Glyphosate’s Suppression of Cytochrome P450 Enzymes and Amino Acid Biosynthesis by the Gut Microbiome: Pathways to Modern Diseases †


Abstract
:1. Introduction
2. Glyphosate’s Pathological Effects: Controlled Studies
3. Gut Dysbiosis, Autism and Colitis
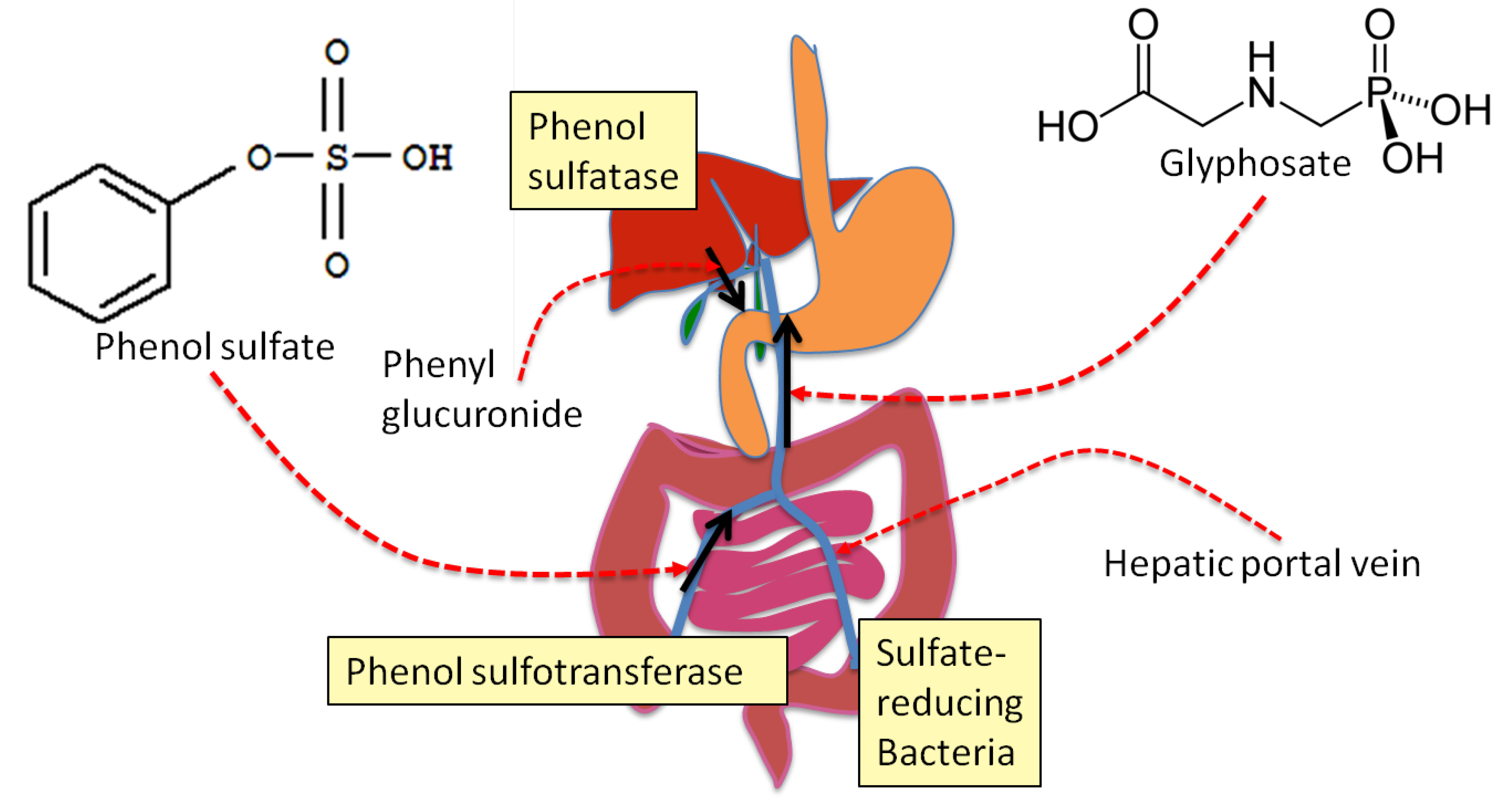
4. Sulfate Transport Impairment and Phenol Synthesis
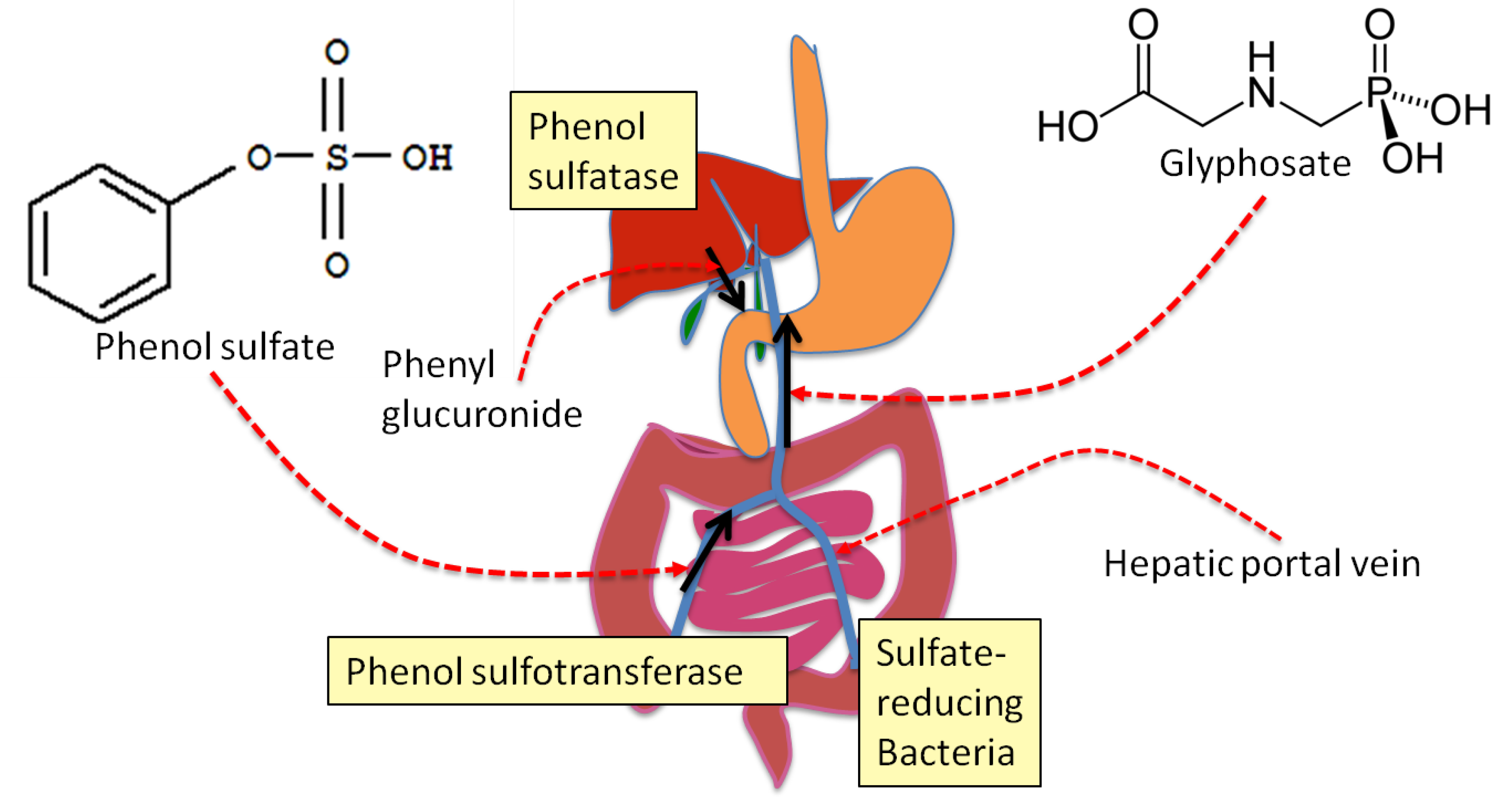
5. Evidence that Glyphosate Inhibits CYP Enzymes
6. The Path to Obesity
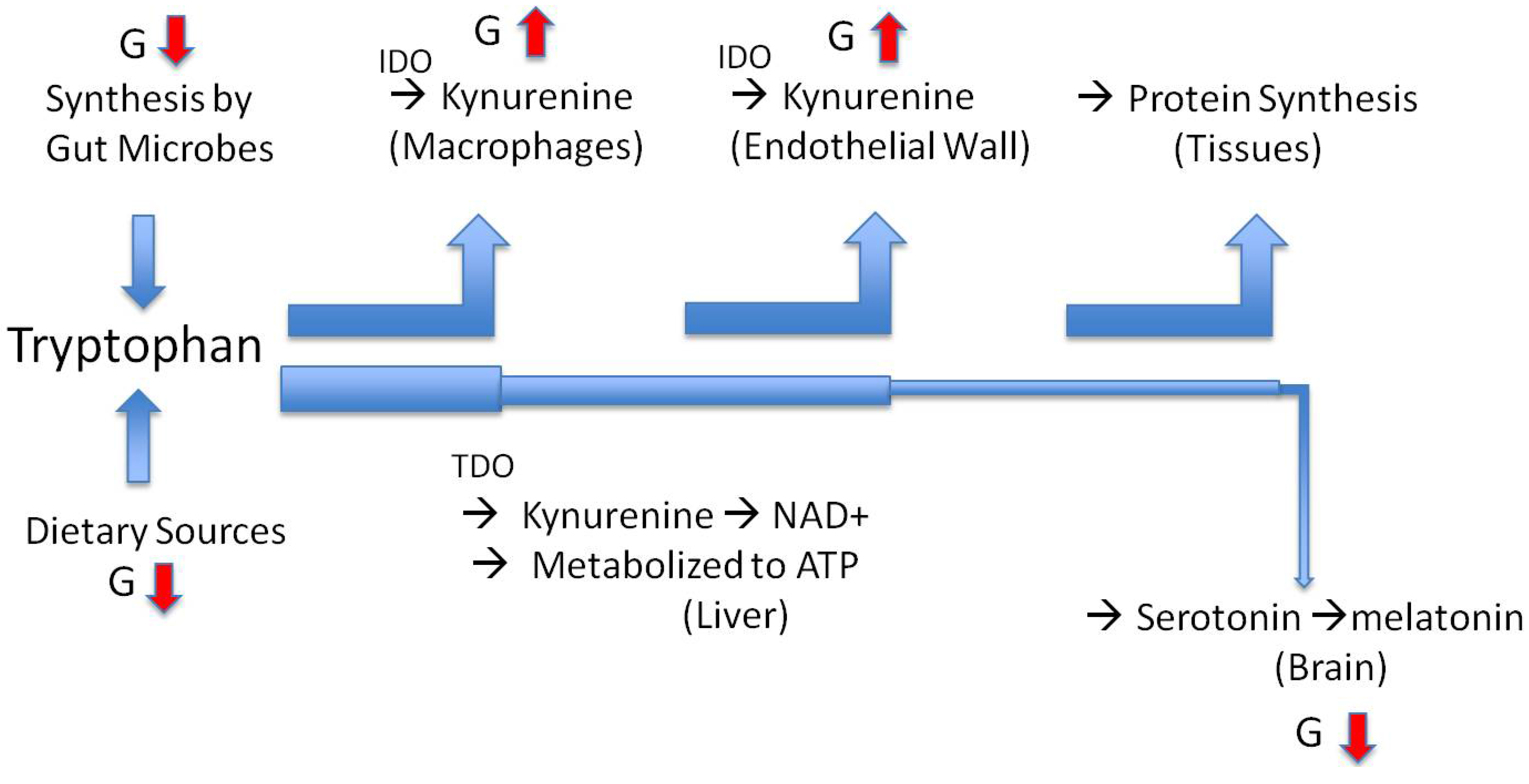
7. The Path to Inflammatory Bowel Disease and Anorexia Nervosa
8. Cytochrome P450 Enzymes
9. Glyphosate’s Potential Role in eNOS Dysfunction
9.1. Lysosomal Dysfunction
9.2. Tetrahydrobiopterin
10. Involvement of the Brain
10.1. Serotonin, Mood Disorders, and Autism
10.2. Ammonia, Autism and Alzheimer’s Disease.
10.3. A Role for Zinc Deficiency.
10.4. Methylation Impairment
10.5. Molecular Mimicry and Multiple Sclerosis
10.6. Dopamine and Parkinson’s Disease
11. Other Adverse Health Effects
11.1. Liver Disease
11.2. Development and Fertility
11.3. Cancer
11.4. Cachexia
12. Glyphosate in Food Sources

{kind=link}
{kind=link}
| Glyphosate usage for the USA (Range in millions of pounds) | ||||
|---|---|---|---|---|
| Year | 2001 | 2003 | 2005 | 2007 |
| Range | 85–90 | 128–133 | 155–160 | 180–185 |
13. Discussion
14. Conclusion
Supplementary Material
Supplementary File 1Acknowledgements
References
- Williams, G.M.; Kroes, R.; Munro, I.C. Safety evaluation and risk assessment of the herbicide roundup and its active ingredient, glyphosate, for humans. Regul. Toxicol. Pharm. 2000, 31, 117–165. [Google Scholar] [CrossRef] [PubMed]
- Battaglin, W.A.; Kolpin, D.W.; Scribner, E.A.; Kuivila, K.M.; Sandstrom, M.W. Glyphosate, other herbicides, and transformation products in midwestern streams, 2002. J. Am. Water Resour. Assoc. 2005, 41, 323–332. [Google Scholar] [CrossRef]
- Shaw, D.R.; Barrentine, W.L. Herbicide combinations for preharvest weed desiccation in early maturing soybean (Glycine max). Weed Technol. 1998, 12, 157–165. [Google Scholar]
- Baur, J.R.; Miller, F.R.; Bovey, R.W. Effects of preharvest desiccation with glyphosate on grain sorghum. Seed 1977, 69, 1015–1018. [Google Scholar] [CrossRef]
- Baig, M.N.; Darwent, A.L.; Harker, K.N.; O’Donovan, J.T. Preharvest applications of glyphosate affect emergence and seedling growth of field pea (Pisum sativum). Weed Technol. 2003, 17, 655–665. [Google Scholar] [CrossRef]
- Duke, S.O.; Powles, S.B. Glyphosate: A once-in-a-century herbicide. Pest. Manag. Sci. 2008, 64, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Weed Science Society of America Committee. Herbicide Handbook of the Weed Science Society of America, 4th ed.; Weed Science Society of America: Champaign, IL, USA, 1979. [Google Scholar]
- Smith, E.A.; Oehme, F.W. The biological activity of glyphosate to plants and animals: A literature review. Vet. Hum. Tocicol. 1992, 34, 531–543. [Google Scholar]
- Séralini, G.-E.; Clair, E.; Mesnage, R.; Gress, S.; Defarge, N.; Malatesta, M.; Hennequin, D.; Spiroux de Vendˆomois, J. Long term toxicity of a Roundup herbicide and a Roundup-tolerant genetically modified maize. Food Chem. Toxicol. 2012, 50, 4221–4231. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.M.; Weaver, L.M. The shikimate pathway. Annu. Rev. Plant. Physiol.Plant. Mol. Biol. 1999, 50, 473–503. [Google Scholar]
- Moco, S.; Martin, F.-P.J.; Rezzi, S. Metabolomics view on gut microbiome modulation by polyphenol-rich foods. J. Proteome Res. 2012, 11, 4781–4790. [Google Scholar] [CrossRef] [PubMed]
- Ganal, S.C.; Sanos, S.L.; Kallfass, C.; Oberle, K.; Johner, C.; Kirschning, C.; Lienen-klaus, S.; Weiss, S.; Staeheli, P.; Aichele, P.; et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 2012, 37, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–483, (The same with ref.160). [Google Scholar] [CrossRef] [PubMed]
- Littman, D.R.; Pamer, E.G. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell. Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Loo, R.L.; Stamler, J.; Bictash, M.; Yap, I.K.; Chan, Q.; Ebbels, T.; De Iorio, M.; Brown, I.J.; Veselkov, K.A.; et al. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008, 453, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Ashorn, M. Gastrointestinal diseases in the paediatric age groups in Europe: epidemicology and impact on healthcare. Aliment. Pharmacol. Ther. 2003, 18, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Bewtra, M.; Su, C.; Lewis, J.D. Trends in hospitalization rates for inflammatory bowel disease in the United States. Clin. Gastroenterol. Hepatol. 2007, 5, 597–601. [Google Scholar] [CrossRef] [PubMed]
- de María, N.; Becerril, J.M.; García-Plazaola, J.I.; Ndez, A.H.; de Felipe, M.R.; Fernández-Pascual, M. New insights on glyphosate mode of action in nodular metabolism: Role of shikimate accumulation. J. Agric. Food Chem. 2006, 54, 2621–2628. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.A.; Dacks, J.B.; Campbell, S.A.; Blanchard, J.L.; Foster, P.G.; McLeod, R.; Roberts, C.W. Evolutionary origins of the eukaryotic shikimate pathway: Gene fusions, horizontal gene transfer, and endosymbiotic replacements. Eukaryot. Cell. 2006, 5, 1517–1531. [Google Scholar] [CrossRef] [PubMed]
- Henry, W.B.; Koger, C.H.; Shaner, D.L. Accumulation of shikimate in corn and soybean exposed to various rates of glyphosate. Crop. Management. 2005. Available online: http://www.plantmanagementwork.org/sub/cm/research/2005/shikimate/ (accessed on 10 February 2013).
- Nafziger, E.D.; Widholm, J.M.; Steinrcken, H.C.; Killmer, J.L. Selection and Characterization of a Carrot Cell Line Tolerant to Glyphosate. Plant. Physiol. 1984, 76, 571–574. [Google Scholar]
- Howles, P.A.; Sewalt, V.J.H.; Paiva, N.L.; Elkind, Y.; Bate, N.J.; Lamb, C.; Dixon, R.A. Overexpression of L-phenylalanine ammonia-lyase in transgenic tobacco plants reveals control points for flux into phenylpropanoid biosynthesis. Plant. Physiol. 1996, 112, 1617–1624. [Google Scholar] [PubMed]
- Guillet, G.; Poupart, J.; Basurco, J.; De Luca, V. Expression of tryptophan decarboxylase and tyrosine decarboxylase genes in tobacco results in altered biochemical and physiological phenotypes. Plant. Physiol. 2000, 122, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Duke, S.O.; Hoagland, R.E.; Elmore, C.D. Effects of glyphosate on metabolism of phenolic compounds V. l-aminooxy-phenylpropionic acid and glyphosate effects on phenylalanine ammonia-lyase in soybean seedlings. Plant Physiol. 1980, 65, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Michalowicz, J.; Duda, W. Phenols sources and toxicity. Polish J. Environ. Stud. 2007, 16, 347–362. [Google Scholar]
- Ortega-García, F.; Peragón, J. Phenylalanine ammonia-lyase, polyphenol oxidase, and phenol concentration in fruits of Olea europaea L. cv. Picual, Verdial, Arbequina, and Frantoio during ripening. J. Agric. Food Chem. 2009, 57, 10331–10040. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, R.E. Effects of glyphosate on metabolism of phenolic compounds: VI. Effects of glyphosine and glyphosate metabolites on phenylalanine ammonia-lyase activity, growth, and protein, chlorophyll, and anthocyanin levels in soybean (Glycine max) seedlings. Weed Sci. 1980, 28, 393–400. [Google Scholar]
- Duke, S.O.; Hoagland, R.E. Effects of glyphosate on metabolism of phenolic compounds I. Induction of phenylalanine ammonia-lyase activity in dark-grown maize roots. Plant Sci. Lett. 1978, 11, 185–190. [Google Scholar] [CrossRef]
- Zhao, J.; Williams, C.C.; Last, R.L. Induction of Arabidopsisl tryptophan pathway enzymes and camalexin by amino acid starvation, oxidative stress, and an abiotic elicitor. Plant Cell 1998, 10, 359–370. [Google Scholar] [PubMed]
- Hernandez, A.; Garcia-Plazaola, J.I.; Becerril, J.M. Glyphosate effects on phenolic metabolism of nodulated soybean (Glycine max L. Merr.). J. Agric. Food Chem. 1999, 47, 2920–2925. [Google Scholar] [CrossRef] [PubMed]
- Moorman, T.B.; Becerril, J.M.; Lydon, J.; Duke, S.O. Production of hydroxybenzoic acids by Bradyrhizobium japonicum strains after treatment with glyphosate. J. Agric. Food Chem. 1992, 289–293. [Google Scholar] [CrossRef]
- Becerra-Moreno, A.; Benavides, J.; Cisneros-Zevallos, L.; Jacobo-Velázquez, D.A. Plants as biofactories: Glyphosate-induced production of shikimic acid and phenolic antioxidants in wounded carrot tissue. J. Agric. Food Chem. 2012, 60, 11378–11386. [Google Scholar] [CrossRef] [PubMed]
- Duke, S.O.; Vaughn, K.C.; Wauchope, R.D. Effects of glyphosate on uptake, translocation, and intracellular localization of metal cations in soybean (Glycine max) seedlings. Pestic. Biochem. Phys. 1985, 24, 384–394. [Google Scholar] [CrossRef]
- Cakmak, I.; Yazici, A.; Tutus, Y.; Ozturk, L. Glyphosate reduced seed and leaf concentrations of calcium, manganese, magnesium, and iron in non-glyphosate resistant soybean. Eur. J. Agron. 2009, 31, 114–119. [Google Scholar] [CrossRef]
- Krüger, M.; Shehata, A.A.; Schrödl, W.; Rodloff, A. Glyphosate suppresses the antagonistic effect of Enterococcus spp. on Clostridium botulinum. Anaerobe 2013, 20, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Shehata, A.A.; Schrödl, W.; Aldin, A.A.; Hafez, H.M.; Krüger, M. The effect of glyphosate on potential pathogens and beneficial members of poultry microbiota in vitro. Curr. Microbiol. 2013, 66, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Shinabarger, D.L.; Braymer, H.D. Glyphosate catabolism by Pseudomonas sp. strain PG2982. J. Bacteriol. 1986, 168, 702–707. [Google Scholar] [PubMed]
- Nie, C.L.; Wang, X.S.; Liu, Y.; Perrett, S.; He, R.Q. Amyloid-like aggregates of neuronal tau induced by formaldehyde promote apoptosis of neuronal cells. BMC Neurosci. 2007, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Li, L.; Chen, M.; Zhou, Z.; Zhang, W.; Ping, S.; Yan, Y.; Wang, J.; Lin, M. Genome-wide transcriptional responses of Escherichia coli to glyphosate, a potent inhibitor of the shikimate pathway enzyme 5-enolpyruvylshikimate-3-phosphate synthase. Mol. Biosyst. 2013, 9, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mallek, A.Y.; Abdel-Kader, M.I.; Shonkeir, A.M. Effect of glyphosate on fungal population, respiration and the decay of some organic matters in Egyptian soil. Microbiol. Res. 1994, 149, 69–73. [Google Scholar] [CrossRef]
- Relyea, R.A. The impact of insecticides and herbicides on the biodiversity and productivity of aquatic communities. Ecol. Appl. 2005, 15, 618–627. [Google Scholar] [CrossRef]
- Paetow, L.J.; McLaughlin, J.D.; Pauli, B.D.; Marcogliese, D.J. Mortality of American bullfrog tadpoles lithobates catesbeianus infected by Gyrodactylus jennyae and experimentally exposed to Batrachochytrium dendrobatidis. J. Aquat. Anim. Health 2013, 25, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.J.; Lips, K.R.; Bermingham, E. Epidemic disease decimates amphibian abundance, species diversity, and evolutionary history in the highlands of central Panama. PNAS 2010, 107, 13777–13782. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.; Najle, R.; Lifschitz, A.; Virkel, G. Effects of sub-lethal exposure of rats to the herbicide glyphosate in drinking water: glutathione transferase enzyme activities, levels of reduced glutathione and lipid peroxidation in liver, kidneys and small intestine. Environ. Toxicol. Pharmacol. 2012, 34, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Mañas, F.J.; Peralta, L.; Garca Ovando, H.; Weyers, A.; Ugnia, L.; Gorla, N. Genotoxicity of glyphosate and AMPA evaluated through comet assay in blood and hepatocytes of treated mice. Biocell. 2009, 33, A80. [Google Scholar]
- Kim, Y.H.; Hong, J.R.; Gil, H.W.; Song, H.Y.; Hong, S.Y. Mixtures of glyphosate and surfactant TN20 accelerate cell death via mitochondrial damage-induced apoptosis and necrosis. Toxicol. In Vitro 2013, 27, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Clair, E.; Linn, L.; Travert, C.; Amiel, C.; Séralini, G.E.; Panoff, J.M. Effects of Roundup and glyphosate on three food microorganisms: Geotrichum candidum, Lactococcus lactis subsp. cremoris and Lactobacillus delbrueckii subsp. bulgaricus. Curr. Microbiol. 2012, 64, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Mariager, T.P.; Madsen, P.V.; Ebbehøj, N.E.; Schmidt, B.; Juhl, A. Severe adverse effects related to dermal exposure to a glyphosate-surfactant herbicide. Clin. Toxicol. (Phila.). 2013, 51, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.P.; Shetty, P. Accidental chemical burns of oral mucosa by herbicide. JNMA J. Nepal Med. Assoc. 2012, 52, 40–42. [Google Scholar] [PubMed]
- Williams, B.L.; Hornig, M.; Buie, T.; Bauman, M.L.; Cho Paik, M.; Wick, I.; Bennettt, A.; Jabado, O.; Hirschberg, D.L.; Lipkin, W.I. Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS One 2011, 6, e24585. [Google Scholar] [CrossRef] [PubMed]
- Horvath, K.; Perman, J.A. Autism and gastrointestinal symptoms. Current Gastroenterology Reports 2002, 4, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Elevated fecal short chain fatty acid and ammonia concentrations in children with autism spectrum disorder. Dig. Dis. Sci. 2012, 57, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- MacFabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: implications in autism spectrum disorders. Microb. Ecol. Health Di. 2012, 23, 19260. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, C.; Finegold, S.M. Real-Time PCR quantitation of Clostridia in feces of autistic children. Appl. Environ. Microbiol. 2004, 70, 6459–6465. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, A.J.; Puleston, J.M.; Montgomery, S.M.; Anthony, A.; O’Leary, J.J.; Murch, S.H. Review article: The concept of enterocolonic encephalopathy, autism and opioid receptor ligands. Aliment. Pharmacol. Ther. 2002, 16, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Shawcross, D.; Jalan, R. The pathophysiologic basis of hepatic encephalopathy: central role for ammonia and inflammation. Cell. Mol. Life Sci. 2005, 62, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, A.; Fernández, A. Hepatic encephalopathy, ammonia, glutamate, glutamine and oxidative stress. Ann. Hepatol. 2009, 8, 95–102. [Google Scholar] [PubMed]
- Romero-Gmez, M.; Jover, M.; Galn, J.J.; Ruiz, A. Gut ammonia production and its modulation. Metab. Brain Dis. 2009, 24, 147–157. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.J.; DCunha, G.B. A modern view of phenylalanine ammonia lyase. Biochem. Cell. Biol. 2007, 85, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Clayton, T.A. Metabolic differences underlying two distinct rat urinary phenotypes, a suggested role for gut microbial metabolism of phenylalanine and a possible connection to autism. FEBS Lett. 2012, 586, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, S.; Seneff, S. Impaired sulfate metabolism and epigenetics: Is there a link in autism? Entropy 2012, 14, 1953–1977. [Google Scholar] [CrossRef]
- Kern, J.K.; Grannemann, B.D.; Trivedi, M.H.; Waring, R.H.; Ramsden, D.B.; Garver, C.R. Abnormal sulfation chemistry in autism. In Trends in Autism Research; Ryaskin, O.T., Ed.; Nova Publishers: Hauppauge, NY, USA, 2004; Chapter XI. [Google Scholar]
- Sivsammye, G.; Sims, H.V. Presumptive identification of Clostridium difficile by detection of p-cresol in prepared peptone yeast glucose broth supplemented with p-hydroxyphenylacetic acid. J. Clin. Microbiol. 1990, 28, 1851–1853. [Google Scholar] [PubMed]
- D’Ari, L.; H.A. Barker, H.A. p-Cresol formation by cell free extracts of Clostridium difficile. Arch. Microbiol. 1985, 143, 311–312. [Google Scholar]
- Kelly, C.P.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile colitis. N. Engl. J. Med. 1994, 330, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.; Vijayapal, A.; Graham, M.B.; Beaulieu, D.B.; Otterson, M.F.; Lundeen, S.; Skaros, S.; Weber, L.R.; Komorowski, R.A.; Knox, J.F.; Emmons, J.; Bajaj, J.S.; Binion, D.G. Impact of Clostridium difficile on inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2007, 5, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Clayton, T.A.; Baker, D.; Lindon, J.C.; Everett, J.R.; Nicholson, J.K. Phar-macometabonomic identification of a significant hostmicrobiome metabolic interaction affecting human drug metabolism. Proc. Natl. Am. Sci. 2009, 106, 14728–14733. [Google Scholar] [CrossRef] [PubMed]
- Altieri, L.; Neri, C.; Sacco, R.; Curatolo, P.; Benvenuto, A.; Muratori, F.; Santocchi, E.; Bravaccio, C.; Lenti, C.; Saccani, M.; et al. Urinary p-cresol is elevated in small children with severe autism spectrum disorder. Biomarkers 2011, 16, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Buckman, N.G.; Hill, J.O.; Magee, R.J.; McCormick, M.J. Separation of substituted phenols, including eleven priority pollutants using high performance liquid chromatography. J. Chromatogr. 1984, 284, 441–446. [Google Scholar] [CrossRef]
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Chari, R.S.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Ozyrskyj, A.L. Gut microbiota of healthy Canadian infants: Profiles by mode of delivery and infant diet at 4 months. Can. Med. Assoc. J. 2013, 185, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.T.; Klonoff-Cohen, H.S.; Wingard, D.L.; Akshoomoff, N.A.; Macera, C.A.; Ji, M.; Bacher, C. Breastfeeding, infant formula supplementation, and autistic disorder: The results of a parent survey. Int. Breastfeed. J. 2006, 1, 16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van der Heiden, C.; Wauters, E.A.K.; Ketting, D.; Duran, M.; Wadman, S.K. Gas chromatographic analysis of urinary tyrosine and phenylalanine metabolites in patients with gastrointestinal disorders. Clin. Chim. Acta. 1971, 34, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Shaw, W. Increased urinary excretion of a 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA), an abnormal phenylalanine metabolite of Clostridia spp. in the gastrointestinal tract, in urine samples from patients with autism and schizophrenia. Nutr. Neurosci. 2010, 13, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Yap, I.K.; Angley, M.; Veselkov, K.A.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Urinary metabolic phenotyping differentiates children with autism from their unaffected siblings and age-matched controls. J. Proteome Res. 2010, 9, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Gatley, S.J.; Sherratt, H.S. The synthesis of hippurate from benzoate and glycine by rat liver mitochondria. Submitochondrial localization and kinetics. Biochem. J. 1977, 166, 39–47. [Google Scholar] [PubMed]
- Ashwood, P.; Anthony, A.; Pellicer, A.A.; Torrente, F.; Walker-Smith, J.A.; Wakefield, A.J. Intestinal lymphocyte populations in children with regressive autism: Evidence for extensive mucosal immunopathology. J. Clin. Immunol. 2003, 23, 504–517. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, B.A.; Waring, R.H. Enzyme and sulphur oxidation deficiencies in autistic children with known food/chemical intolerances. Xenobiotica. 1990, 20, 117–122. [Google Scholar]
- Baldwin, R.L. How Hofmeister ion interactions affect protein stability. Biophys. J. 1996, 71, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, F. Naunyn-Schmiedebergs Zur Lehre von der Wirkung der Salze (Article in German). Arch. Pharmacol. 1888, 24, 247–260. [Google Scholar] [CrossRef]
- Zouaoui, K.; Dulaurent, S.; Gaulier, J.M.; Moesch, C.; Lachâtre, G. Determination of glyphosate and AMPA in blood and urine from humans: About 13 cases of acute intoxication. Forensic Sci. Int. 2013, 226, e20–e25. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Nagrath, D.; Garde, S.; Cramer, S.M. Evaluation of selectivity changes in HIC systems using a preferential interaction based analysis. Biotech. Bioengineer. 2004, 87, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Falany, C.N. Molecular enzymology of human liver cytosolic sulfotransferases. Trends Pharmacol. Sci. 1991, 12, 255–259. [Google Scholar] [CrossRef]
- Berg, N.B.; Young, R.W. Sulfate metabolism in pancreatic acinar cells. J. Cell. Biol. 1971, 50, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.; Claycamp, G.H.; Sweetland, M.A.; Sedlov, A.V.; Tyurin, V.A.; Kisin, E.R.; Tyurina, Y.Y.; Ritov, V.B.; Wenger, S.L.; Grant, S.G.; Kagan, V.E. Myeloperoxidase-catalyzed redox-cycling of phenol promotes lipid peroxidation and thiol oxidation in HL-60 cells. Free Radic. Biol. Med. 1999, 27, 1050–1063. [Google Scholar] [CrossRef]
- Prior, R.L.; Wu, X.; Gu, L. Flavonoid metabolism and challenges to understanding mechanisms of health effects. J. Sci. Food Agric. 2006, 86, 2487–2491. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhou, J.; Yang, C.H.; Xia, B.J.; Hu, M.; Liu, Z.Q. Systematic studies of sulfation and glucuronidation of 12 flavonoids in the mouse liver S9 fraction reveal both unique and shared positional preferences. J. Agric. Food Chem. 2012, 28, 60. 3223–3233. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, F.M.; Yousef, M.I.; Elagamy, E.I. Influence of paraquat, glyphosate, and cadmium on the activity of some serum enzymes and protein electrophoretic behavior (in vitro). J. Environ. Sci. Health B 2001, 36, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Geng, J.; Ren, H.; Xia, X.; Wang, X.; Yu, Y. Physiological and biochemical responses of Microcystis aeruginosa to glyphosate and its Roundup® formulation. J. Hazard. Mater. 2012, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; George, F.; Audhya, T.J. Abnormally high plasma levels of vitamin B6 in children with autism not taking supplements compared to controls not taking supplements. J. Altern. Complement. Med. 2006, 12, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Martineau, J.; Barthelemy, C.; Garreau, B.; Lelord, G. Vitamin B6, magnesium, and combined B6-Mg: Therapeutic effects in childhood autism. Biol. Psych. 1985, 20, 467–478. [Google Scholar] [CrossRef]
- Lelord, G.; Muh, J.P.; Barthelemy, C.; Martineau, J.; Garreau, B. Effects of pyridoxine and magnesium on autistic symptoms—Initial observations. J. Autism Devel. Disord. 1981, 11, 219–230. [Google Scholar] [CrossRef]
- Cohen, B.I. The significance of ammonia/gamma-aminobutyric acid (GABA) ratio for normality and liver disorders. Med. Hypotheses 2002, 59, 757–758. [Google Scholar] [CrossRef]
- Sweeten, T.L.; Posey, D.J.; Shankar, S.; McDougle, C.J. High nitric oxide production in autistic disorder: a possible role for interferon-gamma. Biol. Psychiatry 2004, 55, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Sögüt, S.S.; Zoroglu, S.S.; Özyurt, H.; Ylmaz, H.R.; Ozugurlu, F.; Sivasli, E.; Yetkin, O.; Yanik, M.; Tutkun, H.; Savas, H.A.; et al. Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin. Chim. Acta. 2003, 331, 111–117. [Google Scholar]
- Zoroğlu, S.S.; Yürekli, M.; Meram, I.; Söğüt, S.; Tutkun, H.; Yetkin, O.; Sivasli, E.; Savaş, H.A.; Yanik, M.; Herken, H.; l Akyol, O. Pathophysiological role of nitric oxide and adrenomedullin in autism. Cell. Biochem. Funct. 2003, 21, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.M.; Ferrari, P.; Haimart, M.; Bursztejn, C.; Tabuteau, F.; Braconnier, A.; Pasques- Bondoux, D.; Luong, C. Serotonin Metabolism and other biochemical parameters in infantile autism: A controlled study of 22 autistic children. Neuropsychobiology. 1988, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Al-Yafee, Y.A.; Al-Ayadhi, L.Y.; Haq, S.H.; El-Ansary, A.K. Novel metabolic biomarkers related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia. BMC Neurol. 2011, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Alberti, A.; Pirrone, P.; Elia, M.; Waring, R.H.; Romano, C. Sulphation deficit in “low-functioning” autistic children: A pilot study. Biolog. Psychiat. 1999, 46, 420–424. [Google Scholar] [CrossRef]
- Waring, R.H.; Kovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Finegold, S.M. Therapy and epidemiology of autism–clostridial spores as key elements. Med. Hypotheses 2008, 70, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.H.; MacDonald, T.T.; Walker-Smith, J.A.; Levin, M.; Lionetti, P.; Klein, N.J. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet 1993, 341, 711–714. [Google Scholar] [CrossRef]
- Finegold, S.M. Desulfovibrio species are potentially important in regressive autism. Med. Hypotheses 2011, 77, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.C. Anaerobic degradation of aromatic compounds. Ann. Rev. Microbiol. 1988, 42, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Coates, J.D.; Anderson, R.T.; Lovley, D.R. Oxidation of polycyclic aromatic hydrocarbons under sulfate-reducing conditions. Appl. Environ. Microbiol. 1996, 62, 1099–1101. [Google Scholar] [PubMed]
- Rueter, P.; Rabus, R.; Wilkest, H.; Aeckersberg, F.; Rainey, F.A.; Jannasch, H.W.; Widdel, F. Anaerobic oxidation of hydrocarbons in crude oil by new types of sulphate-reducing bacteria. Nature 1994, 372, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Londry, K.L.; Suflita, J.M.; Tanner, R.S. Cresol metabolism by the sulfate-reducing bacterium Desulfotomaculum sp. strain Groll. Can. J. Microbiol. 1999, 45, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Shangari, N.; Chan, T.S.; O'Brien, P.J. Sulfation and glucuronidation of phenols: Implications in coenyzme Q metabolism. Methods Enzymol. 2005, 400, 342–359. [Google Scholar] [PubMed]
- Gasnier, C.; Dumont, C.; Benachour, N.; Clair, E.; Chagnon, M.C.; Séralini, G.E. Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines. Toxicology 2009, 262, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Moslemi, S.; Sipahutar, H.; Benachour, N.; S ́eralini, G.-E. Differential effects of glyphosate and roundup on human placental cells and aromatase. Environ. Health Perspect. 2005, 113, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Mottier, A.; Kientz-Bouchart, V.; Serpentini, A.; Lebel, J.M.; Jha, A.N.; Costil, K. Effects of glyphosate-based herbicides on embryo-larval development and metamorphosis in the Pacific oyster. Crassostrea gigas. Aquat. Toxicol. 2013, 128–129, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Aulehla, A.; Pourqui, O. Signaling gradients during paraxial mesoderm development. Cold Spring Harb. Perspect. Biol. 2010, 2, a000869. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, A.; Gnazzo, V.; Acosta, H.; Lpez, S.L.; Carrasco, A.E. Glyphosate-based herbicides produce teratogenic effects on vertebrates by impairing retinoic acid signaling. Chem. Res. Toxicol. 2010, 23, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- William J. Ray, W.J.; Gerard Bain, G.; Min Yao, M.; David I. Gottlieb, D.I. CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J. Biol. Chem. 1997, 272, 18702–18708. [Google Scholar]
- Fujii, H.; Sato, T.; Kaneko, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Kelly, D.E.; Hanley, S.Z.; Mehmood, Z.; Kelly, S.L. Glyphosate is an inhibitor of plant cytochrome P450: Functional expression of thlaspi arvensae cytochrome P45071b1/reductase fusion protein in Escherichia coli. Biochem. Biophys. Res. Comm. 1998, 244, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Hietanen, E.; Linnainmaa, K.; Vainio, H. Effects of phenoxyherbicides and glyphosate on the hepatic and intestinal biotransformation activities in the rat. Acta. Pharmacol. Toxicol. 1983, 53, 103–112. [Google Scholar] [CrossRef]
- Khan, S.U.; Young, J.C. N-Nitrosamine formation in soil from the herbicide glyphosate. J. Agric. Food Chem. 1977, 25, 1430–1432. [Google Scholar] [CrossRef] [PubMed]
- Su, K. N-nitrosamine formation in soil from the herbicide glyphosate and its uptake by plants. ACS Symposium Series. 1981, 174, 275–287. [Google Scholar]
- Buchmann, A.; Kuhlmann, W.D.; Schwarz, M.; Kunz, W.; Wolf, C.R.; Moll, E.; Friedberg, T.; Oesch, F. Regulation and expression of four cytochromes P-450 isoenzymes, NADPH-cytochrome P-450 reductase, the glutathione transferases B and C and microsomal epoxide hydrolase in preneoplastic and neoplastic lesions in rat liver. Carcinogenesis 1985, 6, 513–521. [Google Scholar] [PubMed]
- Abass, K.; Turpeinen, M.; Pelkonen, O. An evaluation of the cytochrome P450 inhibition potential of selected pesticides in human hepatic microsomes. J. Environ. Sci. Health B. 2009, 44, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Abass, K.; Lämsä, V.; Reponen, P.; Küblbeck Honkakoski, P.; Mattila, S.; Pelkonen, O.; Hakkola, J. Characterization of human cytochrome P450 induction by pesticides. Toxicology 2012, 294, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; di Carlo Herd, F.J. Human cytochrome P450 enzymes: A status report summarlzlng thelr reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef] [PubMed]
- Schacker, M. A Spring Without Bees: How Colony Collapse Disorder Has Endangered Our Food Supply; Globe Pequot: Guilford, CT. USA, 2008. [Google Scholar]
- Mao, W.; Schuler, M.A.; Berenbaum, M.R. CYP9Q-mediated detoxification of acaricides in the honey bee (Apis mellifera). Proc. Natl. Am. Soi. 2011, 108, 12657–12662. [Google Scholar] [CrossRef] [PubMed]
- Morandin, L.A.; Winston, M.L. Wild bee abundance and seed production in conventional organic, and genetically modified canola. Ecol. Appl. 2005, 15, 871–881. [Google Scholar] [CrossRef]
- Foulk, K.E.; Reeves, C. Identifying the role of glyphosate-containing herbicides on honeybee mortality rates and colony collapse disorder. In Proceedings of Junior Science, Engineering, and Humanities Symposium, Camdenton, MO, USA; 2009; pp. 2–23. [Google Scholar]
- Ratnieks, F.L.W.; Carreck, N.L. Clarity on honey bee collapse? Science 2010, 327, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.; Gawarammana, I.; Robertson, T.A.; Roberts, M.S.; Palangasinghe, C.; Zawahir, S.; Jayamanne, S.; Jegenathen, K.; Eddleston, M.; Buckley, N.; et al. Acute Human self-poisoning with Imidacloprid compound: A neonicotinoid insecticide. Plos One 2009, 4, e5127. [Google Scholar] [CrossRef] [PubMed]
- Baillie-Hamilton, P.F. Chemical toxins: A hypothesis to explain the global obesity epidemic. J. Altern. Complem. Med. 2002, 8, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.C.; McDougle, C.J.; Schumacher, M.; Olcese, J.; Mason, J.W.; Heninger, G.R.; Price, L.H. Effects of acute tryptophan depletion on nocturnal melatonin secretion in humans. J. Clin. Endocr. MeTable 1993, 76, 1160–1164. [Google Scholar]
- Breisch, S.T.; Zemlan, F.P.; Hoebel, B.G. Hyperphagia and obesity following serotonin depletion by intraventricular p-chlorphenylalanine. Science 1976, 192, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; MA ARYAN Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell. Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Espey, M.G.; Namboodiri, M.A. Antibodies to quinolinic acid and the determination of its cellular distribution within the rat immune system. Cell. Tissue. Res. 1994, 278, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Werner-Felmayer, G.; Werner, E.R.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Wachter, H. Induction of indoleamine 2,3-dioxygenase in human cells in vitro. Adv. Exp. Med. Biol. 1991, 294, 505–509. [Google Scholar] [PubMed]
- Yoshida, R.; Nukiwa, T.; Watanabe, Y.; Fujiwara, M.; Hirata, F.; Hayaishi, O. Regulation of indoleamine 2,3-dioxygenase activity in the small intestine and the epididymis of mice. Arch. Biochem. Biophys. 1980, 203, 343–351. [Google Scholar] [PubMed]
- Yoshida, R.; Hayaishi, O. Induction of pulmonary indoleamine 2,3-dioxygenase by in-traperitoneal injection of bacterial lipo-polysaccharide. Proc. Natl. Acad. Sci. USA 1978, 75, 3998–4000. [Google Scholar] [CrossRef] [PubMed]
- Carson, D.A.; Seto, S.; Wasson, D.B.; Carrera, C.J. DNA strand breaks, NAD metabolism, and programmed cell death. Exp. Cell. Res. 1986, 164, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.J.; Stierum, R.H. Niacin, poly (ADP-ribose) polymerase-1 and genomic stability. Mutat. Res. 2001, 475, 45–56. [Google Scholar] [CrossRef]
- Satoh, M.S.; Poirier, G.G.; Lindahl, T. Dual function for poly (ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry 1994, 33, 7099–7106. [Google Scholar] [CrossRef] [PubMed]
- Hayaishi, O. Utilization of superoxide anion by indoleamine oxygenase-catalyzed tryptophan and indoleamine oxidation. Adv. Exp. Med. Biol. 1996, 398, 285–289. [Google Scholar] [PubMed]
- Caballero, B.; Finer, N.; Wurtman, R.J. Plasma amino acids and insulin levels in obesity: response to carbohydrate intake and tryptophan supplements. Metabolism 1988, 37, 672–676. [Google Scholar] [CrossRef]
- Breum, L.; Rasmussen, M.H.; Hilsted, J.; Fernstrom, J.D. Twenty-four hour plasma tryptophan concentrations and ratios are below normal in obese subjects and are not normalized by substantial weight reduction. Am. J. Clin. Nutr. 2003, 77, 1112–1118. [Google Scholar] [PubMed]
- Fei, N.; Liping Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013, 7, 880–884. [Google Scholar]
- Woods, S.C.; Seeley, R.J.; Rushing, P.A.; DAlessio, D.; Tso, P. A controlled high-fat diet induces an obese syndrome in rats. J. Nutr. 2003, 133, 1081–1087. [Google Scholar] [PubMed]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.-H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [PubMed]
- Deckelbaum, R.J.; Williams, C.L. Childhood obesity: The health issue. Obes. Res. 2001, 9, 239S–243S. [Google Scholar] [CrossRef] [PubMed]
- Rolls, B.J. The supersizing of America: Portion size and the obesity epidemic. Nutrition Today 2003, 38, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Popkins, B.M.; Doak, C.M. The obesity epidemic is a worldwide phenomenon. Nutr. Rev. 1998, 56, 106–114. [Google Scholar] [CrossRef]
- Puoane, T.; Steyn, K.; Bradshaw, D.; Laubscher, R.; Fourie, J.; Lambert, V.; Mbananga, N. Obesity in South Africa: the South African demographic and health survey. Obes. Res. 2002, 10, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, S.; Horowitz, L. Converging Networks and Clashing Stories: South Africa’s Agricultural Biotechnology Debate. Africa Today 2004, 51, 325. [Google Scholar]
- Scoones, I. Mobilizing Against GM Crops in India, South Africa and Brazil. J. Agrar. Change 2008, 8, 315–344. [Google Scholar] [CrossRef]
- WHO Global Infobase. Available online: https://apps.who.int/infobase/Indicators.aspx/ (accessed on 18 February 2013).
- Hidaka, H.; Nagatsu, T.; Takeya, K.; Matsumoto, S.; Yagi, K. Inactivation of serotonin by sulfotransferase system. J. Pharmacol. Exp.Ther. 1969, 166, 272–275. [Google Scholar] [PubMed]
- Strott, C.A.; Higashi, Y. Cholesterol sulfate in human physiology: What’s it all about? J. Lipid Res. 2003, 44, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Croonenberghs, J.; Spaas, K.; Wauters, A.; Verkerk, R.; Scharpe, S.; Deboutte, D.; Maes, M. Faulty serotonin--DHEA interactions in autism: Results of the 5-hydroxytryptophan challenge test. Neuro. Endocrinol. Lett. 2008, 29, 385–390. [Google Scholar] [PubMed]
- Hernández-Morante, J.J.; Pérez-de-Heredia, F.; Luján, J.A.; Zamora, S.; Garaulet, M. Role of DHEA-S on body fat distribution: Gender- and depot-specific stimulation of adipose tissue lipolysis. Steroids 2008, 73, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Santos, C.; Hernández-Morante, J.J.; Tébar, F.J.; Granero, E.; Garaulet, M. Differential effect of oral dehydroepiandrosterone-sulphate on metabolic syndrome features in pre- and postmenopausal obese women. Clin. Endocrinol. 2012, 77, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, J.; Milewicz, A.; Thijssen, J.H.H.; Blankenstein, M.A.; Daroszewski, J. Concentration of sex steroids in adipose tissue after menopause. Steroids 1998, 63, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. The Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Anzenbacher, P.; Anzenbacherova, E. Cytochromes p450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Stiles, A.R.; McDonald, J.G.; Bauman, D.R.; Russell, D.W. CYP7B1: One cytochrome P450, two human genetic diseases, and multiple physiological functions. J. Biol. Chem. 2009, 284, 28485–28489. [Google Scholar] [CrossRef] [PubMed]
- Wikvall, K. Cytochrome P450 enzymes in the bioactivation of vitamin D to its hormonal form (review). Int. J. Mol. Med. 2001, 7, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Schuster, I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim. Biophys. Acta 2011, 1814, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Ginde, A.A.; Liu, M.C.; A. Camargo, C.A. Demographic Differences and Trends of Vitamin D Insufficiency in the US Population, 1988–2004. JAMA Internal Medicine 2009, 169, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. P450 oxidoreductase deficiency: a disorder of steroidogenesis with multiple clinical manifestations. Sci. Signal. 2012, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Sarachana, T.; Xu, M.; Wu, R.C.; Hu, V.W. Sex hormones in autism: Androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. Plos One 2011, 6, e17116. [Google Scholar] [CrossRef] [PubMed]
- Baron-Cohen, S. The extreme male brain theory of autism. Trends Cog. Sci. 2002, 6, 248–254. [Google Scholar] [CrossRef]
- Andreola, F.; Fernandez-Salguero, P.M.; Chiantore, M.V.; Petkovich, M.P.; Gonzalez, F.J.; De Luca, L.M. Aryl hydrocarbon receptor Ahr(−/−) knockout mice exhibit liver retinoid accumulation and reduced retinoic acid metabolism. Cancer Res. 1997, 57, 2835–2838. [Google Scholar] [PubMed]
- Jetten, A.M.; George, M.A.; Pettit, G.R.; Herald, C.L.; Rearick, J.I. Action of phorbol esters, bryostatins, and retinoic acid on cholesterol sulfate synthesis: Relation to the multistep process of differentiation in human epidermal keratinocytes. J. Invest. Dermatol. 1989, 93, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Lorbek, G.; Lewinska, M.; Rozman, D. Cytochrome P450s in the synthesis of cholesterol and bile acids–from mouse models to human diseases. FEBS J. 2012, 279, 1516–1533. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Stegherr, J.; Latz, W.; Koch, W.; Mehilli, J.; Dörrler, K.; Morath, T.; Schömig, A.; Kastrati, A.; von Beckerath, N. Cytochrome P450 2C19 loss-of-function polymorphism and stent thrombosis following percutaneous coronary intervention. Eur. Heart J. 2009, 30, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, Y.-T.; Verdo, A.; Qi, W.-G.; Zhang, D.-F.; Hu, B. Relationship between cytochrome P450 2C19*2 polymorphism and stent thrombosis following percutaneous coronary intervention in Chinese patients receiving clopidogrel. The J. Int. Med. Res. 2011, 39, 2012–2019. [Google Scholar] [CrossRef] [PubMed]
- Slofstra, S.H.; Spek, C.A.; ten Cate, H. Disseminated intravascular coagulation. Hematol. J. 2003, 4, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Gorren, A.C.; Mayer, B. Nitric-oxide synthase: A cytochrome P450 family foster child. Biochim. Biophys. Acta. 2007, 1770, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Lauritzen, A.; Davidson, R.; Lentz-Marino, L. Is endothelial nitric oxide synthase a moonlighting protein whose day job is cholesterol sulfate synthesis? Implications for cholesterol transport, diabetes and cardiovascular disease. Entropy 2012, 14, 2492–2530. [Google Scholar] [CrossRef]
- Cryle, M.J.; De Voss, J.J. Is the ferric hydroperoxy species responsible for sulfur oxidation in cytochrome P450s? Angew. Chem. Int. Ed. 2006, 45, 8221–8223. [Google Scholar] [CrossRef] [PubMed]
- Engelberg, H. Endogenous heparin activity deficiency: The missing link in atherogenesis? Atherosclerosis 2001, 159, 253–260. [Google Scholar] [CrossRef]
- Khalili, H.; Huang, E.S.; Ananthakrishnan, A.N.; Higuchi, L.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Geographical variation and incidence of inflammatory bowel disease among US women. Gut 2012, 61, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Ruijter, G.J.G.; van Diggelen, O.P. Sanfilippo syndrome: A minireview. J. Inherit. Metab. Dis. 2008, 31, 240252. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of -Synuclein and LRRK2 in the brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. The involvement of lysosomes in myocardial aging and disease. Curr. Cardiol. Rev. 2008, 4, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Miyata, S.; Kawase, Y.; Okada, H.; Maruyama, R.; Fujiwara, H. Autophagic Degeneration and Death of Cardiomyocytes in Heart Failure. Autophagy 2006, 2, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Gustafsson, B.; Brunk, U.T. The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem. Biol. Interact. 2006, 163, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sun, X.; Sharma, S.; Aggarwal, S.; Ravi, K.; Fineman, J.R.; Black, S.M. GTP cyclohydrolase I expression is regulated by nitric oxide: role of cyclic AMP. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L309–L317. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Dikalov, S.; Price, R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Werner-Felmayer, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Wachter, H. Parallel induction of tetrahydrobiopterin biosynthesis and indoleamine 2,3-dioxygenase activity in human cells and cell lines by interferon-gamma. Biochem. J. 1989, 262, 861–866. [Google Scholar] [PubMed]
- McCully, K.S. Chemical pathology of homocysteine V: Thioretinamide, thioretinaco, and cystathionine synthase function in degenerative diseases. Ann. Clin. Lab. Sci. 2011, 41, 300313. [Google Scholar]
- McCully, K.S. Homocysteine, vitamins, and vascular disease prevention. Am. J. Clin. Nutr. 2007, 86, 1563S–1568S. [Google Scholar] [CrossRef] [PubMed]
- Vasan, R.S.; Beiser, A.; D’Agostino, R.B.; Levy, D.; Selhub, J.; Jacques, P.E.; Rosenberg, I.H.; Wilson, P.W.F. Plasma homocysteine and risk for congestive heart failure in adults without prior myocardial infarction. J. Am. Med. Assoc. 2003, 289, 1251–1257. [Google Scholar] [CrossRef]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.F.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- van Guldener, C.; Stam, F.; Stehouwer, C.D. Homocysteine metabolism in renal failure. Kidney Int. Suppl. 2001, 78, S234–S237. [Google Scholar] [CrossRef] [PubMed]
- van Guldener, C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol. Dial. Transplant. 2006, 21, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Cowen, P.J. Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol. Sci. 2008, 29, 433–436. [Google Scholar] [CrossRef] [PubMed]
- McDougle, C.J.; Naylor, S.T.; Cohen, D.J.; Aghajanian, G.K.; Heninger, G.R.; Price, L.H. Effects of tryptophan depletion in drug-free adults with autistic disorder. Arch. Gen. Psychiatry 1996, 53, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; van der Schyf, C.J. Role of serotonin in Alzheimer’s disease: A new therapeutic target? CNS Drugs 2011, 25, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, C.C.; Smith, G.; DeKosky, S.T.; Pollock, B.G.; Mathis, C.A.; Moore, R.Y.; Kupfer, D.J.; Reynolds, C.F., III. Serotonin in aging, late-life depression, and Alzheimer’s disease: The emerging role of functional imaging. Neuropsychopharmacology 1998, 18, 407–430. [Google Scholar] [CrossRef]
- Lansdowne, A.T.G.; Provost, S.C. Vitamin D3 enhances mood in healthy subjects during winter . Psychopharmacology 1998, 135, 319–323. [Google Scholar]
- Maes, M.; Kubera, M.; Leunis, J.-C. The gut-brain barrier in major depression: Intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuroendocrin. Lett. 2008, 29, 117–124. [Google Scholar] [CrossRef]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The inflammatory and neurodegenerative (I&ND) hypothesis of depression: Leads for future research and new drug developments in depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar] [PubMed]
- Song, C.; Lin, A.; Bonaccorso, S.; Heide, C.; Verkerk, R.; Kenis, G.; Bosmans, E.; Scharpe, S.; Whelan, A.; Cosyns, P.; de Jongh, R.; Maes, M. The inflammatory response system and the availability of plasma tryptophan in patients with primary sleep disorders and major depression. J. Affect. Disord. 1998, 49, 211–219. [Google Scholar] [CrossRef]
- Hallikainen, T.; Saito, T.; Lachman, H.M.; Volavka, J.; Pohjalainen, T.; Ryynnen, O.P.; Kauhanen, J.; Syvlahti, E.; Hietala, J.; Tiihonen, J. Association between low activity serotonin transporter promoter genotype and early onset alcoholism with habitual impulsive violent behavior. Mol. Psychiatr. 1999, 4, 385–388. [Google Scholar] [CrossRef]
- Anderson, M.; Kaufman, J.; Simon, T.R.; Barrios, L.; Paulozzi, L.; Ryan, G.; Hammond, R.; Modzeleski, W.; Feucht, T.; Potter, L. School-associated violent deaths in the United States, 1994–1999. J. Am. Medical Assoc. 2001, 286, 2695–2702. [Google Scholar] [CrossRef]
- Retz, W.; Retz-Junginger, P.; Supprian, T.; Thome, J.; Rösler, M. Association of serotonin transporter promoter gene polymorphism with violence: relation with personality disorders, impulsivity, and childhood ADHD psychopathology. Behav. Sci. Law 2004, 22, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Shiva, V.; Jafri, A.H.; Emani, A.; Pande, M. Seeds of Suicide: the Ecological and Human Costs of Globalisation of Agriculture; Zed Books: London, UK, 2005. [Google Scholar]
- Roy, A.; Linnoila, M. Suicidal behavior, impulsiveness and serotonin. Acta Psychiatr. Scand. 1988, 78, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Delahanty, R.J.; Prasad, H.C.; McCauley, J.L.; Han, Q.; Jiang, L.; Chun Li, C.; Folstein, S.E.; Blakely, R.D. Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid-compulsive behaviors. Am. J. Hum. Genet. 2005, 77, 265–279. [Google Scholar] [CrossRef] [PubMed]
- D’Eufemia, P.; Finocchiaro, R.; Celli, M.; Viozzi, L.; Monteleone, D.; Giardini, O. Low serum tryptophan to large neutral amino acids ratio in idiopathic infantile autism. Biomed. Pharmacother. 1995, 49, 288–292. [Google Scholar] [CrossRef]
- Veenstra-VanderWeele, J.; Muller, C.L.; Iwamoto, H.; Sauer, J.E.; Owens, W.A.; Shah, C.R.; Cohen, J.; Mannangatti, P.; Jessen, T.; J. Thompson, B.J.; et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc. Natl. Acad. Sci. USA 2012, 109, 5469–5474. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; BaHammam, A.S.; Brown, G.M.; Spence, D.W.; Bharti, V.K.; Kaur, C.; Hardeland, R.; Cardinali, D.P. Melatonin antioxidative defense: Therapeutical implications for aging and neurodegenerative processes. Neurotox. Res. 2013, 23, 267–300. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Bentez-King, G.A.; Rosales-Corral, S.A.; Pacheco-Moiss, F.P.; Velzquez-Brizuela, I.E. Cellular and biochemical actions of melatonin which protect against free radicals: Role in neurodegenerative disorders. Curr. Neuropharmacol. 2008, 6, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.N.; Jamieson, S.; Graham, A.J.; Shneerson, J.M. REM sleep behaviour disorder treated with melatonin in a patient with Alzheimers disease. Clin. Neurol. Neurosurg. 2008, 110, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Asayama, K.; Yamadera, H.; Ito, T.; Suzuki, H.; Kudo, Y.; Endo, S. Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. J. Nippon Med. Sch. 2003, 70, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Antolin, I.; Mayo, J.C.; Sainz, R.M.; del Brio, M.L.; Herrera, F.; Martin, V.; Rodríıguez, M.V. Protective effect of melatonin in a chronic experimental model of Parkinsons disease. Brain Res. 2002, 943, 163–173. [Google Scholar] [CrossRef]
- Borah, A.; Mohanakumar, K.P. Melatonin inhibits 6-hydroxydopamine production in the brain to protect against experimental parkinsonism in rodents. J. Pineal. Res. 2009, 47, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, A.J. The Gut-Brain Axis in Childhood Developmental Disorders. JPGN 2002, 34, S14–S17. [Google Scholar] [CrossRef] [PubMed]
- Basile, A.S.; Jones, E.A. Ammonia and GABA-ergic neurotransmission: Interrelated factors in the pathogenesis of hepatic encephalopathy. Hepatology. 1997, 25, 1303–1305. [Google Scholar] [CrossRef] [PubMed]
- Seiler, N. Ammonia and Alzheimer’s disease. Neurochem. Int. 2002, 41, 189–207. [Google Scholar] [CrossRef]
- Caulfield, L.E.; Black, R.E. Zinc deficiency. In Comparative Quantification of Health Risks: Global and Regional Burden of Disease Attributable to Selected Major Risk Factors; Ezzati, M., Lopez, A.D., Rodgers, A.A., Murray, C.J.L., Eds.; World Health Organization: Geneva, Swiss, 2004; Chapter 5. [Google Scholar]
- Famularo, G.; de Simone, C.; Pandey, V.; Sahu, A.R.; Minisola, G. Probiotic lactobacilli: an innovative tool to correct the malabsorption syndrome of vegetarians? Med. Hypotheses 2005, 65, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Watt, N.T.; Whitehouse, I.J.; Hooper, N.M. The role of zinc in Alzheimers disease. Int. J. Alz. Dis. 2011, 2011, 971021. [Google Scholar]
- Yasuda, H.; Yoshida, K.; Yasuda, Y.; Tsutsui, T. Infantile zinc deficiency: Association with autism spectrum disorders. Scientific Reports 2011, 1, 129. [Google Scholar] [CrossRef] [PubMed]
- Akhondzadeh, S.; Mohammadi, M.R.; Khademi, M. Zinc sulfate as an adjunct to methylphenidate for the treatment of attention deficit hyperactivity disorder in children: a double blind and randomised trial. BMC Psychiatr. 2004, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.E.; Bozzolo, H.; Hollway, J.; Cook, A.; DiSilvestro, R.A.; Bozzolo, D.R.; Crowl, L.; Ramadan, Y.; Williams, C. Serum zinc correlates with parent- and teacher-rated inattention in children with attention-deficit/hyperactivity disorder. J. Child. Adolesc. Psychopharmacol. 2005, 15, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1736. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Copper excess, zinc deficiency, and cognition loss in Alzheimer’s disease. Biofactors. 2012, 38, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Potocnik, F.C.; van Rensburg, S.J.; Hon, D.; Emsley, R.A.; Moodie, I.M.; Erasmus, R.T. Oral zinc augmentation with vitamins A and D increases plasma zinc concentration: Implications for burden of disease. Metab. Brain Dis. 2006, 21, 139–147. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [PubMed]
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain s-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J. Neurochem. 1996, 67, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Ejim, L.J.; D’Costa, V.M.; Elowe, N.H.; Concepcíon Loredo-Osti, J.; Malo, D.; Wright, G.D. Cystathionine-Lyase is important for virulence of salmonella enterica serovar typhimurium. Infect. Immun. 2004, 72, 3310–3314. [Google Scholar] [CrossRef] [PubMed]
- Alkhawajah, M.M.; Caminero, A.B.; Freeman, H.J.; Oger, J.J. Multiple sclerosis and inflammatory bowel diseases: What we know and what we would need to know! Mult. Scler. 2013, 19, 259–265. [Google Scholar]
- Westall, F.C. Molecular Mimicry Revisited: Gut Bacteria and Multiple Sclerosis. J. Clin. Microbiol. 2006, 44, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Noonan, C.W.; Kathman, S.J.; White, M.C. Prevalence estimates for MS in the United States and evidence of an increasing trend for women. Neurology 2002, 58, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, A.J.; McTavish, S.F.B.; Cowen, P.J.; Grasby, P.M. Reduction of brain dopamine concentration with dietary tyrosine plus phenylalanine depletion: An [11C] Raclopride PET study. Am. J. Psychiatry 2003, 160, 1887–1889. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.; Cockburn, M.; Bronstein, J.; Zhang, X.; Ritz, B. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am. J. Epidemiol. 2009, 169, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Negga, R.; Rudd, D.A.; Davis, N.S.; Justice, A.N.; Hatfield, H.E.; Valente, A.L.; Fields, A.S.; Fitsanakis, V.A. Exposure to Mn/Zn ethylene-bis-dithiocarbamate and glyphosate pesticides leads to neurodegeneration in Caenorhabditis elegans. Neurotoxicology 2011, 32, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Heafield, M.T.; Fearn, S.; Steventon, G.B.; Waring, R.H.; Williams, A.C.; Sturman, S.G. Plasma cysteine and sulphate levels in patients with Motor neurone, Parkinsons and Alzheimers disease. Neurosci. Lett. 1990, 110, 216–220. [Google Scholar] [CrossRef]
- Carter-Kent, C.; Zein, N.N.; Feldstein, A.E. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: implications for treatment. Am. J. Gastroenterol. 2008, 103, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Peraldi, P.; Hotamisligil, G.S.; Buurman, W.A.; White, M.F.; Spiegelman, B.M. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J. Biol. Chem. 1996, 271, 13018–13022. [Google Scholar] [PubMed]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Uysal, K.T.; Becherer, J.D.; Arner, P.; Hotamisligil, G.S. Altered tumor necrosis factor-alpha (TNF-alpha) processing in adipocytes and increased expression of transmembrane TNF-alpha in obesity. Diabetes 2002, 51, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Langlais, J.; Zollinger, M.; Plante, L.; Chapdelaine, A.; Bleau, G.; Roberts, K.D. Localization of cholesterol sulfate in human spermatozoa in support of a hypothesis for the mechanism of capacitation. Proc. Natl. Acad. Sci. USA 1981, 78, 7266–7270. [Google Scholar] [CrossRef] [PubMed]
- Hidiroglou, M.; Knipfel, J.E. Zinc in mammalian sperm: a review. J. Dairy Sci. 1984, 67, 1147–1156. [Google Scholar] [CrossRef]
- Mose, T.; Kjaerstad, M.B.; Mathiesen, L.; Nielsen, J.B.; Edelfors, S.; Knudsen, L.E. Placental passage of benzoic acid, caffeine, and glyphosate in an ex vivo human perfusion system. J. Toxicol. Environ. Health A 2008, 71, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Davidson, R.M.; Liu, J. Is cholesterol sulfate deficiency a common factor in preeclampsia, autism, and pernicious anemia? Entropy 2012, 14, 2265–2290. [Google Scholar] [CrossRef]
- Robin, M.-M. Argentina: The Soybeans of Hunger. Chapter 13 in The World According to Monsanto. English Translation, Translated from French by George Holoch; The New Press: New York, NY, USA, 2010. [Google Scholar]
- Cerdeira, A.L.; Gazziero, D.L.; Duke, S.O.; Matallo, M.B.; Spadotto, C.A. Review of potential environmental impacts of transgenic glyphosate-resistant soybean in Brazil. J. Environ. Sci. Health B 2007, 42, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.F.; Santos, I.S.; Barros, A.J.D.; Matijasevich, A.; Barros, F.C.; Victora, C.G. Increase in preterm births in Brazil: Review of population-based studies. Rev. Saúde. Pública. 2008, 42, 1–7. [Google Scholar]
- Arbuckle, T.E.; Lin, Z.; Mery, L.S. An exploratory analysis of the effect of pesticide exposure on the risk of spontaneous abortion in an Ontario farm population. Environ. Health Persp. 2001, 109, 851–857. [Google Scholar] [CrossRef]
- Hamilton, B.E.; Martin, J.A.; Ventura, S.J. Births: Preliminary data for 2011. In National Vital Statistics Reports; National Center for Health Statistics: Hyattsville, MD, USA, 2012; Volume 61. [Google Scholar]
- Clair, E.; Mesnage, R.; Travert, C.; Séralini, G.E. A glyphosate-based herbicide induces necrosis and apoptosis in mature rat testicular cells in vitro, and testosterone decrease at lower levels. Toxicol. In Vitro 2012, 26, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.P.; McCormick, C.; Martin, C.; Stocco, D.M. Roundup inhibits steroidogenesis by disrupting steroidogenic acute regulatory (StAR) protein expression. Environ. Health Persp. 2000, 108, 769–776. [Google Scholar] [CrossRef]
- Motoyama, N.; Naka, K. DNA damage tumor suppressor genes and genomic instability. Curr. Opin. Genet. Dev. 2004, 14, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Marc, J.; Mulner-Lorillon, O.; Boulben, S.; Hureau, D.; Durand, G.; Bellé, R. Pesticide roundup provokes cell division dysfunction at the level of CDK1/cyclin B activation. Chem. Res. Toxicol. 2002, 15, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Marc, J.; Bellé, R.; Morales, J.; Cormier, P.; Mulner-Lorillon, O. Formulated glyphosate activates the DNA-response checkpoint of the cell cycle leading to the prevention of G2/M transition. Toxicol. Sci. 2004, 82, 436–442. [Google Scholar] [CrossRef] [PubMed]
- de Roos, A.J.; Blair, A.; Rusiecki, J.A.; Hoppin, J.A.; Svec, M.; Dosemeci, M.; Sandler, D.P.; Alavanja, M.C. Cancer incidence among glyphosate-exposed pesticide applicators in the agricultural health study. Environ. Health Persp. 2005, 113, 49–54. [Google Scholar] [CrossRef]
- Walters, D.K.; Wu, X.; Tschumper, R.C.; Arendt, B.K.; Huddleston, P.M.; Henderson, K.J.; Dispenzieri, A.; Jelinek, D.F. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia 2011, 25, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.D.; Mink, P.J.; Adami, H.-O.; Cole, P.; Mandel, J.S.; Oken, M.M.; Trichopoulos, D. Multiple myeloma: A review of the epidemiologic literature. Int. J. Cancer 2007, 120, 4061. [Google Scholar] [CrossRef] [PubMed]
- Troussard, X.; Avet-Loiseau, H.; Macro, M.; Mellerin, M.P.; Malet, M.; Roussel, M.; Sola, B. Cyclin D1 expression in patients with multiple myeloma. Hematol. J. 2000, 1, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Yong, M.; Schwartz, S.M.; Atkinson, C.; Makar, K.W.; Thomas, S.S.; Newton, K.M.; Bowles, E.J.A.; Holt, V.L.; Leisenring, W.M.; Lampe, J.W. Associations between polymorphisms in glucuronidation and sulfation enzymes and mammographic breast density in premenopausal women in the United States. Cancer Epidemiol. Biomarkers Prev. 2010, 537, 537–546. [Google Scholar] [CrossRef] [PubMed]
- McCormack, V.A.; dos Santos Silva, I. Breast density and parenchymal patterns as markers of breast cancer risk: A meta-analysis. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-C.; Tang, B.-K.; Hammond, G.L.; Tritchler, D.; Yaffe, M.; Boyd, N.F. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: A cross-sectional study. Breast Cancer Res. 2004, 6, R352–R365. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, L.M.; White, E.; Chen, Z.; Chlebowski, R.T.; Hays, J.; Kuller, L.; Lopez, A.M.; Manson, J.; Margolis, K.L.; Muti, P.C.; et al. Obesity, body size, and risk of postmenopausal breast cancer: the Women’s Health Initiative (United States). Cancer Cause Control. 2002, 13, 741–751. [Google Scholar]
- Hakkak, R.; Holley, A.W.; MacLeod, S.L.; Simpson, P.M.; Fuchs, G.J.; Jo, C.H.; Kieber-Emmons, T.; Korourian, S. Obesity promotes 7,12-dimethylbenz(a)anthracene-induced mammary tumor development in female zucker rats. Breast Cancer Res. 2005, 7, R627–R633. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P.; Kopelovich, L.; Hudis, C.A.; Dannenberg, A.J. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev. Res. (Phila.) 2011, 4, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P.; Grossmann, M.E. Minireview: Obesity and breast cancer: The estrogen connection. Endocrinology 2009, 150, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Jagoe, R.T.; Goldberg, A.L. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.; Donaldsson, D.; Kiely, T.; Wu, L. Pesticides Industry Sales and Usage: 2006 and 2007 Market Estimates; U.S. Environmental Protection Agency: Washington, DC, USA, 2011. [Google Scholar]
- Johnson, R.J.; Perez-Pozo, S.E.; Sautin, Y.Y.; Manitius, J.; Sanchez-Lozada, L.G.; Feig, D.I.; Shafiu, M.; Segal, M.; Glassock, R.J.; Shimada, M.; Roncal, C.; Nakagawa, T. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocr. Rev. 2009, 30, 96–116. [Google Scholar] [CrossRef] [PubMed]
- Vivancos, P.D.; Driscoll, S.P.; Bulman, C.A.; Ying, L.; Emami, K.; Treumann, A.; Mauve, C.; Noctor, G.; Foyer, C.H. Perturbations of amino acid metabolism associated with glyphosate-dependent inhibition of shikimic acid metabolism affect cellular redox homeostasis and alter the abundance of proteins involved in photosynthesis and photorespiration. Plant Physiol. 2011, 157, 256–268. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.; McBride, W. The transformation of U.S. livestock agriculture: Scale, efficiency, and risks. In Economic Information Bulletin No. (EIB-43); USDA Economic Research Service: Washington, DC, USA, 2009. [Google Scholar]
- European Food Safety Authority. Modification of the existing MRL for glyphosate in lentils. EFSA J. 2012, 10, 2550–2575. [Google Scholar]
- Seneff, S.; Lauritzen, A.; Davidson, R.M.; Lentz-Marino, L. Is encephalopathy a mechanism to renew sulfate in autism? Entropy 2013, 15, 372–406. [Google Scholar] [CrossRef]
- Dietert, R.R.; Dietert, J.M. Early-life immune insult and developmental immunotoxicity (DIT)-associated diseases: Potential of herbal- and fungal-derived medicinals. Curr. Med. Chem. 2007, 14, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R. Role of developmental immunotoxicity and immune dysfunction in chronic disease and cancer. Reprod. Toxicol. 2011, 31, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Leifer, C.A.; Dietert, R.R. Early life environment and developmental immunotoxicity in inflammatory dysfunction and disease. Toxicol. Environ. Chem. 2011, 93, 1463–1485. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Liu, J.; Davidson, R. Empirical data confirm autism symptoms related to aluminum and acetaminophen exposure. Entropy 2012, 14, 2227–2253. [Google Scholar] [CrossRef]
- Chen, M.X.; Cao, Z.Y.; Jiang, Y.; Zhu, Z.W. Direct determination of glyphosate and its major metabolite, aminomethylphosphonic acid, in fruits and vegetables by mixed-mode hydrophilic interaction/weak anion-exchange liquid chromatography coupled with electrospray tandem mass spectrometry. J. Chromatogr. A. 2013, 1272, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Arul, S.A.; Sreenivasa, M.A.; Manonmani, H.K. Enzyme-linked immunoassay for the detection of glyphosate in food samples using avian antibodies. Food Agri. Immunol. 2011, 22, 217–228. [Google Scholar]
- Sun, Y.; Wang, C.; Wen, Q.; Wang, G.; Wang, H.; Qu, Q.; Hu, X. Determination of glyphosate and aminomethylphosphonic acid in water by LC using a new labeling reagent, 4-methoxybenzenesulfonyl fluoride. Chromatographia. 2010, 72, 679–686. [Google Scholar] [CrossRef]
- Sullivan, T.P.; Sullivan, D.S. The effects of glyphosate herbicide on food preference and consumption in black-tailed deer. Can. J. Zool. 1979, 57, 1406–1412. [Google Scholar] [CrossRef]
- Pesticide residues in food. In FAO/WHO. Evaluations Part I: Residues, 1st ed.; Volume 78, In Proceeedings of the Joint Meeting of the FAO Panel of Experts Residues in Food and the Environment and the WHO Expert Group on Pesticide Residues; Rome, Italy, 29 September–8 October, 1986, Food and Agriculture Organization of the United Nations: Rome, Italy, 1986; FAO Plant Production and Protection Paper.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Samsel, A.; Seneff, S. Glyphosate’s Suppression of Cytochrome P450 Enzymes and Amino Acid Biosynthesis by the Gut Microbiome: Pathways to Modern Diseases. Entropy 2013, 15, 1416-1463. https://doi.org/10.3390/e15041416
Samsel A, Seneff S. Glyphosate’s Suppression of Cytochrome P450 Enzymes and Amino Acid Biosynthesis by the Gut Microbiome: Pathways to Modern Diseases. Entropy. 2013; 15(4):1416-1463. https://doi.org/10.3390/e15041416
Chicago/Turabian StyleSamsel, Anthony, and Stephanie Seneff. 2013. "Glyphosate’s Suppression of Cytochrome P450 Enzymes and Amino Acid Biosynthesis by the Gut Microbiome: Pathways to Modern Diseases" Entropy 15, no. 4: 1416-1463. https://doi.org/10.3390/e15041416
APA StyleSamsel, A., & Seneff, S. (2013). Glyphosate’s Suppression of Cytochrome P450 Enzymes and Amino Acid Biosynthesis by the Gut Microbiome: Pathways to Modern Diseases. Entropy, 15(4), 1416-1463. https://doi.org/10.3390/e15041416