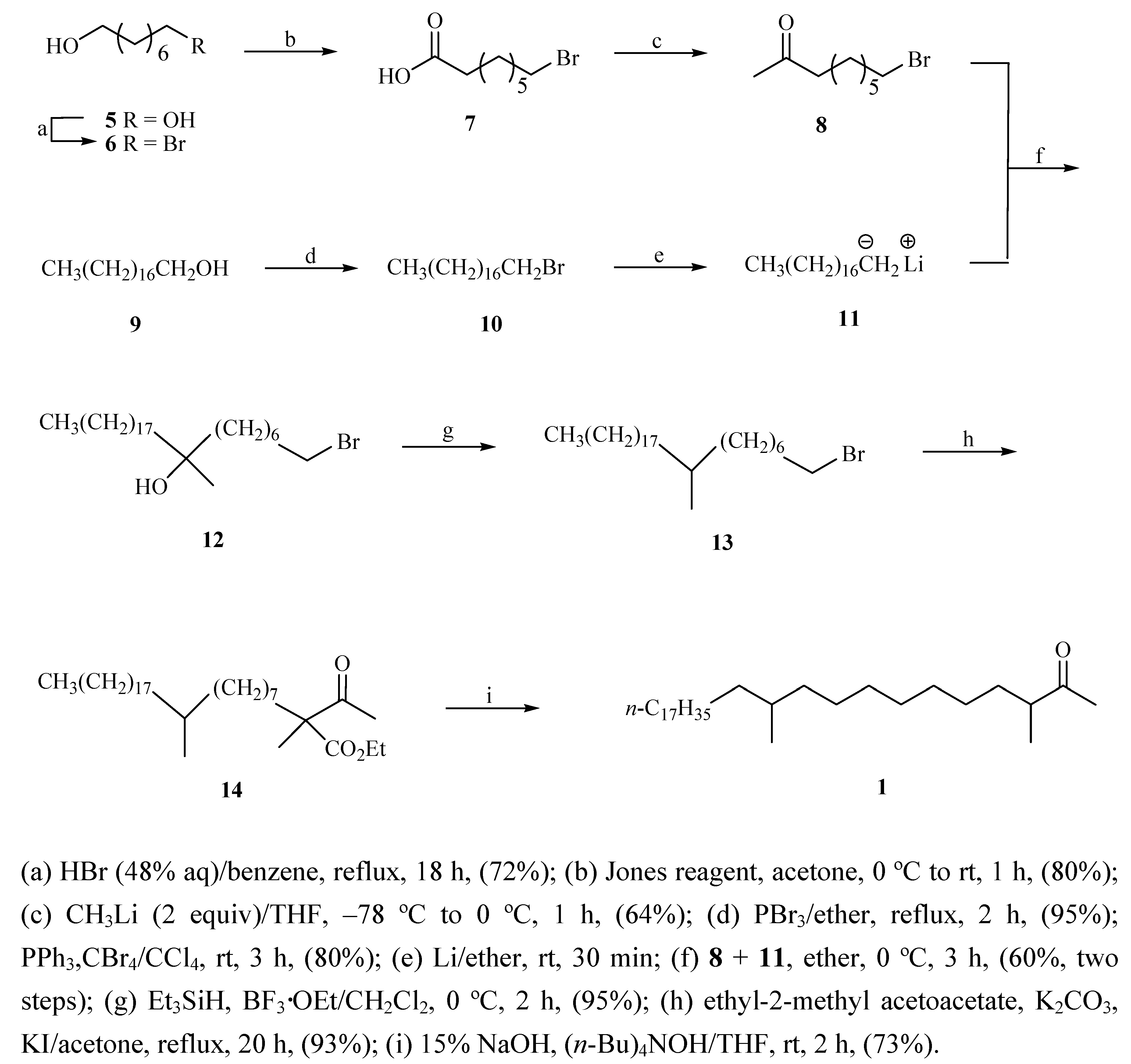Introduction
Many insects emit precise chemical odors to attract their mates (sex pheromones). Pheromones are naturally occurring, environmentally friendly and species specific compounds that do not result in the development of insecticide resistance [
1]. The Nishida group [
2] reported the isolation of the female–produced sex pheromone of the German cockroach,
Blattella germanica, from its cuticular waxes in 1974. The compound, which elicited typical courting behavior in males including wing–raising [
3,
4], was identified as 3,11-dimethylnonacosan-2-one (
1,
Figure 1). Since the synthesis of the mixture of diastereomers of
1 was first achieved by Nishida
et al. [
3], a number of syntheses yielding a diastereomeric mixture of
1 were reported [
5]. Bioassay of these four stereoisomers of
1 showed that they were equally active [
6]. Most of these methods for the generation of 3,11-dimethylnonacosan-2-one (
1) are linear synthetic approaches involving expensive reagents and inconvenient handling [
5a-b,
5f].
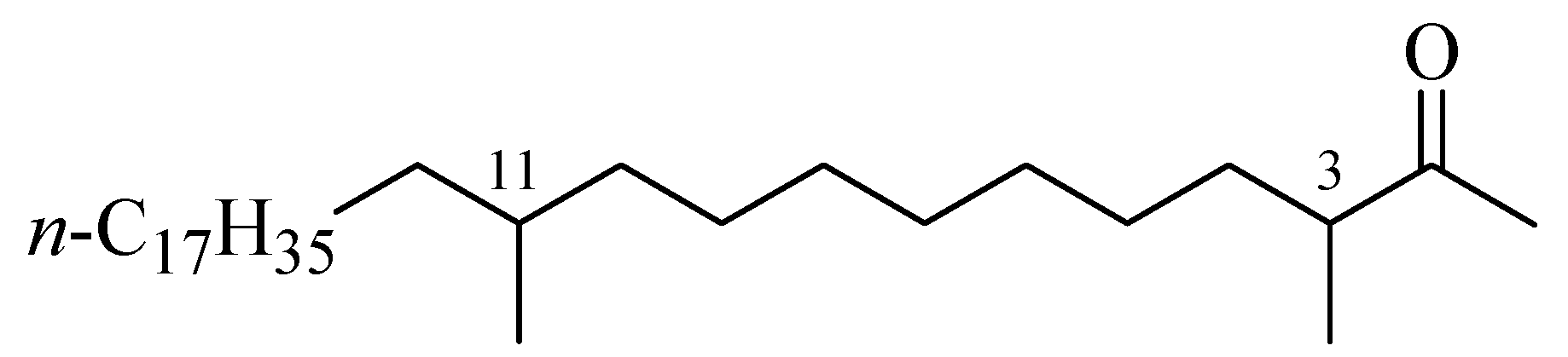
Figure 1.
Structure of 3,11-dimethylnonacosan-2-one (1)
Figure 1.
Structure of 3,11-dimethylnonacosan-2-one (1)
Our research required large quantities of 1 and key fragments 2–4 to serve as potential scaffolds for modification of 1. We therefore became interested in studying a synthesis of 1 suitable for large scale production. We wish to report herein a convenient new approach to the synthesis of this target molecule via C–alkylation–ionic hydrogenation and C–alkylation–decarboxylation reactions starting from 1,8-octane-diol (5) and 1-octadecanol (9).
Results and Discussion
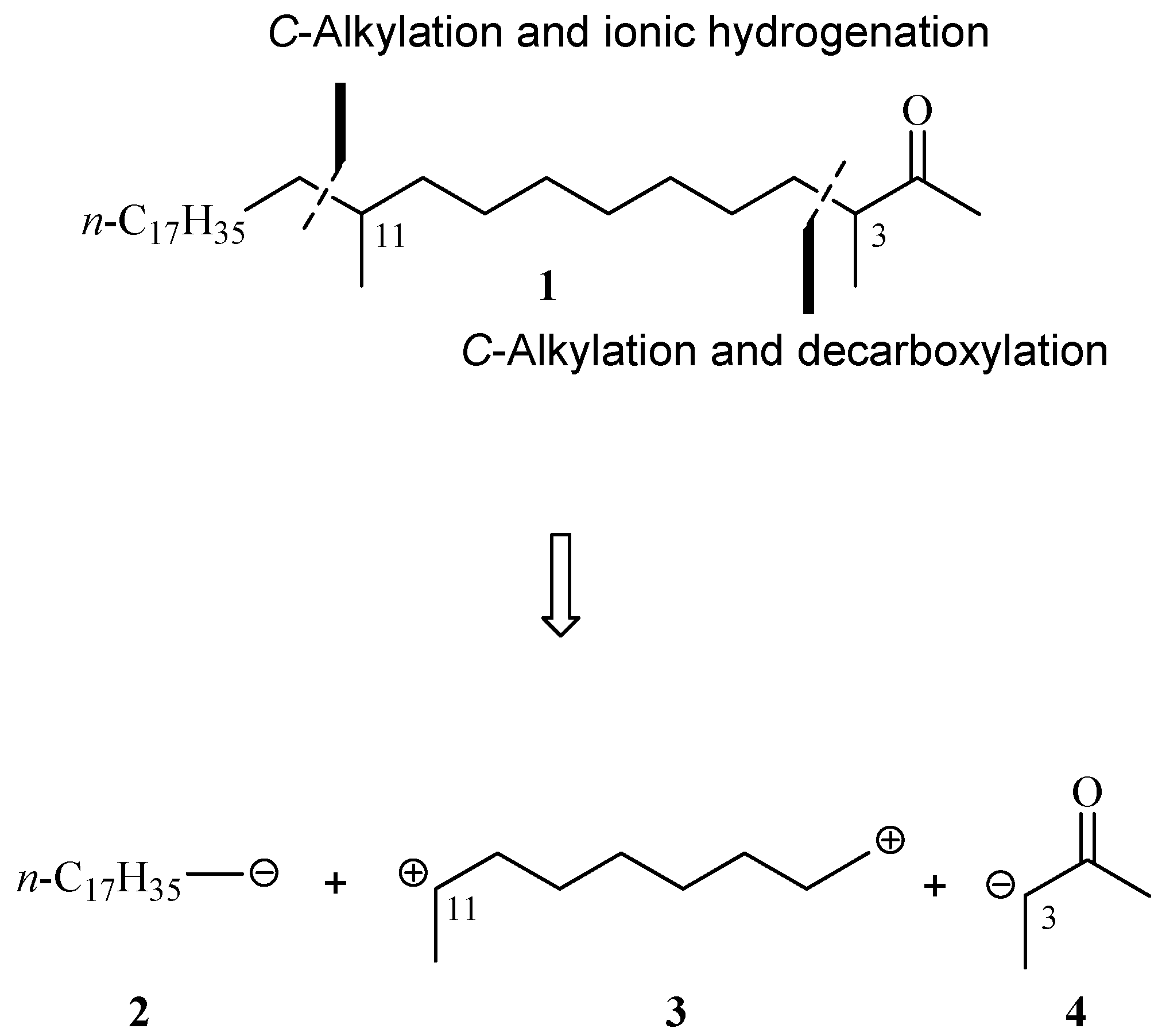
The retrosynthetic analysis for 3,11-dimethylnonacosan-2-one (
1) is summarized in
Scheme 1. The synthetic key strategies involve
C–alkylation–decarboxylation reaction of unit
3 with acetate anion
4 in order to form the C3–C4 bond, and
C–alkylation–ionic hydrogenation of alkyl anion
2 with unit
3 for generation of the C11–C12 bond. Units
2 and
4 can be smoothly derived from freshly prepared bromide
10 [
7] and commercially available ethyl 2-methylacetoacetate in the presence of base. The electrophilic unit
3 was generated via bromination, oxidation, and methylation of 1,8-octanediol (
5).
Scheme 1.
Retrosynthetic analysis of 3,11-dimethylnonacosan-2-one (1).
Scheme 1.
Retrosynthetic analysis of 3,11-dimethylnonacosan-2-one (1).
The synthesis of key fragments
8 and
11 was accomplished as depicted in
Scheme 2. Commercially available
5 underwent bromination with aqueous hydrogen bromide (48%) in benzene using a Dean–Stark apparatus to afford a 6:1 ratio (by G.L.C. analysis) of monobromo alkanol
6 and dibromo alkane [
8]. Compound
6 was oxidized by Jones reagent at 0 °C in acetone to generate acid
7 in 80% yield [
9], which was then treated with methyllithium (1.4 M solution in ether) in THF to give 9-bromononan-2-one (
8) in 64% yield [
10]. Bromination of alcohol
9 was performed with phosphorus tribromide in ether to produce a 95% yield of bromide
10, which was treated with Li metal in dry THF to yield anion
11, which was coupled
in situ with bromide
8 [11] in DMF to generate tertiary alcohol
12 in 60% yield for two steps. In this bromination reaction the alternate CBr
4/PPh
3 method proved to be relatively low yielding (80%) [
12]. Compound
12 was treated with Et
3SiH in the presence of BF
3·OEt in CH
2Cl
2 to afford the product
13 [
5a] in 95% yield. Chain–elongation of
13 with ethyl 2-methylacetoacetate was executed employing potassium carbonate as the base to give
14 in 93% yield. Hydrolysis of
14 with concomitant decarboxylation of the resulting acid furnished the final product, 3,11-dimethylnonacosan-2-one (
1) in 73% yield [
5].
Scheme 2.
Synthesis of 3,11-dimethylnonacosan-2-one (1).
Scheme 2.
Synthesis of 3,11-dimethylnonacosan-2-one (1).
Experimental
General
Reactions requiring anhydrous conditions were performed following the usual precautions for rigorous exclusion of air and moisture. Tetrahydrofuran was distilled from sodium benzophenone ketyl prior to use. Thin layer chromatography (TLC) was performed on precoated silica gel G and GP uniplates (Analtech) and visualized with 254–nm UV light. Flash chromatography was carried out on silica gel 60 [Scientific Adsorbents Incorporated (SAI), particle size 32–63
µm, pore size 60 Å].
1H-NMR,
13C-NMR, and 2D-NMR spectra were recorded in CDCl
3 on a Bruker DPX 400 instrument operating at 400 MHz (
1H) and 100 MHz (
13C), respectively. The chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane. Infrared (IR) spectra were obtained on an ATI Mattson FT/IR spectrometer. Mass spectra were recorded with a Waters Micromass ZQ LC–Mass system and high resolution mass spectra (HRMS) were measured with a Bruker BioApex FTMS system by direct injection using an electrospray interface (ESI). When necessary, chemicals were purified according to the reported procedures [
13].
8-Bromooctan-1-ol (6)
To a stirred solution of 1,8-octanediol (
5, 31.2 g, 213.5 mmol) in benzene (500 mL) was added 48% aqueous HBr (37.5 mL) and the mixture was refluxed for 18 h, while the water formed was trapped using a Dean–Stark water separator. The reaction mixture was washed successively with 6N aqueous NaOH solution (200 mL), 10% HCl aqueous solution (250 mL), water (300 mL) and brine (300 mL). The organic layer was separated, dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The residue was purified by Kugelrohr distillation (b.p. 66–70 ºC/3 torr, lit. [
8c] b.p. 110–120 ºC/2 torr) to give
6 (32.0 g, 72%) as a colorless oil. IR (neat, NaCl) 3455, 2984, 2875, 1142, 1048, 852 cm
-1;
1H-NMR δ 3.72 (t,
J = 6.8 Hz, 2H, CH
2Br), 3.45 (t,
J = 7.0 Hz, 2H, CH
2OH), 1.25–1.15 (m, 12H, 6×CH
2); HRMS calcd. for C
8H
18OBr: 209.0541 [M+H]
+, found: 209.0547.
9-Bromononan-2-one (8)
A stirred solution of acid 7 (45.0 g, 201.0 mmol) in dry THF (1.4 L) was cooled to –78 ºC and methyllithium (287.5 mL, 402.0 mmol, 1.4 M solution in ether) was added dropwise over 20 min. The reaction mixture was allowed to warm to 0 ºC for 1 h, and quenched with sat’d aqueous NH4Cl solution (250 mL), followed by extraction with ether (3×300 mL). The organic layer was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 15% ethyl acetate in hexanes) to give 8 (28.5 g, 64%) as a colorless oil. Rf = 0.4 (20% ethyl acetate in hexanes); IR (neat, NaCl) 2977, 2876, 1712, 1518, 1452, 1250, 1068, 781 cm-1; 1H-NMR δ 3.38 (t, J = 6.8 Hz, 2H, CH2Br), 2.40 (t, J = 7.6 Hz, 2H, CH2CO), 2.11 (s, 3H, CH3), 1.87–1.77 (m, 2H, CH2), 1.58–1.49 (m, 2H, CH2), 1.44–1.36 (m, 2H, CH2), 1.35–1.18 (m, 4H, 2×CH2); 13C-NMR δ 208.9 (CO), 43.5 (CH2CO), 33.8 (CH2Br), 31.9 (CH3), 30.2 (CH2), 28.5 (CH2), 28.1 (CH2), 26.8 (CH2), 23.4 (CH2); HRMS calcd. for C5H10OBr: 164.9915 [M+H]+, found: 164.9908.
1-Bromomethylhexacosan-8-ol (12)
Anhydrous ether (150 mL) was placed in a three–necked flask equipped with a dropping funnel, a N2 gas inlet and a septum. After the apparatus was swept with dry N2, lithium wire (1.5 g, 216.0 mmol) was cut into small pieces, which were allowed to fall into the reaction flask. With stirring, a solution of 1-bromooctadecane (10, 30.0 g, 90.0 mmol) in anhydrous ether (150 mL) was added. After a period of 1 h, the mixture became slightly cloudy. The reaction mixture then was cooled to –10 ºC. The remainder of the 1-bromooctadecane solution was added over a period of 3 h at the same temperature. After addition was completed, the reaction mixture was kept at the same temperature for additional 1 h and then was allowed to warm up to 10 ºC with stirring. Over a period of 30 min, the reaction mixture was cannulated to a two–necked flask in which bromide 8 (15.0 g, 60.0 mmol) in ether (75 mL) at 0 ºC was placed and the resulting reaction mixture was stirred at 0 ºC for 3 h. The reaction mixture was quenched by addition of 50% aqueous NH4Cl solution (150 mL) and aqueous phase was extracted with ether (180 mL). The combined organic phases were washed with brine (150 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, 20% hexane in ethyl acetates) to give alcohol 12 (25.5 g, 60%) as a colorless oil. Rf = 0.3 (20% hexane in ethyl acetates); IR (neat, NaCl) 3520–2916, 2848 cm-1; 1H-NMR δ 3.64 (t, J = 6.8 Hz, 2H, CH2Br), 1.65–1.48 (m, 6H, 3×CH2), 1.32–1.20 (m, 44H, 22×CH2), 0.88 (t, J = 7.2 Hz, 3H, CH3); HRMS calcd. for C27H56OBr: 475.3515 [M+H]+, found: 475.3521.
1-Bromo-8-methylhexacosane (13)
To a stirred solution of 1-bromo-8-methylhexacosan-8-ol (12, 24.0 g, 50.0 mmol) in dichloromethane (600 mL) was added triethylsilane (12.0 g, 120.0 mmol), followed by addition of BF3·OEt (20.0 g, 150.0 mmol) at 0 ºC and the mixture was stirred for 2 h. The reaction mixture was diluted with CH2Cl2 (400 mL) and washed with sat’d aqueous NH4Cl solution (500 mL) and brine (500 mL). The organic layer was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 5% ethyl acetate in hexanes) to give bromide 13 (22.0 g, 95%) as a colorless oil. Rf = 0.5 (5% ethyl acetate in hexanes); IR (neat, NaCl) 2978, 1446, 1062, 838 cm-1; 1H-NMR δ 3.43 (t, J = 6.8 Hz, 2H, CH2Br), 1.61–1.39 (m, 7H), 1.35–1.21 (m, 43H), 0.89 (t, J = 7.0 Hz, 3H, CH3); HRMS calcd. for C27H56Br: 459.3565 [M+H]+, found: 459.3573.
3,11-Dimethyl 3-ethoxycarbonylnonacosan-2-one (14)
To a stirred solution of ethyl 2-methylacetoacetate (9.0 g, 63.0 mmol) in acetone (150 mL) was added K2CO3 (18.6 g, 135.0 mmol) and KI (3.0 g, 18.0 mmol) at room temperature. After 10 min, a solution of bromide 13 (27.6 g, 60.0 mmol) in acetone (60 mL) was added and the reaction mixture was rapidly brought to reflux by heating in oil bath. After 20 h, the resultant mixture was cooled to room temperature, diluted with ether (300 mL) and filtered. The residue was washed with sat’d aqueous NH4Cl solution (150 mL) and brine (150 mL). The organic layer was separated, and the aqueous phase was extracted with ether (150 mL). The combined organic phases were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 10% ethyl acetate in hexanes) to give acetate 14 (28.5 g, 93%) as a colorless oil. Rf = 0.3 (10% ethyl acetate in hexanes); IR (neat, NaCl) 2924, 2853, 1735 (CO), 1716 (COO), 1458, 1265, 1120 cm-1; 1H-NMR δ 4.18 (t, J = 7.0 Hz, 2H, CH2), 2.03 (s, 3H, CH3), 1.86–1.72 (m, 2H, CH2), 1.48–1.21 (m, 51H, CH, 25×CH2), 0.85–1.02 (m, 6H, 2×CH3); HRMS calcd. for C33H65O3: 509.4934 [M+H]+, found: 509.4922.
3,11-Dimethylnonacosan-2-one (1)
To a stirred solution of 3,11-dimethyl-3-ethoxycarbonylnonacosan-2-one (14, 15.2 g, 30.0 mmol) in THF (500 mL) was added 15% aqueous NaOH solution (300 mL), followed by addition of tetrabutylammonium hydroxide solution (38.0 g, 150.0 mmol, 1.0 M sol. in water) and the mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with ethyl acetate (600 mL) and washed with sat’d aqueous NH4Cl solution (600 mL) and brine (400 mL). The organic layer was separated, and the aqueous phase was extracted with ethyl acetate (300 mL). The combined organic phases were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 10% ethyl acetate in hexanes) to give 1 (9.8 g, 73%) as a colorless oil. Rf = 0.3 (10% ethyl acetate in hexanes); IR (neat, NaCl) 2957, 1729 (CO), 1446, 1285, 1024, 837 cm-1; 1H-NMR δ 2.55–1.41 (m, 1H, CH), 2.06 (s, 3H, CH3), 1.72–1.22 (m, 55H), 0.84 (t, J = 7.0 Hz, 3H, CH3); HRMS calcd. for C31H63O: 451.4879 [M+H]+, found: 451.4887.

{kind=link}
{kind=link}
{kind=link}