxicity, and it can be characterized by analysing exchange reactions with other ions. Thus, the de-coordination of the paramagnetic ion and interaction of the ligand with endogenous cations, such as Zn(II), Ca(II) and Mg(II), to form mono- or binuclear complexes, gives rise to the release of Gd(III), a highly toxic metal. Assuming that the first step of the de-coordination process is the rate-limiting step [
11], we have calculated the transition structures involved (
Figure 5).
Figure 5.
Optimized transition structures for the first step of the dissociation process of the Gd(III)-dtpa (left) and Gd(III)-dota (right) complexes.
Figure 5.
Optimized transition structures for the first step of the dissociation process of the Gd(III)-dtpa (left) and Gd(III)-dota (right) complexes.
The computed activation energy values suggest that the first step of the complex dissociation is kinetically more difficult for the macrocyclic system (
Table 4). The results are in agreement with the experimental evidence suggesting a higher kinetic stability, inertness, for the rigid macrocyclic as compared with the acyclic ligands.
Table 4.
Activation energy (in kcal/mol, DFT level) for the first step of the dissociationprocess of the Gd(III)-dtpa and Gd(III)-dota complexes.
Table 4.
Activation energy (in kcal/mol, DFT level) for the first step of the dissociationprocess of the Gd(III)-dtpa and Gd(III)-dota complexes.
| Complex | Gas Phase | Aqueous Phasea |
|---|
| Gd(III)-dtpa | 18.48 | 12.49 |
| Gd(III)-dota | 30.06 | 22.26 |
Experimental
General
Melting points were measured on a microscope hot stage and are uncorrected. Elemental analyses were performed with Perkin-Elmer 240 apparatus. Mass spectra were carried out VGAutoSpec mass spectrometer. IR spectra were determined on a Bruker Vector 22 spectrophotometer equipped with an ATR accessory. NMR spectra were recorded with a Bruker DRX-400 instrument, operating at 400.13 MHz for 1H, and 100.033 MHz for 13C. 1H and 13C chemical shifts (δ) in CDCl3 are given relative to an internal tetramethylsilane standard and 13C δ in D2O are given relative to an external DMSO-d6 signal with an accuracy of ± 0.01 for 1H and ± 0.1 ppm for 13C. The residual water signal in 1H-NMR spectra in D2O solution was suppressed when necessary using a 1 s (low power, 0.5 watts) presaturating pulse applied with decoupler. 1H-1H coupling constants (J) are accurate to ± 0.2 Hz for 1H-NMR spectra. TLC chromatography was performed on DC-Aulofolien/Kieselgel 60 F245 (Merck) and column chromatography though silica gel Merck 60 (230-400 mesh). D2O (99.9 D) was purchased from Apollo Scientific (Stockport, Great Britain). The rest of the products and dry solvents were obtained from Aldrich.
(2-{(2-tert-butoxycarbonylaminoethyl)-[2-(3,5-dimethylpyrazol-1-yl)-ethyl]amino}ethyl) carbamic acid tert-butyl ester (3)
A mixture of the bromoethyl-3,5-dimethylpyrazole (345 mg; 1.7 mmol) and N,N’-Boc-diethylentriamine (1g; 3.3 mmol) was heated at 80 °C for 15 h. After cooling, CH2Cl2 (10 mL) was added. The solid formed was subsequently filtered and the solvent was evaporated in vacuo. Purification of the residue by column chromatography on silica gel (98:2 CH2Cl2-EtOH) gave 3 (700 mg; 97 %) as a white solid (m.p. 106-108 °C, from hexane-EtOH). 1H-NMR (CDCl3): δ 5.81 (s, 1 H, H4), 5.29 (br s, 2 H, NH-BOC), 3.99 (apparent t, 2 H, 3J (H,H) = 5.6, 5.5 Hz, CH2-N(azole)), 3.01 (m, 4 H, CH2-NH-BOC), 2.73 (t, 2 H, 3J (H,H) = 5.5 Hz, CH2-N), 2.52 (apparent t, 4 H, 3J (H,H) = 5.6, 5.3 Hz, CH2-N), 2.26 (s, 3 H, CH3), 2.23 (s, 3 H, CH3), 1.49 (s, 18 H, CH3); 13C-NMR (CDCl3, 300 K): δ 156.2, 138.5, 105.1, 78.7, 53.6, 46.6, 38.5, 28.4, 13.3, 10.9; IR (ATR): ν 3396, 3264, 1698, 1505, 1458, 1249, 1164 cm-1; Elemental analysis: calcd. (%) for (C21H39N5O4): C, 59.27; H, 9.24; N, 16.46; found C, 59.30; H, 9.00; N, 16.46.
[(2-{[2-(Bis-carboxymethylamino)ethyl]-[2-(3,5-dimethylpyrazol-1-yl)ethyl]amino}ethyl)carboxy-methylamino] acetic acid tetrasodium salt (1)
A solution of the corresponding carbamate 3 (800 mg; 1.88 mmol) in HCl-MeOH (6 N, 10 mL) was stirred at room temperature for 1 h. The solvent was then evaporated in vacuo to give a white solid. A suspension of the residue obtained, methyl bromoacetate (1.44 g; 9.4 mmol) and K2CO3 (2.6 g; 18.8 mmol) in acetonitrile (30 mL) was next refluxed for 17 h. After cooling, the solid formed was filtered and the solvent was evaporated in vacuo. Purification of the residue by column chromatography on silica gel (95:5 CH2Cl2-EtOH) gave the corresponding tetramethyl ester as a yellow oil (817 mg; 85 %); 1H-NMR (CDCl3, 300 K): δ 5.71 (s, 1 H, H4), 3.96 (apparent t, 2 H, 3J (H,H) = 6.8, 6.7 Hz, CH2-N(azole)), 3.68 (s, 12 H, OCH3), 3.51 (s, 8 H, CH2CO2Me), 2.33 (apparent t, 2 H, 3J (H,H) = 6.6, 6.5 Hz, CH2-N), 2.72 (m, 4 H, N-CH2CH2-N), 2.58 (m, 4 H, N-CH2CH2-N), 2.20 (s, 3 H, CH3), 2.17 (s, 3 H, CH3); 13C-NMR (CDCl3, 300 K): δ 171.5, 147.2, 139.0, 104.6, 55.0, 54.5, 53.3, 52.2, 51.3, 47.1, 13.3, 10.9; IR (ATR): ν 1733, 1434, 1198, 1174, 881 cm-1; HRMS (FAB) calcd. (C23H40N5O8) [M+] 514.2877; found 514.2882. A suspension of the obtained ester (696 mg; 1.36 mmol) and NaOH (218 mg; 5.44 mmol) in H2O (MQ, 0.6 %) was stirred at room temperature until total consumption of the starting material was detected by TLC. The crude product was washed with CH2Cl2 and the solvent was evaporated in vacuo to give compound 1 (662 mg, 89 %) as a yellow solid; 1H-NMR (D2O, 300 K): δ 5.86 (s, 1 H; H4), 4.02 (apparent t, 2 H, 3J (H,H) = 7.1, 7.0 Hz, CH2-N(azole)), 3.12 (s, 8 H; CH2CO2Na), 2.80 (apparent t, 2 H, 3J (H,H) = 7.1, 7.0 Hz, CH2-N), 2.61 (m, 8 H, N-CH2CH2-N), 2.20 (s, 3 H, CH3), 2.09 (s, 3 H, CH3); 13C-NMR (D2O, 300 K): δ 176.7, 148.0, 140.8, 104.3, 57.4, 51.5, 50.3, 49.9, 44.4, 11.2, 9.4; IR (ATR): ν 1579, 1404, 1103 cm-1.
4,7-Bis-carboxymethyl-10-[2-(3,5-dimethylpyrazol-1-yl)-ethyl]-1,4,7,10-tetraazacyclododec-1-yl-acetic acid trisodium salt (2)
A mixture of cyclen·2H2SO4 (460 mg; 2.67 mmol), bromoethyl-3,5-dimethylpyrazole (542 mg; 2.67 mmol) and Na2CO3 (1.42 g; 13.35 mmol) in CHCl3 (50 mL) was refluxed for 16 h. After cooling the solid salts were filtered off and the organic solvent was evaporated in vacuo. Next a suspension of the residue, methyl bromoacetate (1.37 g; 8.98 mmol) and Na2CO3 (952 mg; 8.98 mmol) in acetronitrile (20 mL) was refluxed for 15 h. After cooling the solid salts were filtered and the organic solvent was evaporated in vacuo. Purification of the residue by column chromatography on silica gel (95:5 CH2Cl2-EtOH) gave the corresponding trimethyl ester as a yellow oil (752 mg, 55 %); 1H-NMR (DMSO-d6): δ 5.79 (s, 1 H, H4), 4.03 (t apparent, 2 H, J = 7.0, 6.5 Hz, CH2-N(azole)), 3.66 (s, 9 H, OCH3), 3.30 (s, 6 H, N-CH2CO2Me), 2.75 (br s, 2 H, CH2-N), 2.74-2.18 (m, 16 H, N-CH2-CH2-N), 2.21 (s, 3 H, CH3), 2.10 (s, 3 H, CH3). The obtained ester (696 mg; 1.36 mmol) and NaOH (218 mg; 5.44 mmol) in H2O (MQ, 0.6 %) was stirred at room temperature until total consumption of the starting material was detected by TLC. The reaction mixture was then washed with CH2Cl2 and the solvent was evaporated in vacuo to give compound 2 (662 mg, 89 %) as a yellow solid; IR (ATR): ν 1579, 1404, 1103 cm-1; 1H-NMR (D2O): δ 5.86 (s, 1 H, H4), 4.02 (apparent t, 2 H, J = 7.1, 7.0 Hz, CH2-N(azole)), 3.12 (s, 8 H, CH2CO2Na), 2.80 (apparent t, 2 H, J = 7.1, 7.0 Hz, CH2-N), 2.61 (m, 8 H, N-CH2CH2-N), 2.20 (s, 3 H, CH3), 2.09 (s, 3 H, CH3); 13C-NMR (D2O): δ 181.5, 181.3, 181.24, 149.9, 142.8, 106.8, 61.1, 60.42, 60.24, 54.2, 53.0-51.1, 45.9, 13.8, 11.8
T1 and T2 measurements
1H-NMR relaxation times
T1 and
T2 of aqueous solutions of
1 and
2 and the corresponding gadolinium complexes were measured to determine the relaxivity values r
1 and r
2. All measurements were made with 0.3 mL solutions contained in 10 mm tubes using a Bruker Minispec NMR spectrometer operating at 60 MHz. The temperature was controlled using a HAAKE K15 unit and was in a range between 15-37 °C. Before each measurement the spectrometer was tuned and calibrated.
T1 values were determined by the inversion-recovery method (d1-1800(ph1)-τ-900(ph1)-aq) and
T2 values were determined by the Carr-Purcell-Maiboom-Gill sequence (d1-90o(ph1)-[τ-180o(ph2)-τ]n-aq), using, in both cases, no less than three different τ values. Three different measurements of
T1 or
T2 were performed in every sample. Typically, compounds
1 and
2 (1 mM) and dtpa were dissolved in Tris/HCl (100 mM), NaCl (150 mM) and the final pH was adjusted with HCl and NaOH solutions. These pH values were directly read as measured on a Crison micropH 2002 equipped with a glass electrode at 20 °C. The glass electrode was calibrated by measuring the electromotive force of standard buffers at pH 4.000 and 7.020. The paramagnetic contribution was measured using the corresponding complex under the same conditions. Relaxivities r
1(2) were calculated according to the expression:
where for every complexone, Δ is the difference in relaxation rates (1 / T
1(2)) of the water protons in the presence and absence of Gd(III)-complex, and [GdL] the molar concentration of Gd(III)-complex.
17O-NMR measurements
Hydration number (q) was determined by 17O-NMR using aqueous solutions of the corresponding dysprosium(III)-complexes. The 17O-NMR measurements were carried out with 0.5 mL samples in 5 mm tubes on a Bruker-DRX-400 spectrometer operating at 54.26 MHz and with a 34 ms pulse at 0 dB (pre-acquisition delay 100 ms), equipped with a broadband probe and using the field-frequency lock (D2O ° 20 %). The temperature was maintained at 310 K. Dy(III)-complexes were synthesized by mixing the corresponding ligand and DyCl3·6H2O using an excess of the ligand (1:0.9) to avoid the presence of the free dysprosium ions. The concentrations of the complex were between 15 and 100 mM in water solution (MQ water) containing D2O in 20% and the pH solutions was adjusted between 6-6.5 using HCl and NaOH water solutions.
Theoretical calculations
Geometry optimisations were performed with Gaussian 98 [
12] at the HF/3-21G/CPCM level. For the metal atom, we applied a quasi-relativistic ECP as described by Dolg
et al., which includes 46+4f n electrons in the core, leaving the outermost 11 electrons to be treated explicitly by a (7s6p5d)/[5s4p3d] Gaussian basis set [
13]. Solvent effects were evaluated by the polarizable continuum model CPCM. For Gd atom, the previously parameterized radii were used [
14], while other options for the solvation model were selected by default. Then, single-point energy calculations were carried out on the optimized geometries at the density functional theory level (mPW1PW91 functional), using the 6-31+G** basis sets for the ligand and the CPCM model for taking into account the solvent effects.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}