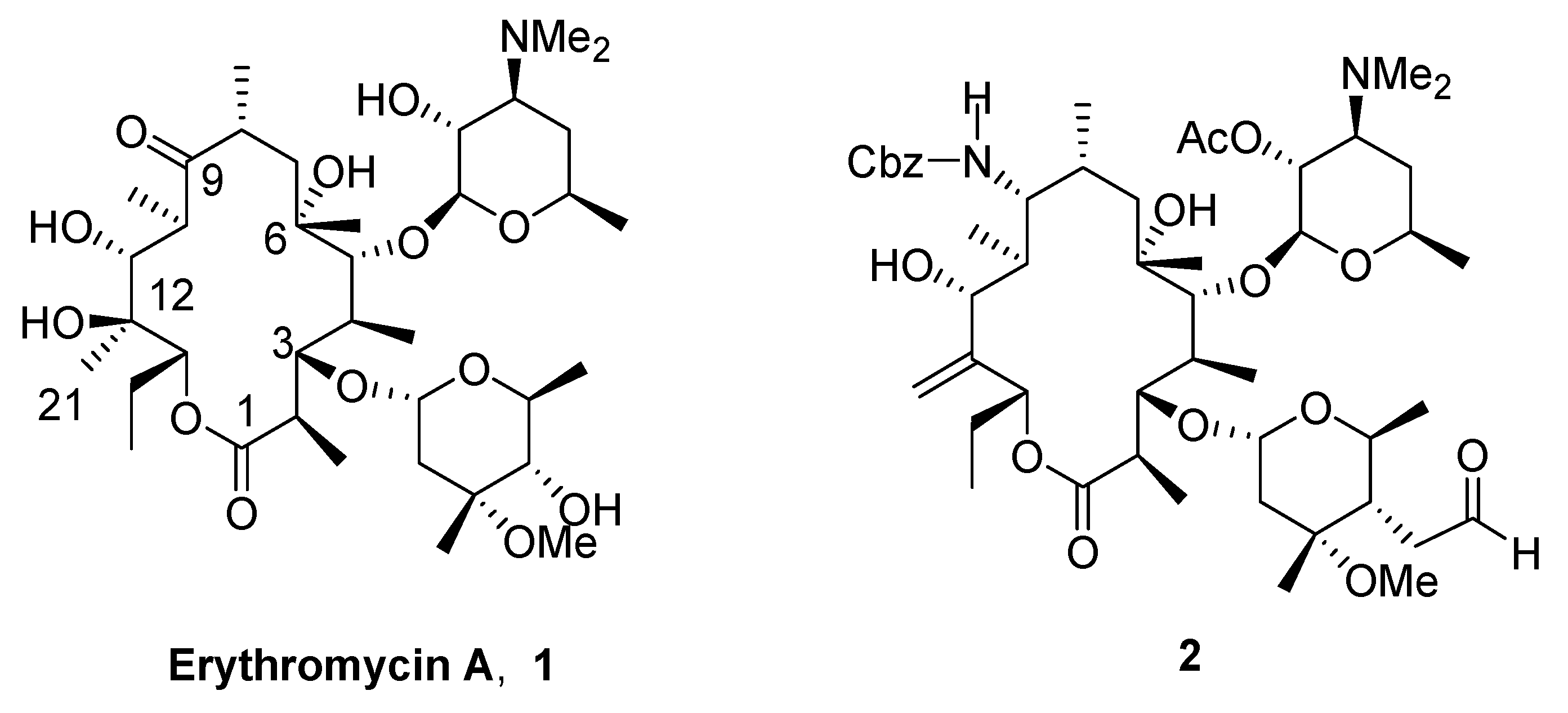
Experimental Section
General
All reagents and solvents were reagent grade. Further purification and drying by standard methods were employed when necessary. Erythromycin A and its derivatives were pre-dried azeotropically from benzene. THF was distilled from sodium benzophenone ketyl. CH2Cl2 and EtOAc were distilled from CaH2. DMF, DMSO and HMPA were redistilled and stored in screw-cap vials with molecular sieve (4A). All organic solvents were evaporated under reduced pressure with a rotary evaporator. The plates used for thin-layer chromatography (TLC) were E. Merck silica gel 60F254 (0.1 mm thickness) precoated on aluminum plates, and they were visualized under both long (365 nm) and short (254 nm) UV light. Compounds on TLC plates were visualized with a spray of 5% dodocamolybdophosphoric acid in ethanol and subsequent heating. Column chromatography was performed using E. Merck silica gel (230-400 mesh). Melting points were measured using Electrothermal IA9100 digital melting point apparatus and are uncorrected. NMR spectra were recorded at 300K in deuterated chloroform solution on a Bruker DPX-300 spectrometer (operating at 300.13 MHz for 1H and 75.47 MHz for 13C). Chemical shifts are reported as parts per million (ppm) in the δ scale relative to the resonance of CDCl3 (7.26 ppm in the 1H, 77.00 ppm for the central line of the triplet in the 13C modes, respectively). Coupling constants (J) are reported in Hz. Splitting patterns are described by using the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. 1H-NMR data is reported in this order: chemical shift; multiplicity; coupling constant(s), number of protons. Mass spectra (ERMS and HRMS) were obtained with a Thermo Finnigan MAT95XL spectrometer or a API 2000 LC/MS/MS system. Relevant data are tabulated as m/z. Elemental analyses were performed at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, China.
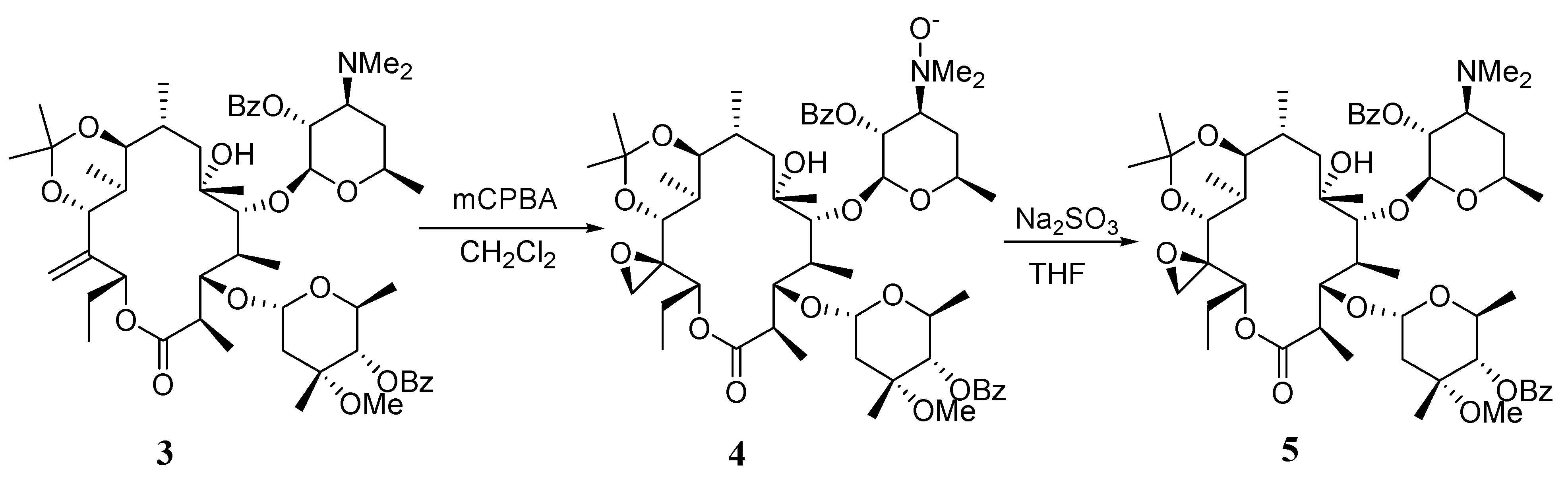
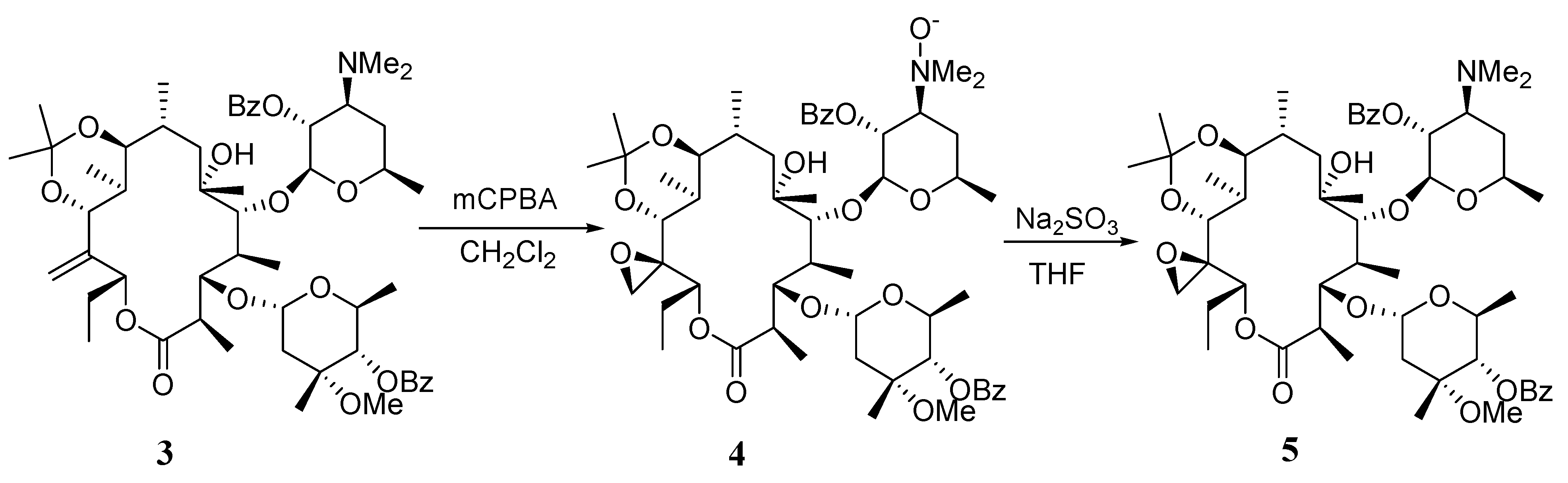
(9S)-9,11-O-Isopropylidene-2',4''-O-bis(benzoyl)- (12R)-12,21-epoxy-9-dihydroerythromycin A (5).
To a solution of compound 3 (6.0 g, 6.2 mmol) in dichloromethane (60 mL) at 0°C was added m-CPBA (5.4 g, 31 mmol, 5 equiv). The mixture was stirred at r.t. for 19 hrs. The reaction was quenched by adding cyclohexene (2.6 mL) and stirred for another 4 hrs. The reaction was worked up by pouring into sat. aq. NaHCO3 (100 mL). The organic layer was separated and the aqueous layer was extracted with dichloromethane (60 mL×2). The combined organic layer was washed with brine (100 mL), dried over Na2SO4 and concentrated in vacuo to give a residue 4, which was used in the next step without further purification. The crude material 4 was dissolved in THF (90 mL). To this was added a solution of Na2SO3 (10.8 g) in water (45 mL). The mixture was stirred for 20 hrs. and then worked up by separating the organic layer, extracting the aqueous layer with EtOAc (50 mL) and combining the organic portions, which were washed with brine (100 mL), dried over Na2SO4 and concentrated in vacuo. The crude material was purified with a silica gel column (200 g, eluted with 4:1 hexanes- acetone containing 0.5% triethylamine) to give 5 (4.2 g, 69% from 3); mp: 126-127°C; IR (KBr) cm-1 3528 (w), 1725 (s), 1268 (s); 1H-NMR (CDCl3) δ 0.68 (d, J =7.2 Hz, 3H), 0.80 (t, J =7.2 Hz, 3H), 0.91 (d, J =5.7 Hz, 3H), 1.00 (m, 6H), 1.09 (d, J =7.2 Hz, 3H), 1.17 (m, 10H), 1.30 (2s, 6H), 1.4 (m, 1H), 1.5-1.8 (m, 5H), 2.0 (m, 2H), 2.33 (s, 6H), 2.35-2.60 (m, 2H), 2.63 (m, 1H), 2.99 (m, 2H), 3.34 (m, 1H), 3.52 (m, 1H), 3.56 (s, 3H), 3.92 (s, 2H), 4.06 (s, 1H), 4.06 (s, 1H), 4.5 (m, 1H), 4.70 (d, J =4.5 Hz, 1H), 4.85 (m, 1H), 4.92 (d, J =9.6 Hz, 1H), 5.03 (d, J =9.6 Hz, 1H), 5.20 (m, 1H), 7.41 (m, 4H), 7.54 (m, 2H), 8.00 (m, 4H); 13C-NMR (CDCl3) δ 9.5, 11.1, 12.5, 16.0, 17.6, 18.0, 20.9, 21.2, 21.7, 23.6, 27.5, 31.9, 32.4, 40.8, 42.9, 43.2, 49.3, 59.2, 63.4, 63.7, 65.3, 68.2, 72.1, 73.3, 73.8, 76.5, 78.7, 79.8, 85.3, 95.2, 99.7, 101.1, 128.1, 128.3, 129.4, 129.9, 130.6, 132.6, 133.2, 165.4, 166.0, 175.2; MS m/z 982 (MH+); Anal. Calcd for C54H79NO15: C, 66.03; H, 8.11; N, 1.43. Found: C, 65.45; H, 8.06; N, 1.39.
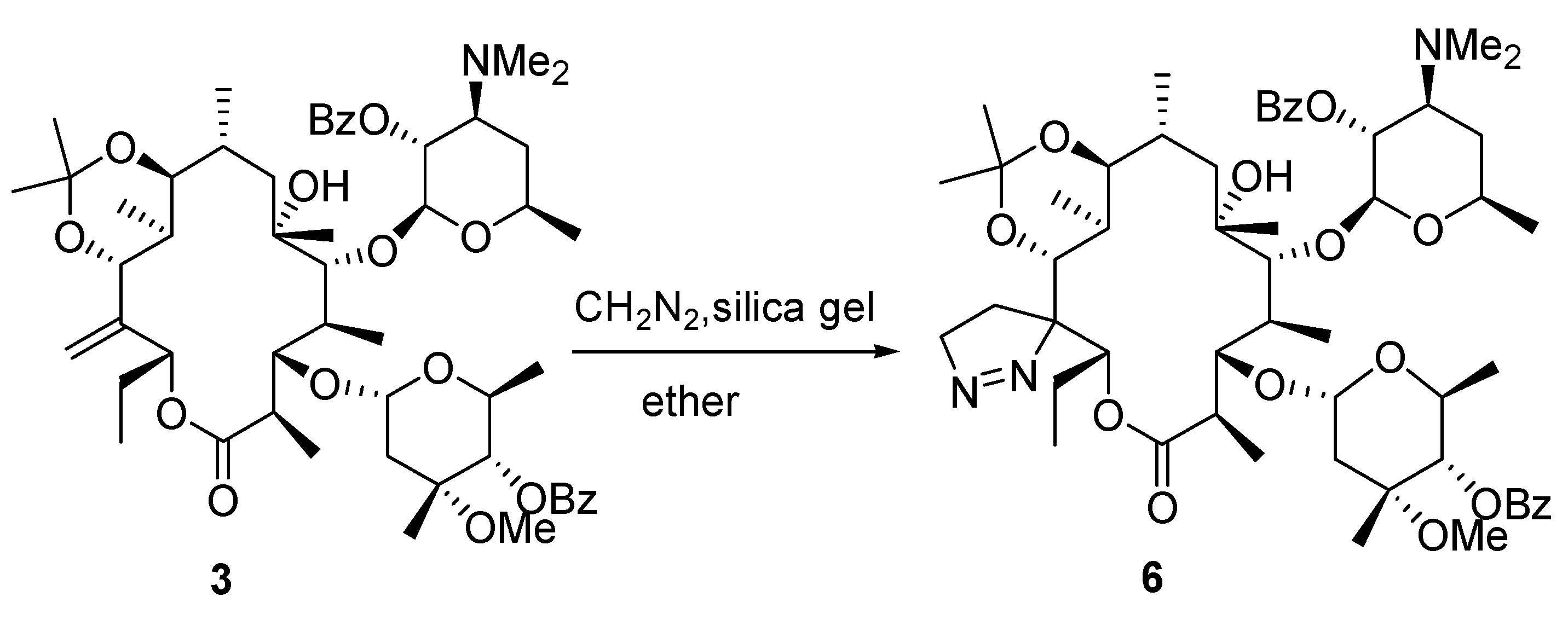
(9S)-9,11-O-Isopropylidene-2',4''-O-bis(benzoyl)-12-(Δ1-pyrazoline)-12-dehydroxyl-21-demethyl-9- dihydroerythromycin A (6).
To a solution of compound 3 (1.0 g, 1 mmol) in ether (10 mL) cooled with ice-salt bath, was added diazomethane (~10 mmol), freshly prepared by reacting CH3N(NO)CONH2 (3.0 g, 20 mmol) with KOH (2.0 g, 36 mmol) in H2O (3 mL) and ether (20 mL) solution and silica gel (100 mg). The mixture was stirred at r.t. for 44 hr. The ether was removed. To the residue was added EtOAc (50 mL) and the mixture was filtered. The filtrate was concentrated and purified on a silica gel column (50 g, elution with 5:1 hexanes-ethyl acetate) to give compound 6 (0.13 g, 13%). Starting material 3 (0.64 g) was also recovered, so the yield based on reacted substrate was 80%. IR (KBr) cm-1 3528 (w), 1727 (s), 1268 (s); 1H-NMR (CDCl3) δ 0.71 (d, J =7.2 Hz, 3H), 0.80 (m, 3H), 0.92-1.02 (m, 9H), 1.11-1.27 (m, 23H), 1.30-1.50 (m, 4H), 1.74-1.79 (m, 3H), 2.05 (br s, 2H), 2.35 (s, 6H, N(CH3)2), 2.39 (ss, 1H), 2.48 (m,1H), 3.02 (m, 1H), 3.34 (m, 1H), 3.56 (s, 3H, -O-CH3), 3.92 (m,1H), 4.02 (m,1H), 4.29 (m,2H), 4.45-4.53 (m, 3H), 4.75 (s, 1H), 4.92 (d, J=9.9 Hz,1H), 5.02 (d, J=7.5, 1H), 5.20 (m,1H), 7.44 (m, 4H), 7.55 (m,2H), 7.99 (m,4H); 13C- NMR (CDCl3) δ 9.9, 11.1, 12.5, 14.0, 16.5, 17.8, 18.1, 20.7, 21.0, 21.2, 23.7, 24.4, 27.7, 29.2, 29.6, 31.7, 32.1, 32.5, 35.2, 40.9, 43.1, 49.4, 63.5, 63.7, 68.2, 68.7, 72.1, 73.3, 73.9, 78.9, 79.0, 80.1, 85.4, 95.6, 99.3, 99.9, 128.2, 128.3, 129.6, 130.0, 130.7, 132.6, 133.2, 165.5, 166.0, 174.8; MS m/z 1008 (MH+), HRMS (FAB) Calcd for C55H82N3O14 (MH+): 1008.5791. Found: 1008.5813.
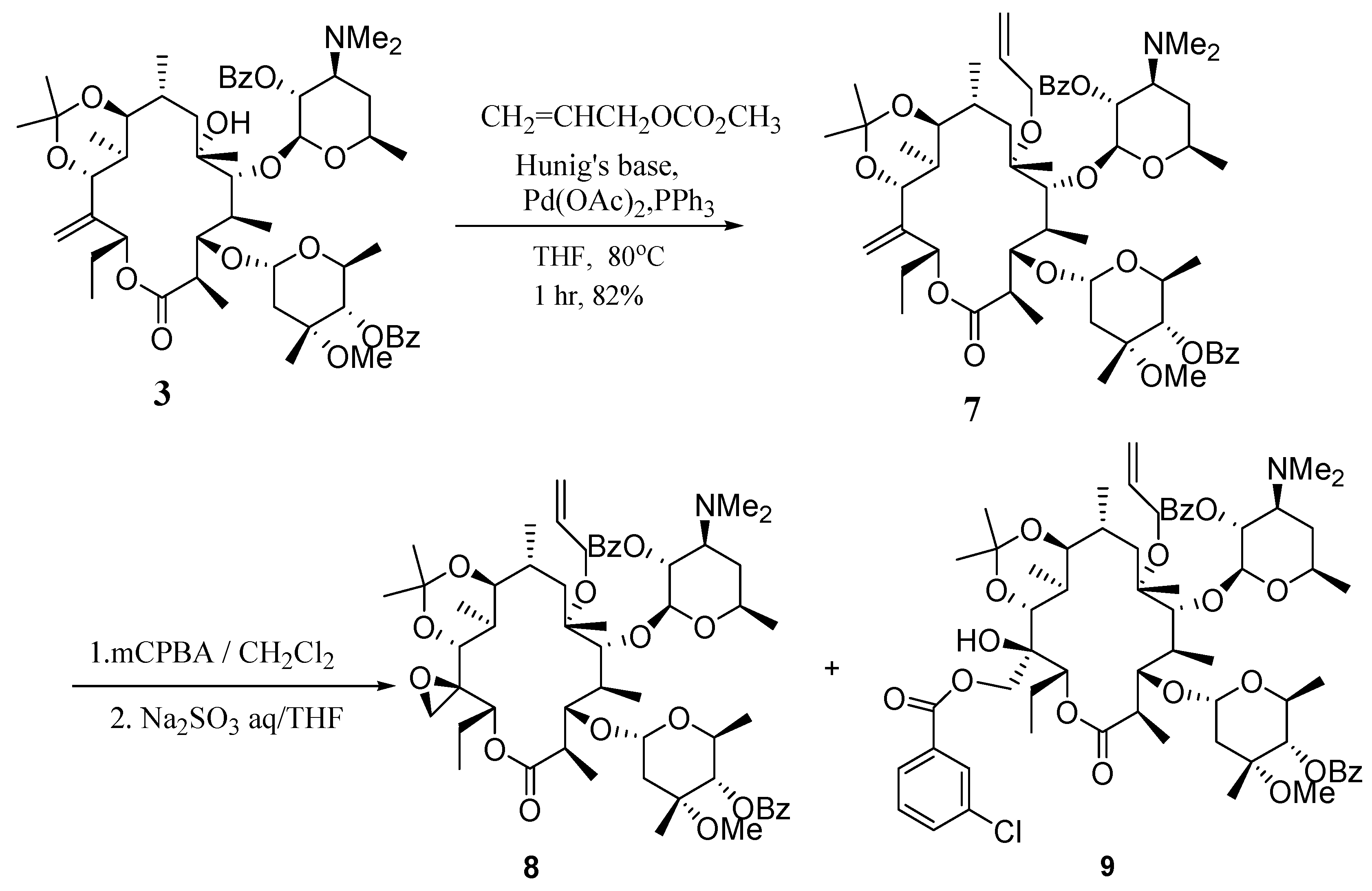
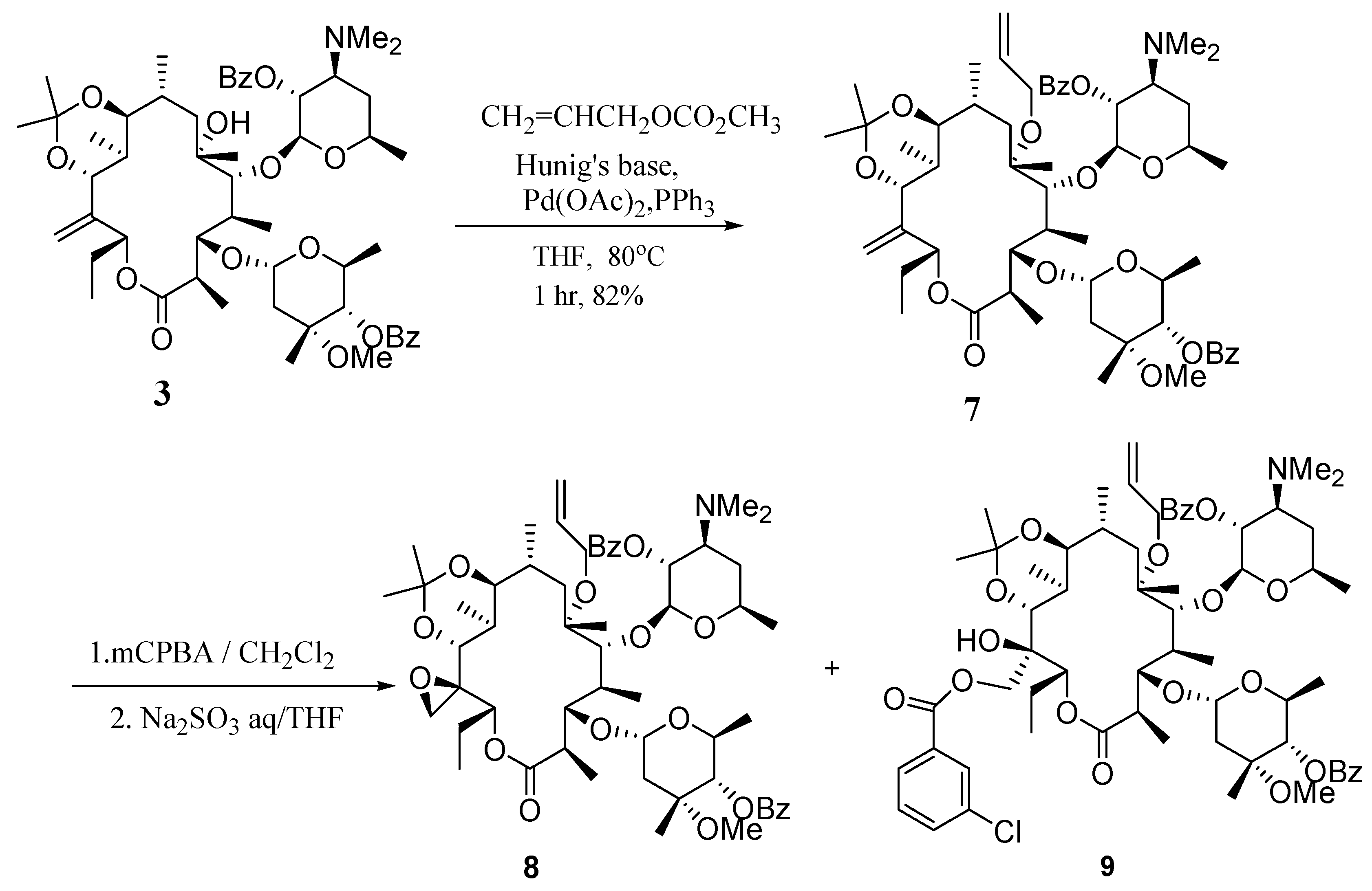
(9S)-9,11-O-Isopropylidene-6-O-allyl-2',4''-O-bis(benzoyl)-12,21-anhydro-9-dihydroerythromycin A (7).
To a solution of compound 3 (0.50 g, 0.52 mmol) in anhydrous THF (5 mL) was added allyl methyl carbonate (0.12 mL, 1.04 mmol) and Hünig’s base (0.18 mL, 1.04 mmol). The mixture was degassed with nitrogen for 30 min. This solution was transferred to a mixture of palladium (II) acetate (18 mg, 15 mol %) and triphenylphosphine (42 mg, 25 mol %). The reaction mixture was heated to 80°C for 1 hr. Worked up by diluting with EtOAc (20 mL), washed with sat. aq. NaHCO3 (20 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by a silica gel column (100 g, hexanes-acetone 4:1 with 0.5% TEA) to give 6-O-allyl product 7 (430 mg, 82%); IR (KBr) cm-1 1726 (s), 1268(s); 1H-NMR (CDCl3) δ 0.66 (d, J =6.6 Hz, 3H), 0.76 (d, J =6.9 Hz, 3H), 0.88 (m, 1H), 0.99 (d, J =6.9 Hz, 3H), 1.00-1.25 (m, 15H), 1.29 (s, 3H), 1.34 (s, 3H), 1.41 (m, 2H), 1.61 (m, 5H), 1.8 (m, 2H), 1.9-2.1 (m, 3H), 2.31 (s, 6H, N(CH3)2), 2.36 (d, J =7.5 Hz, 1H), 2.38 (s, 0.5H), 2.85 (m, 1H), 2.95 (m, 1H), 3.40 (s, 3H, -O-CH3), 3.55 (d, J =3.6 Hz, 1H), 3.77 (s, 1H), 3.70-3.85 (m, 2H), 3.95 (s, 1H), 4.50 (m, 2H), 4.54 (d, J =6 Hz, 2H), 4.73 (d, J =4.5 Hz, 2H), 4.88 (m, 2H), 5.00 (s, 1H), 5.08 (s, 1H), 5.10-5.35 (m, 3H), 5.65 (m, 1H), 5.80-6.00 (m, 2H), 7.46 (m, 4H), 7.58 (m, 2H), 8.03 (m, 4H); 13C- NMR (CDCl3) δ 10.7, 11.2, 13.2, 17.2, 17.5, 18.4, 20.9, 21.3, 22.4, 23.7, 25.2, 31.0, 31.9, 37.5, 40.8, 41.3, 49.5, 63.1, 63.3, 65.3, 68.6, 68.8, 72.1, 73.1, 74.5, 78.2, 78.9, 80.1, 82.8, 95.3, 99.9, 100.5, 112.6, 118.6, 124.1, 128.1, 128.3, 129.6, 129.8, 131.2, 132.1, 132.5, 133.2, 143.5, 165.4, 166.3, 175.2; MS m/z 1006(MH+); Anal. Calcd for C57H83NO14: C, 68.03; H, 8.31; N, 1.39. Found: C, 67.90; H, 8.35; N, 1.44.
(9S)-9,11-O-Isopropylidene-6-O-allyl-2',4''-O-bis(benzoyl)-12,21-epoxy-9-dihydroerythromycin A (8) and (9S)-9,11-O-isopropylidene-6-O-allyl-2',4''-O-bis(benzoyl)-12-hydroxy-21-(m-chlorobenzoyloxy)- 9-dihydroerythromycin A (9).
To a solution of compound 7 (0.19 g, 0.19 mmol) in dichloromethane (5 mL) at 0°C was added m-CPBA (96 mg, 0.39 mmol, 2.05 equiv). The mixture was stirred at r.t. for 13 hrs. The reaction was worked up by pouring into sat. aq. NaHCO3 (15 mL). The organic layer was separated and the aqueous layer was extracted with dichloromethane (15 mL×2). The combined organic layers were washed with brine (15 mL), dried over Na2SO4 and concentrated in vacuo to give a residue which was used without further purification. The crude material was dissolved in THF (3 mL) and to this was added a solution of Na2SO3 (0.36 g) in water (1.5 mL). The mixture was stirred for 18 h, then worked up by separating the organic layer and extracting the aqueous layer was with EtOAc (5 mL). The organic portions were combined, washed with brine (5 mL), dried over Na2SO4 and concentrated in vacuo. The crude material was purified with a silica gel column (50 g, hexanes-acetone 4:1 with 0.5% TEA) to give 8 (130 mg, 67% from 7) and 9 (20 mg, 8.9%). Compound 8: IR (KBr) cm-1 1727 (s), 1269 (s); 1H-NMR (CDCl3) δ 0.71 (m, 3H), 0.80 (m, 3H), 0.96 (m, 5H), 1.04 (d, J =6 Hz, 3H), 1.10-1.26 (m, 15H), 1.27 (s, 3H), 1.32 (s, 3H), 1.39 (s, 3H), 1.50-2.00 (m, 7H), 2.30 (s, 6H, NMe2), 2.4 (ss, 1H), 2.80 (m, 1H), 2.95 (m, 1H), 3.41 (s, 3H, O-Me), 3.51 (m, 2H), 3.65 (s, 1H), 3.80-4.00 (m,2H), 4.2 (m, 1H), 4.4 (m, 1H), 4.52 (d, J =5.7 Hz, 2H), 4.73 (m, 1H), 4.88 (m, 2H), 5.05-5.25 (m, 4H), 5.90 (m, 1H), 7.34-7.56 (m, 6H), 7.99 (m, 4H); 13C-NMR (CDCl3) δ 10.3, 11.6, 13.5, 17.0, 17.5, 17.7, 20.9, 21.3, 21.8, 23.7, 25.3, 32.0, 35.5, 40.8, 41.0, 41.4, 49.5, 57.3, 57.7, 63.1, 65.3, 69.5, 72.0, 73.1, 74.5, 78.2, 78.8, 83.0, 95.3, 100.5, 101.5, 110.4, 118.5, 128.1, 128.3, 129.6, 129.8, 130.5, 132.0, 132.5, 133.2, 143.2, 143.3, 165.3, 166.2, 175.0; MS m/z 1022 (MH+), HRMS (FAB) Calcd for C57H84NO15(MH+): 1022.5835. Found: 1022.5841. Compound 9: IR (KBr) cm-1 1727 (s), 1267 (s); 1H-NMR (CDCl3) δ 0.71 (m, 5H), 0.83 (m, 2H), 0.96 (d, J=6.9 Hz, 3H), 1.04 (d, J=5.7 Hz, 3H), 1.10 (m, 12H), 1.20 (m, 1H), 1.28 (s,3H), 1.32 (s, 3H), 1.38 (s, 1H), 1.44 (d, J=6 Hz, 3H), 1.60-1.80 (m, 3H), 1.90 (m,1H), 2.0 (m, 2H), 2.29 (s, 6H, NMe2), 2.39 (d, J=13.2 Hz, 1H), 2.75 (m, 1H), 2.95 (m, 1H), 3.41 (s, 3H, O-Me), 3.42 (m,1H), 3.49 (s, 1H), 3.79 (m, 2H), 3.90 (m,1H), 4.19 (m,1H), 4.42 (m, 2H), 4.52 (d, J=5.7 Hz, 2H), 4.73 (m, 1H), 4.87 (dd, J=9.9, 9.6 Hz, 2H), 5.10 (m, 1H), 5.20 (d, J=10.5, 1H), 5.28 (d J=15.6 Hz,1H), 5.58 (d, J=10.5 Hz,1H), 5.90 (m, 2H), 7.30-7.60 (m, 8H), 7.87 (m,1H), 7.90-8.05 (m, 5H); 13C-NMR (CDCl3) δ 10.4, 11.6, 13.6, 17.4, 17.6, 20.9, 21.2, 21.9, 24.1, 25.6, 31.2, 31.9, 34.6, 35.5, 38.0, 40.6, 40.8, 41.5, 45.9, 49.5, 59.1, 63.1, 63.2, 65.3, 68.5, 68.8, 70.1, 72.1, 73.2, 74.5, 78.7, 78.9, 80.2, 82.8, 95.3, 99.7, 100.5, 118.6, 126.1, 127.6, 128.1, 128.3, 129.5, 129.7, 130.5, 131.9, 132.1, 132.5, 132.7, 133.2, 134.3, 140.5, 164.9, 165.4, 166.2, 175.1; MS m/z 1178 (MH+), HRMS (FAB) Calcd for C64H88Cl1N1O17 (MH+): 1178.5814. Found: 1178.5841.
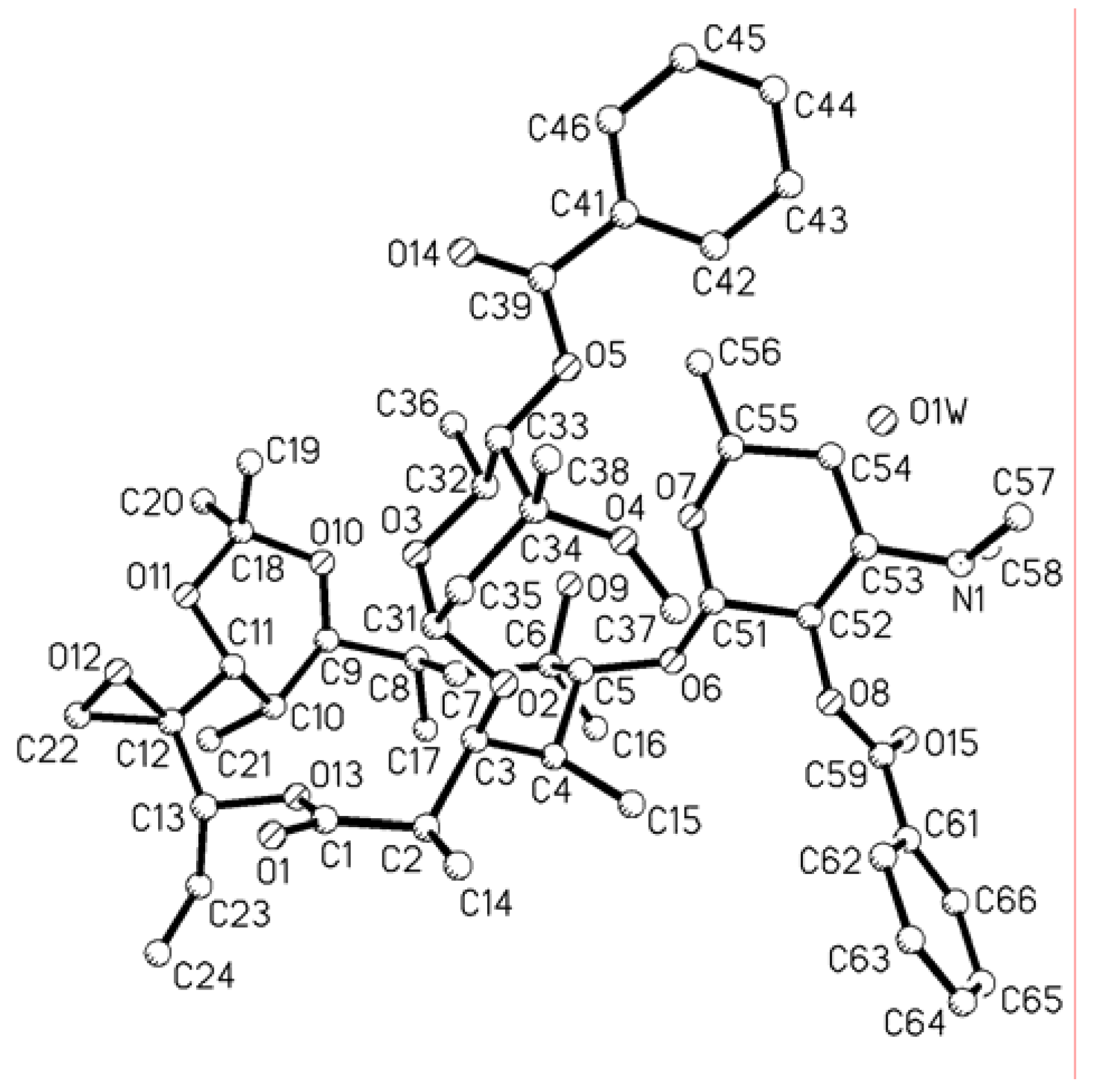
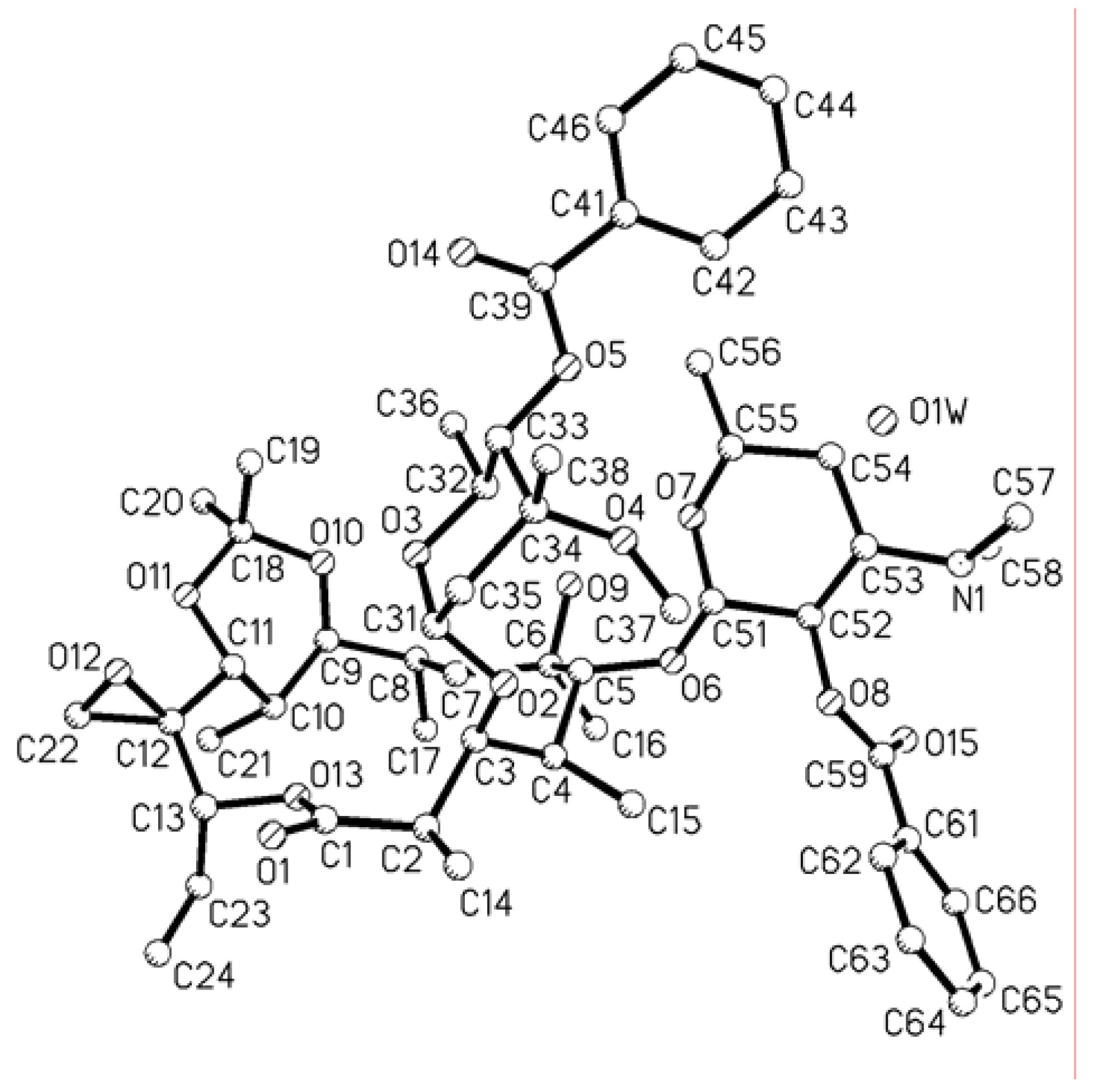
Single crystal X-ray structure analyses of compound 5 [14].
The structure of 5 was determined by X-ray crystallography of a crystal that measured 1.522 × 1.319 × 0.743 mm. Diffraction measurements were made on a Bruker SMART CCD diffractometer with graphite monochromated Mo Kα radiation. Preliminary indications of the unit cell based on randomly selected reflections revealed orthorhombic symmetry with the following lattice parameters: a =1 1.4313(9) Å, b = 16.2380(13) Å, c = 30.192(3) Å, with α = β = γ =90.0°. The space group was P212121, Z = 4 with one molecule of composition C54H79NO15·1/4H2O. The calculated density was 1.164 g/cm3. There were 30258 reflections collected with θ rang from 1.42 to 25.00°, of those reflections 9866 with I>2σ(I) were adjudged observed. The structure was solved by SHELXS-97.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}