Modeling Structures and Motions of Loops in Protein Molecules


Abstract
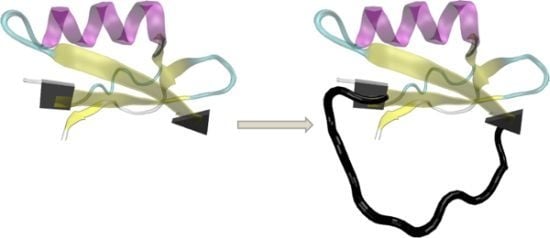
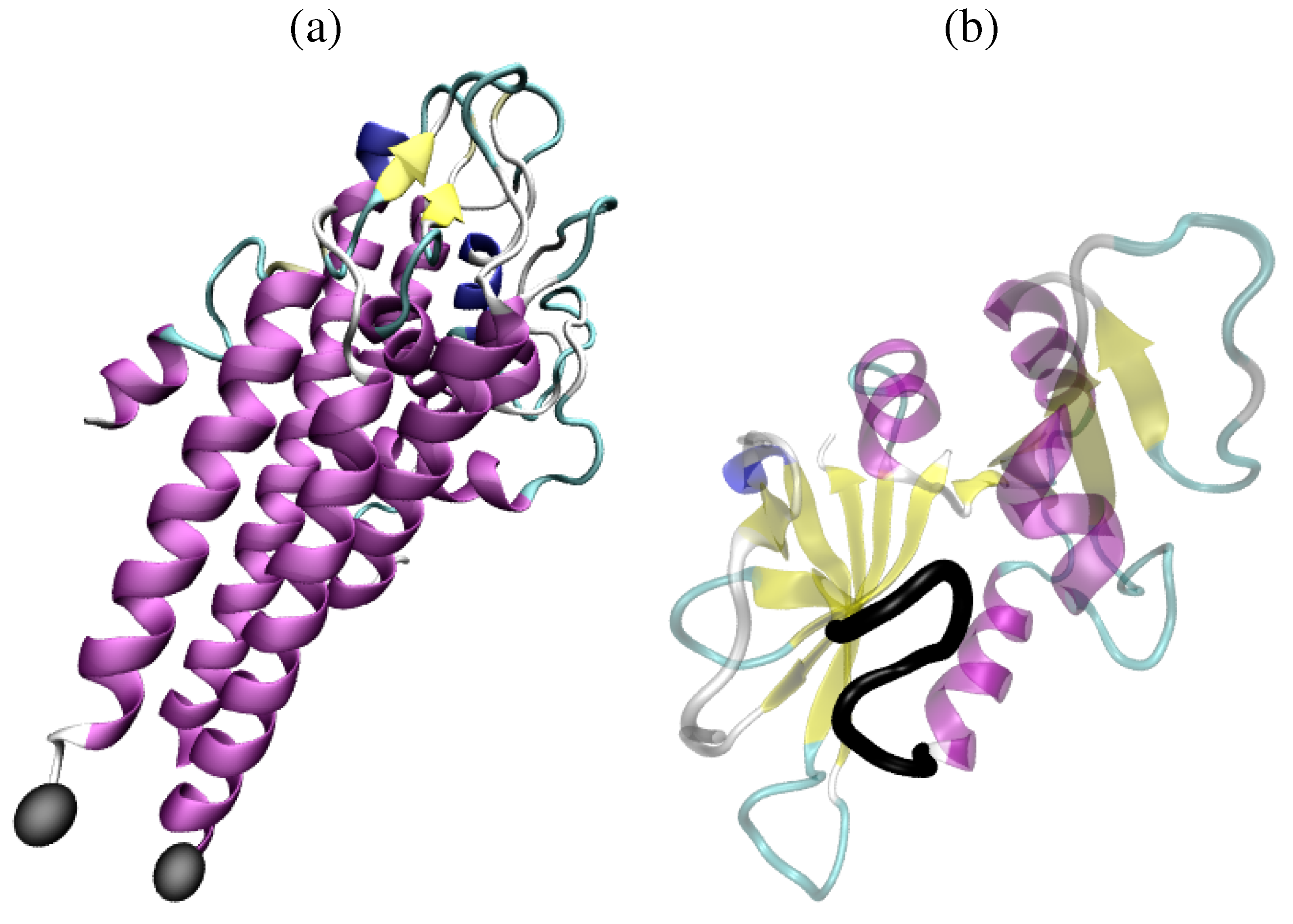
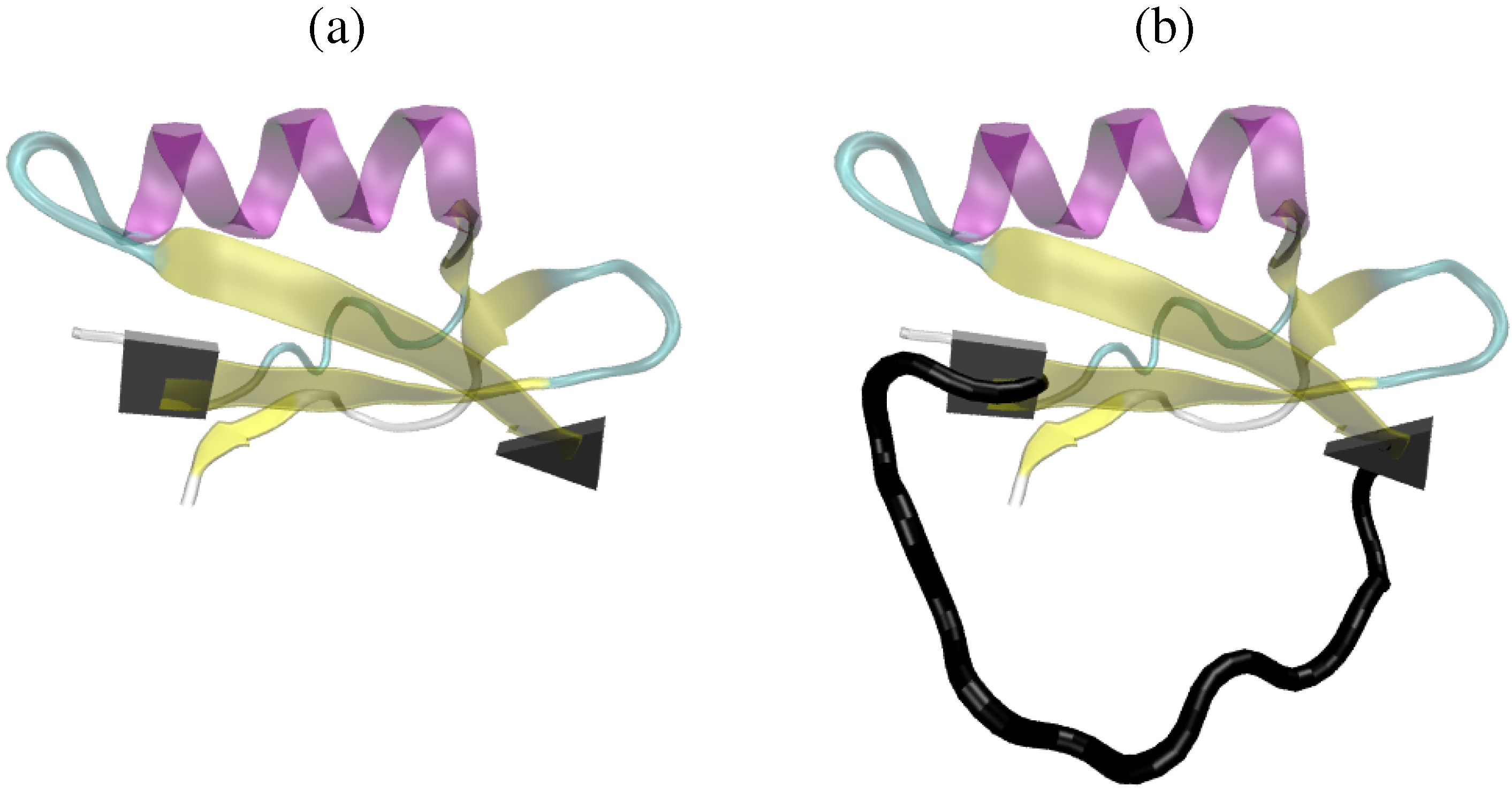
:
{kind=link}
{kind=link}
{kind=link}
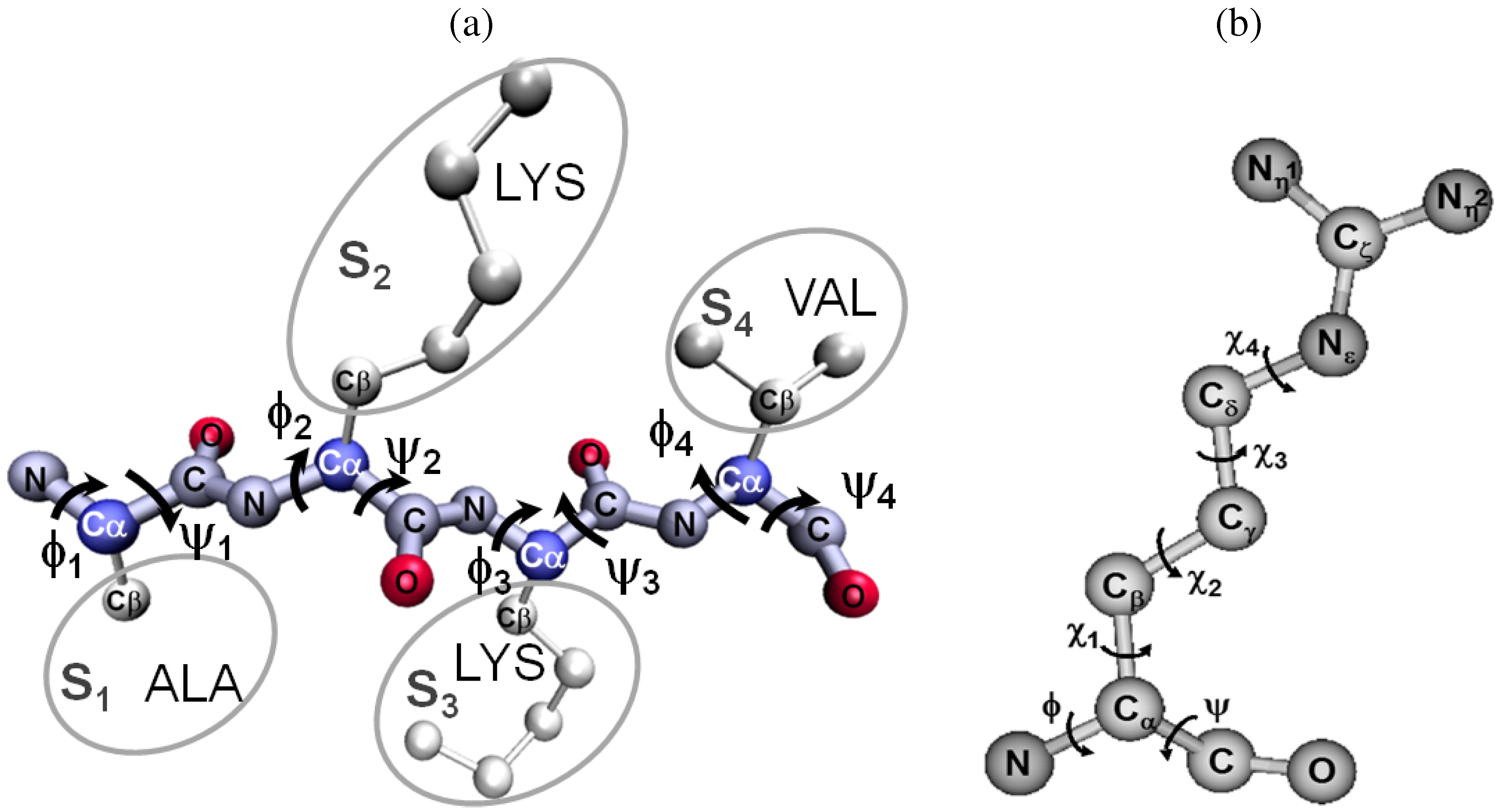{kind=link}
{kind=link}
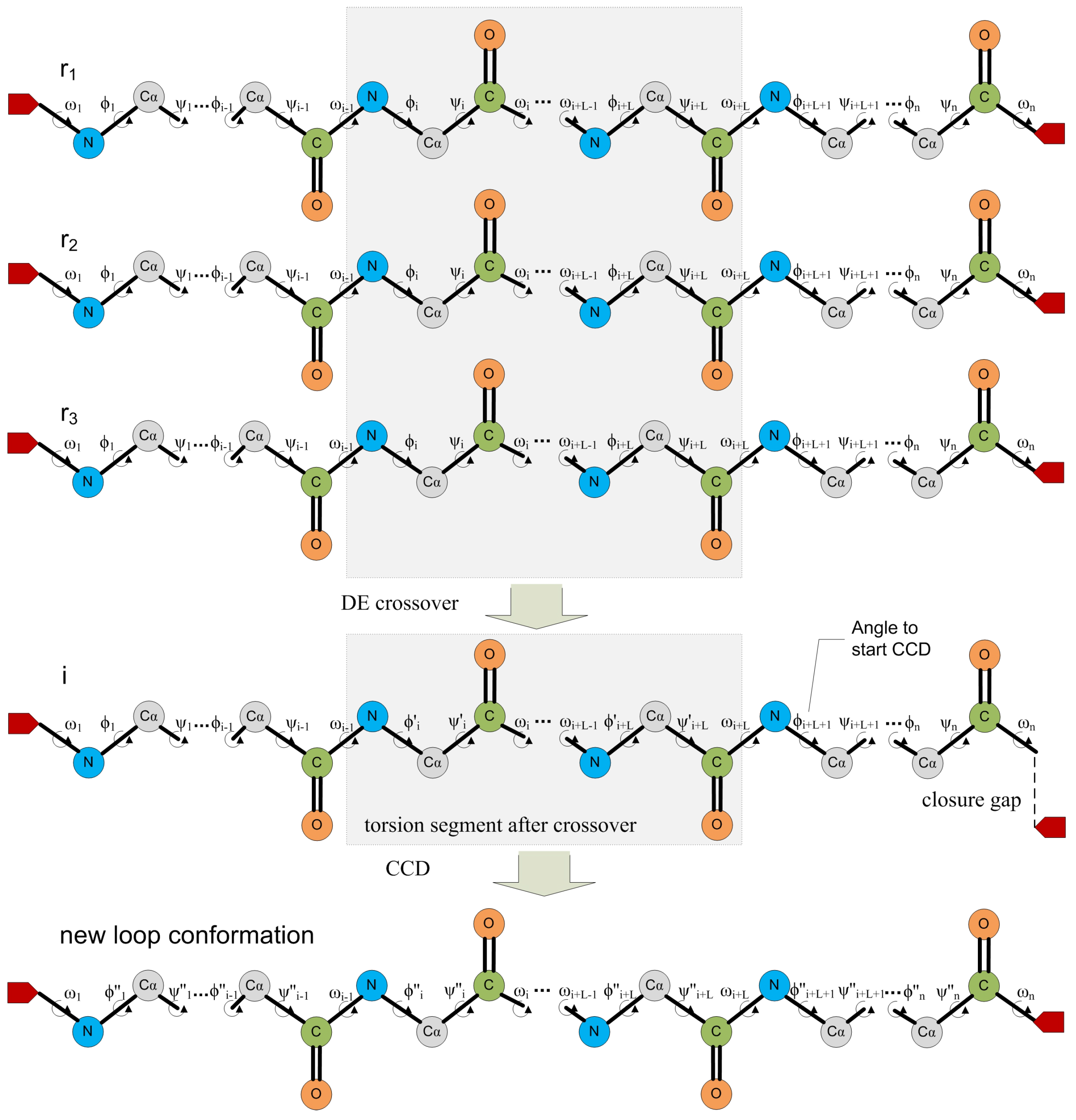{kind=link}
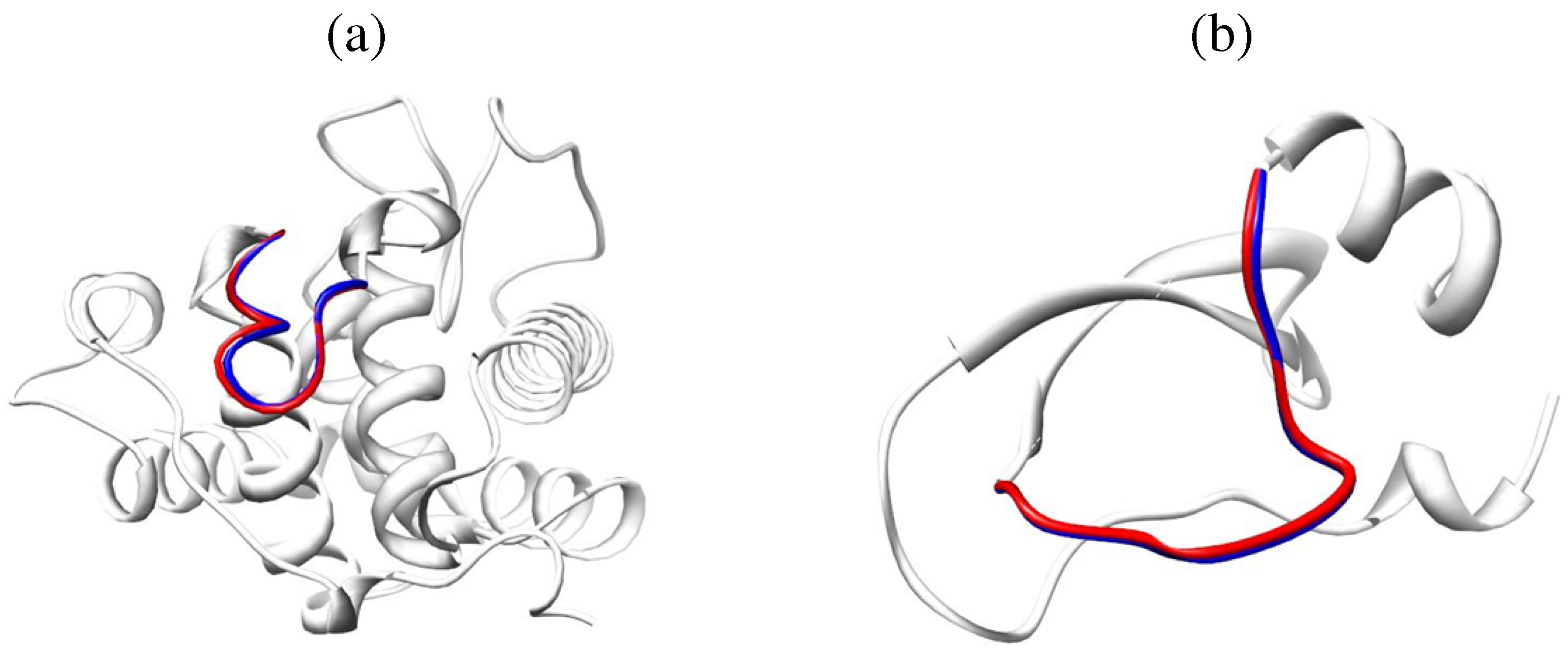{kind=link}
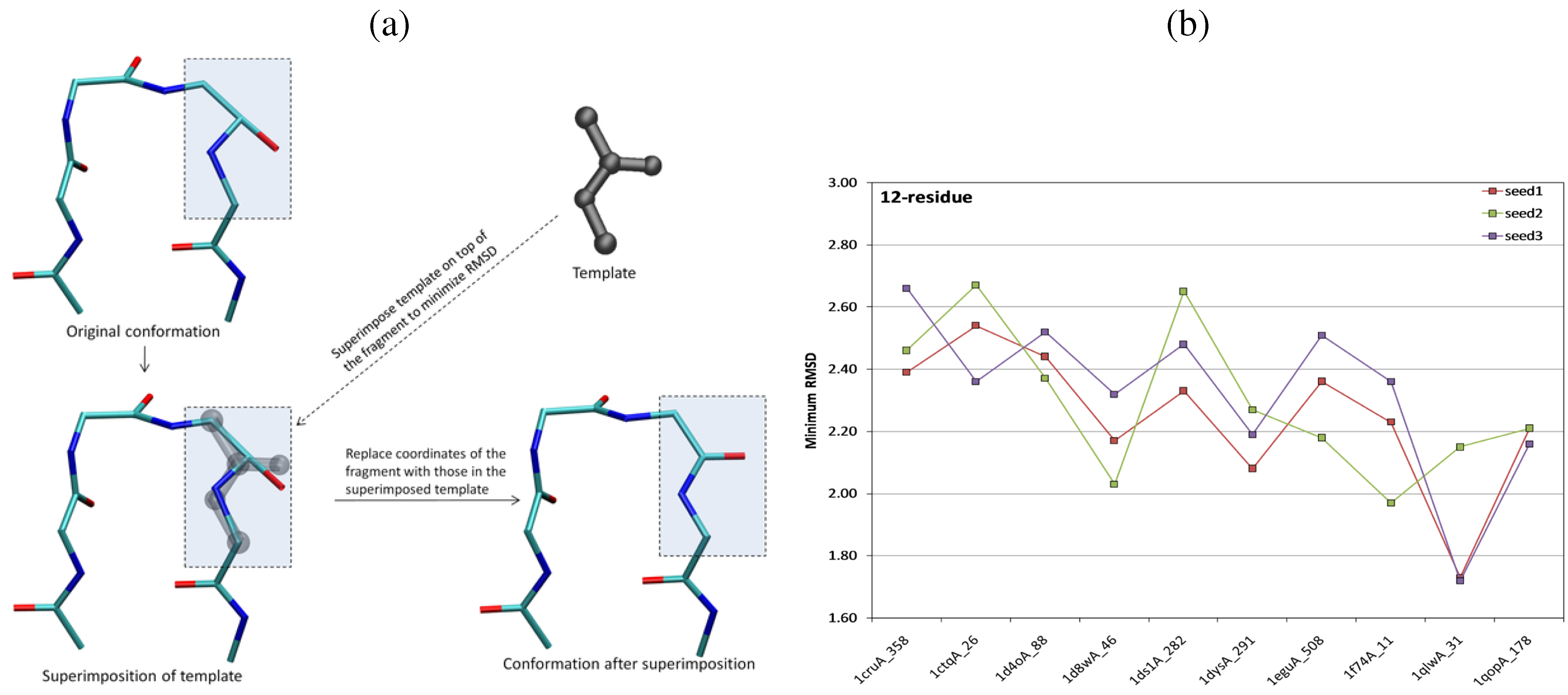{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
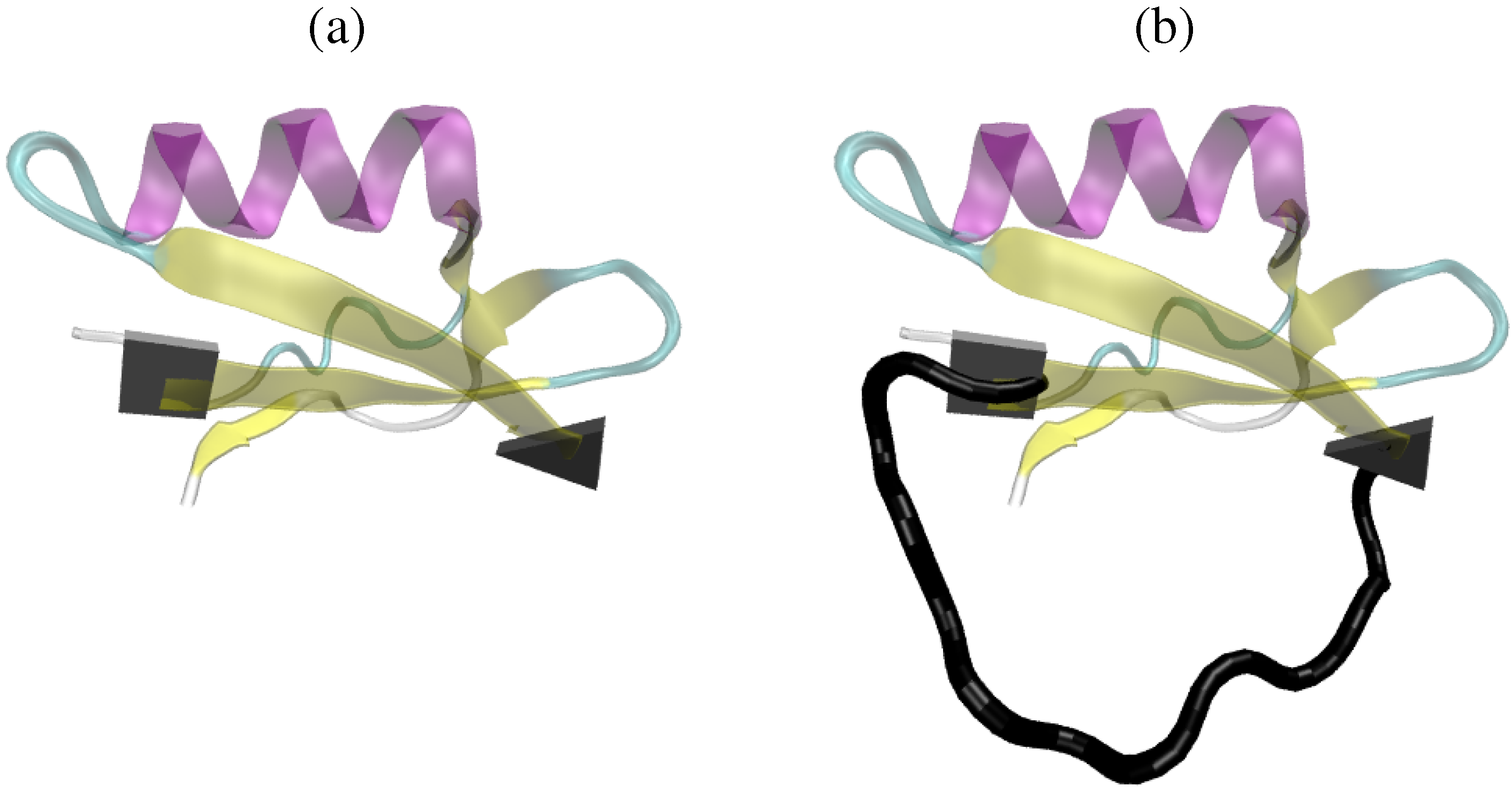
2. Short Primer on Protein Modeling
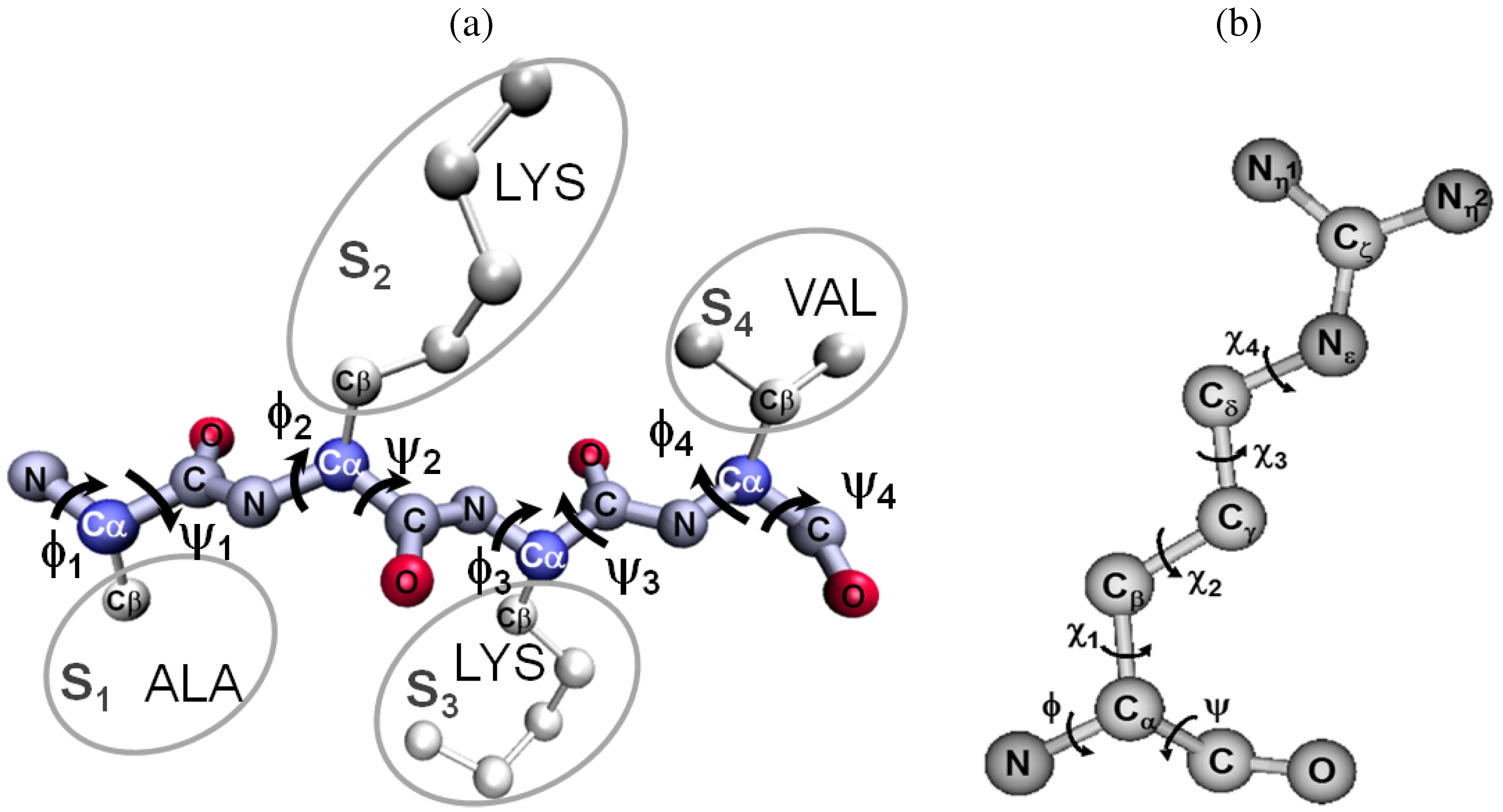
3. Inverse Kinematics Methods
3.1. Exact IK Techniques
3.2. Optimization-Based IK Techniques
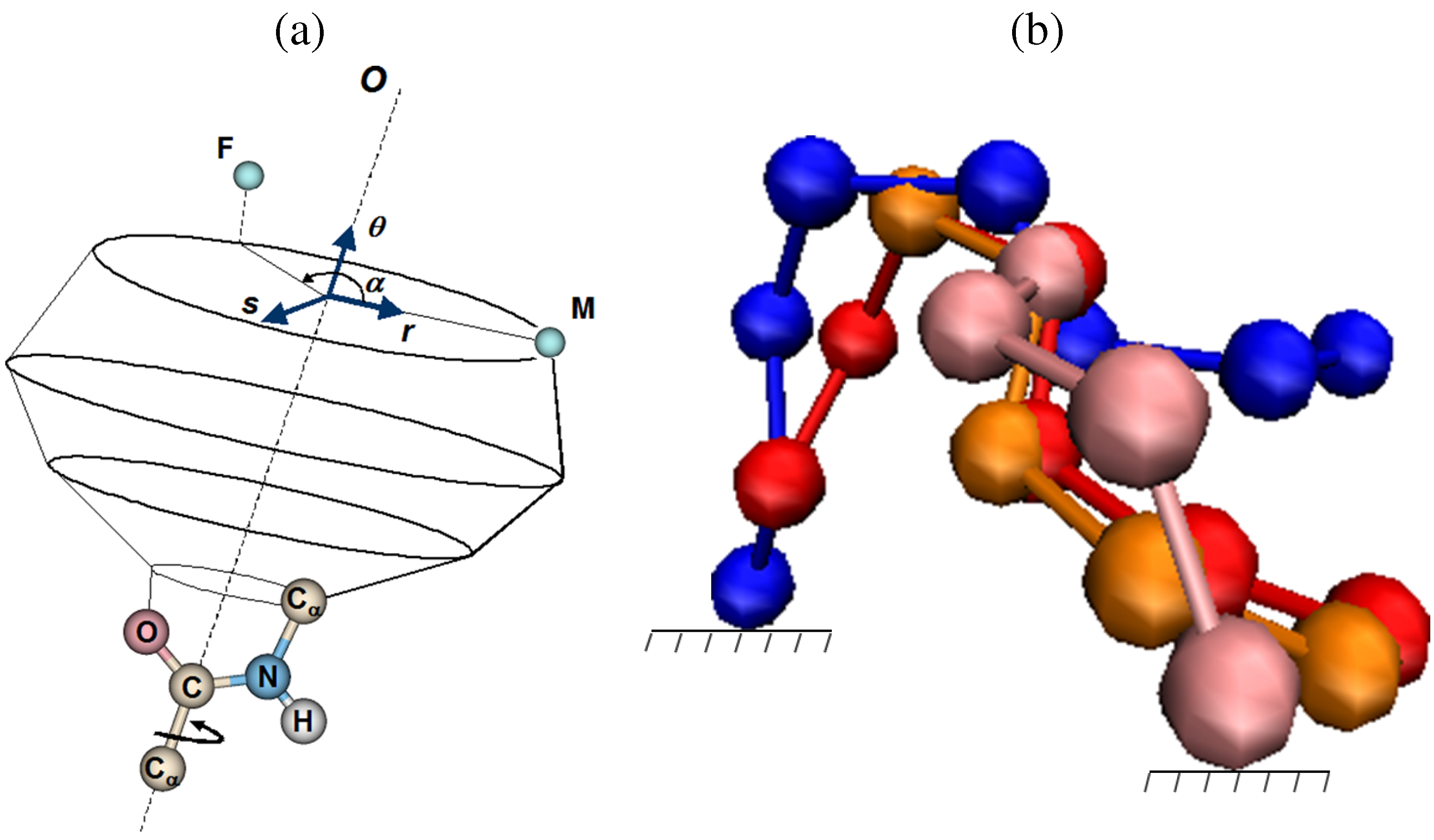
3.3. Remaining Challenges Related to IK Techniques
4. Database Methods
5. Search-Based Methods
5.1. Generate-and-Close Methods
5.1.1. Energy-Based Approaches
5.1.2. Geometry-Based Approaches
5.2. Close-and-Relax Methods
5.3. Accuracy and Time Demands
6. Highlights of Selected Representative Methods
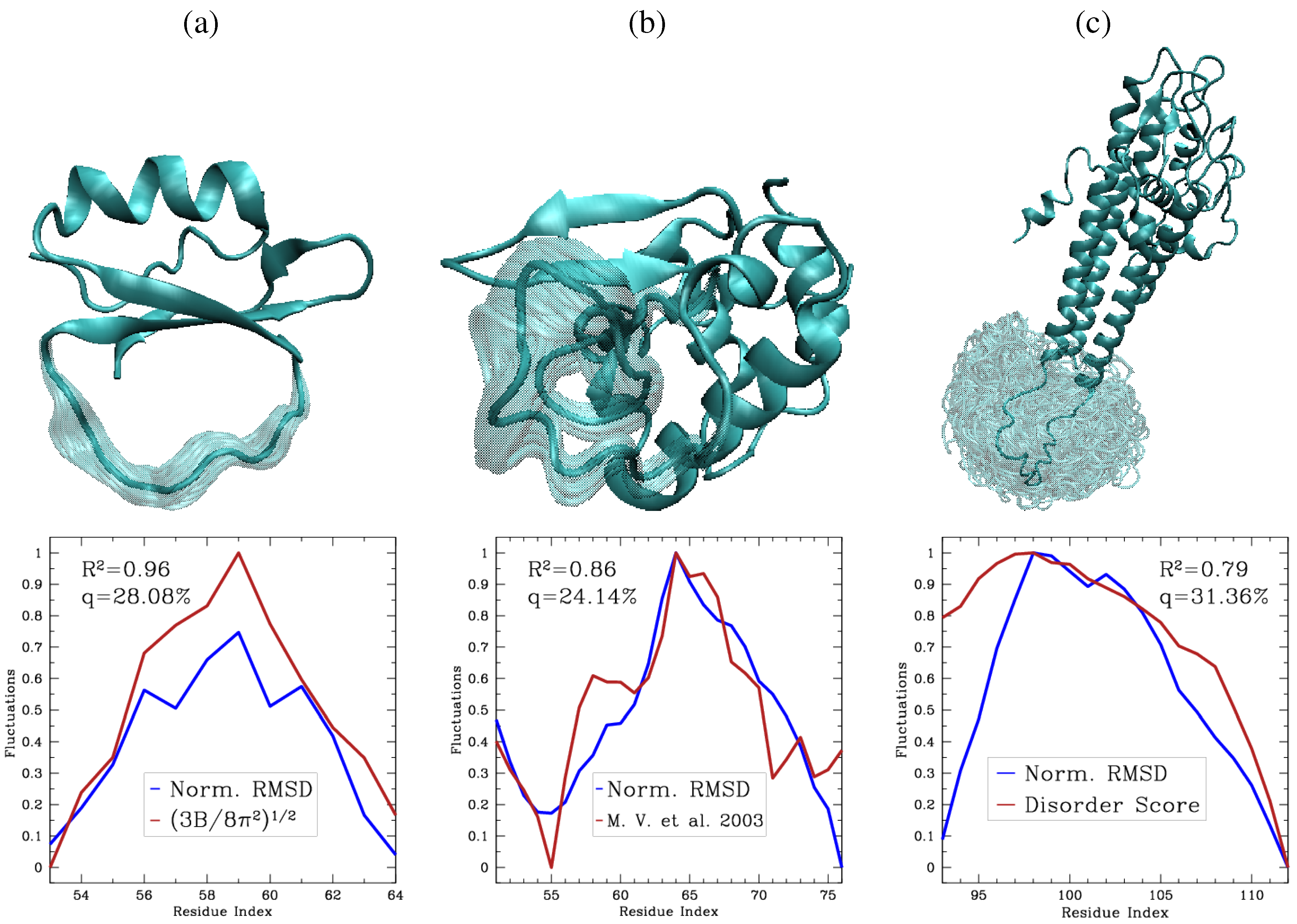
6.1. Pareto Optimal Sampling Method
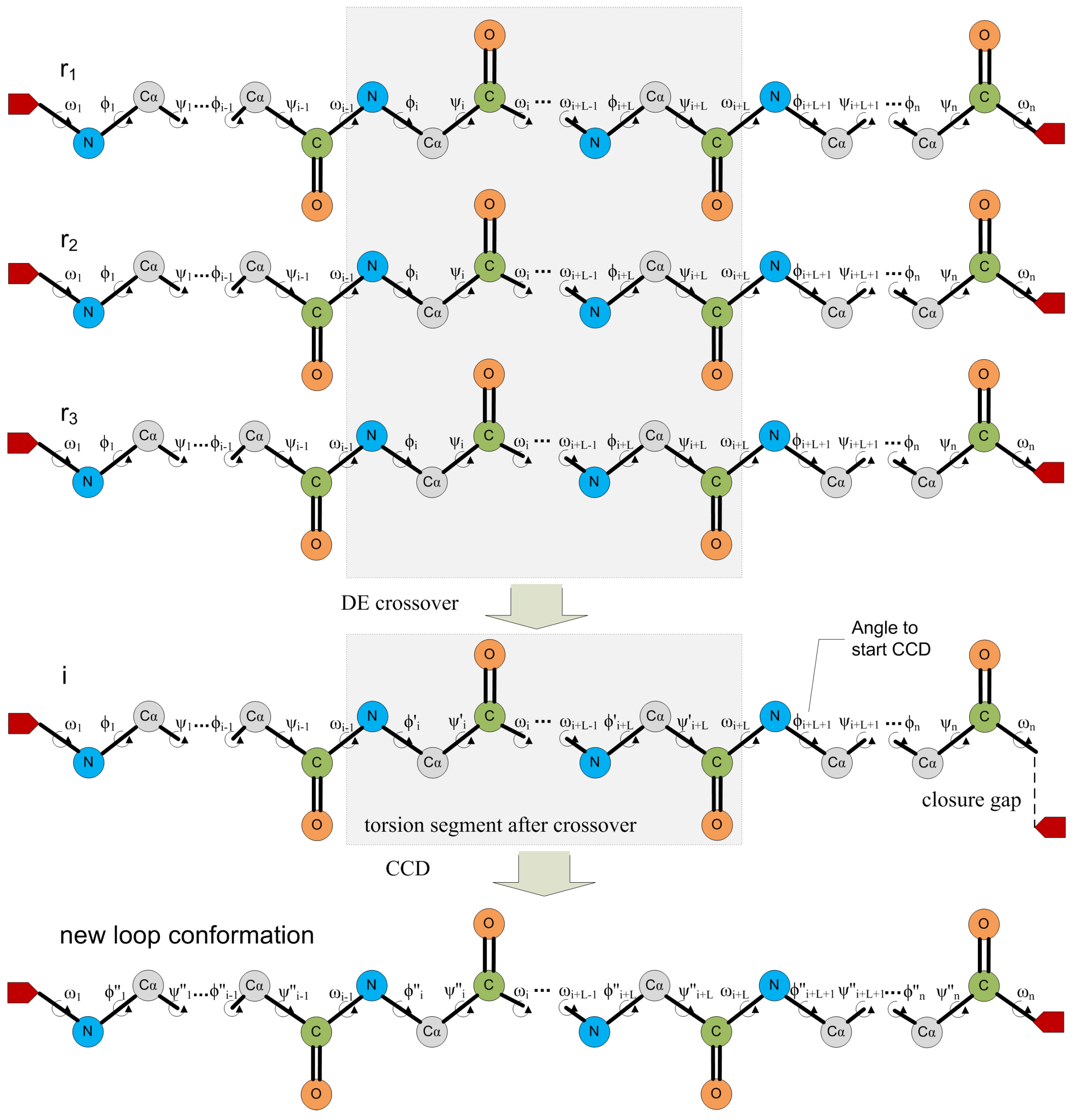
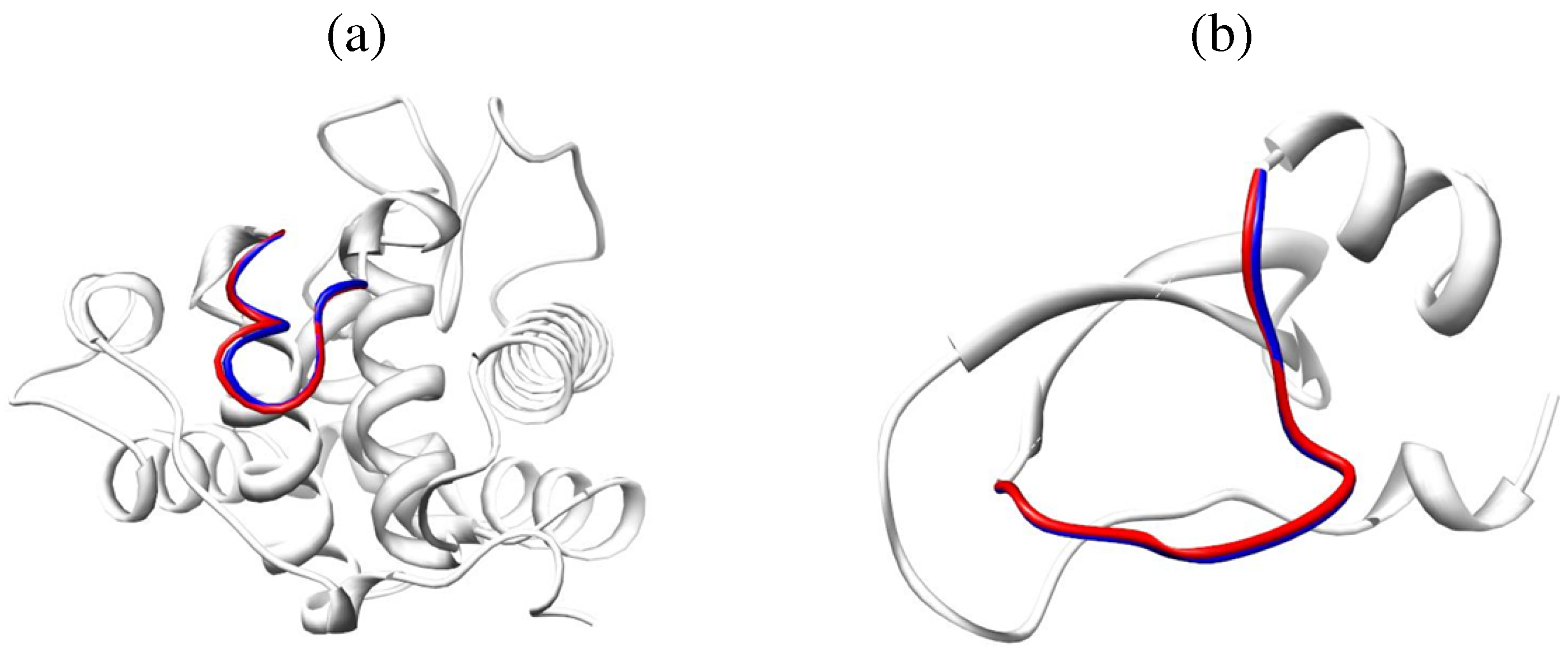
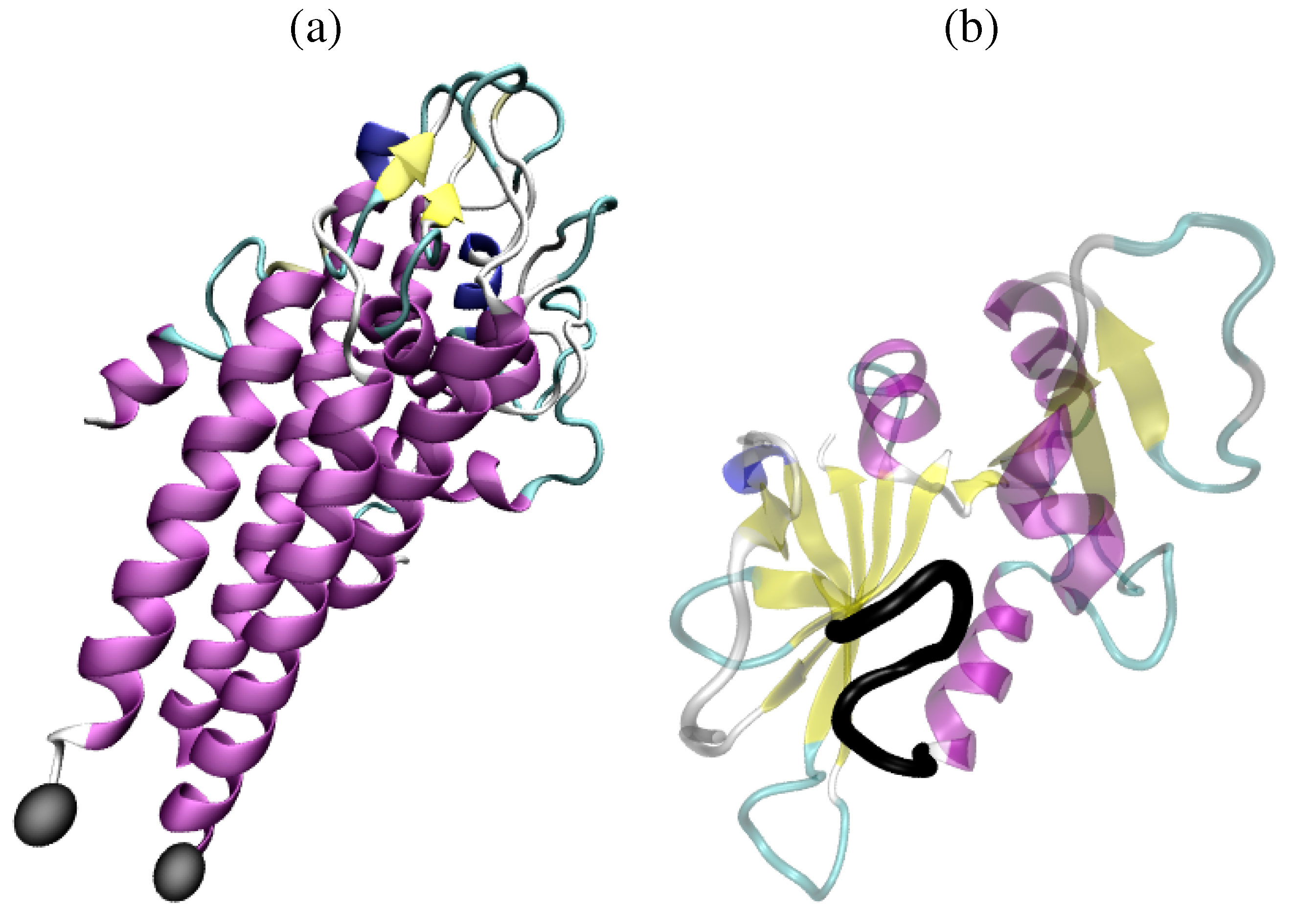
6.2. Self-Organizing Method

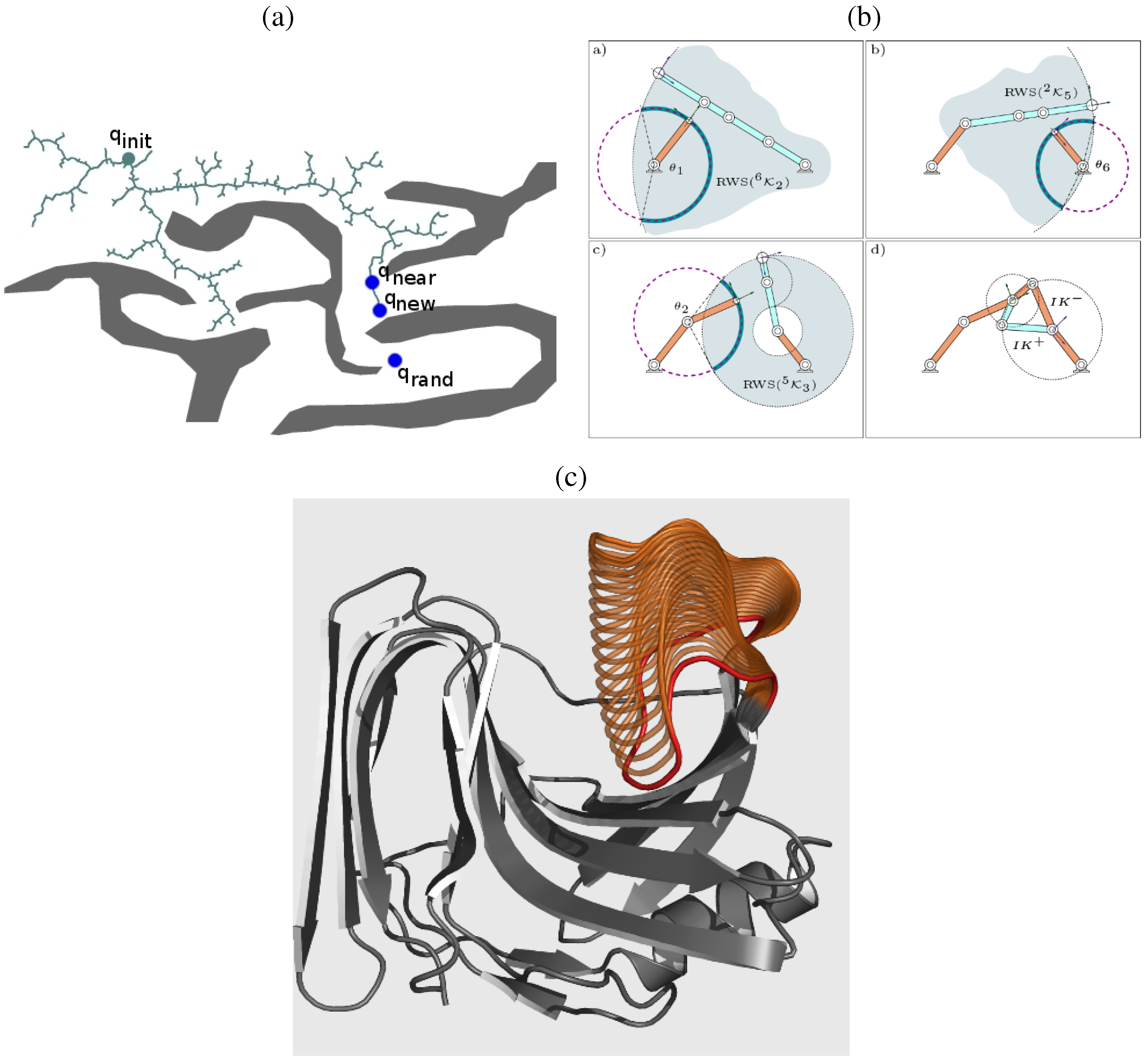
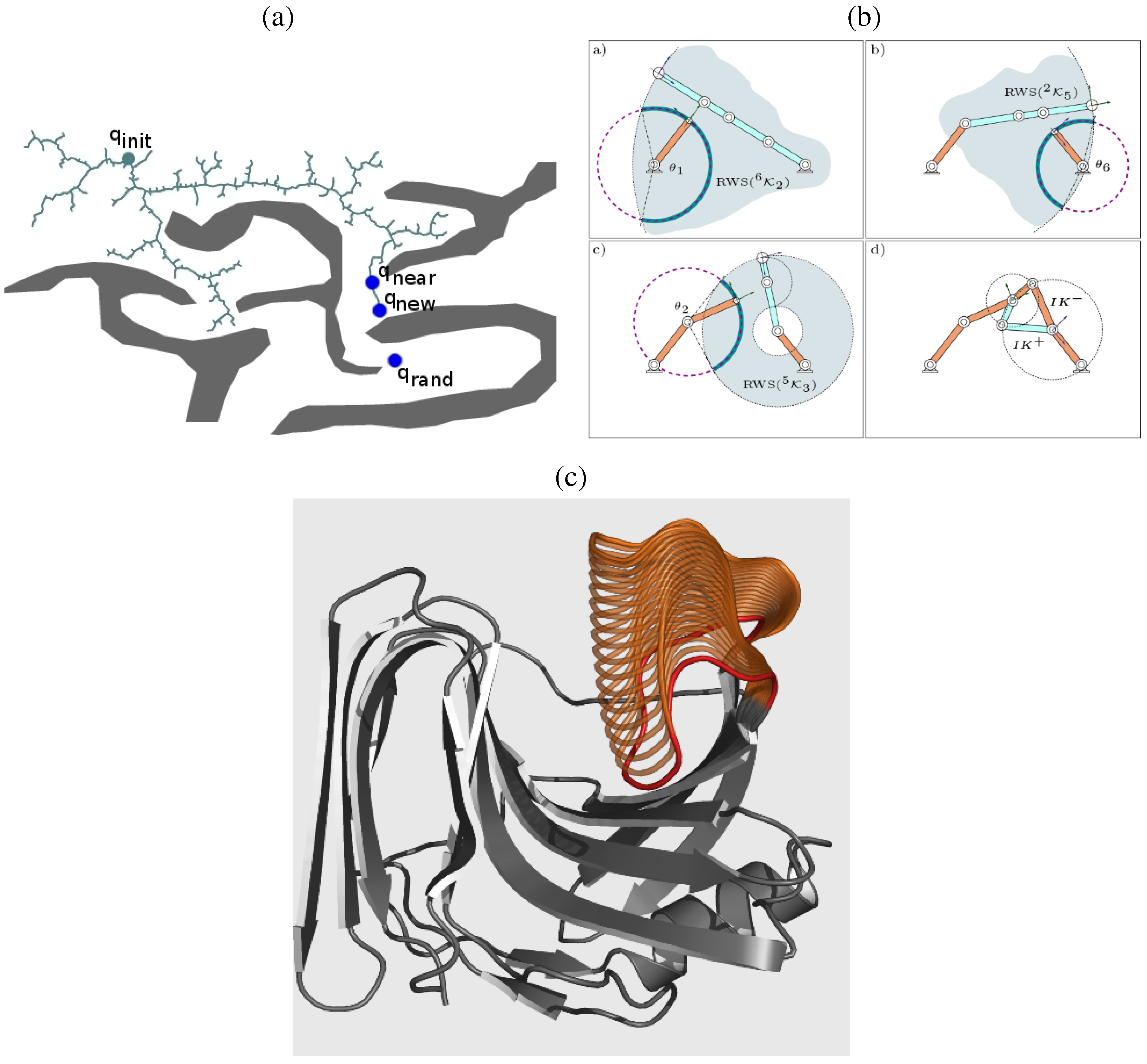
6.3. A Robotics-Inspired Method for Sampling the Loop Closure Space
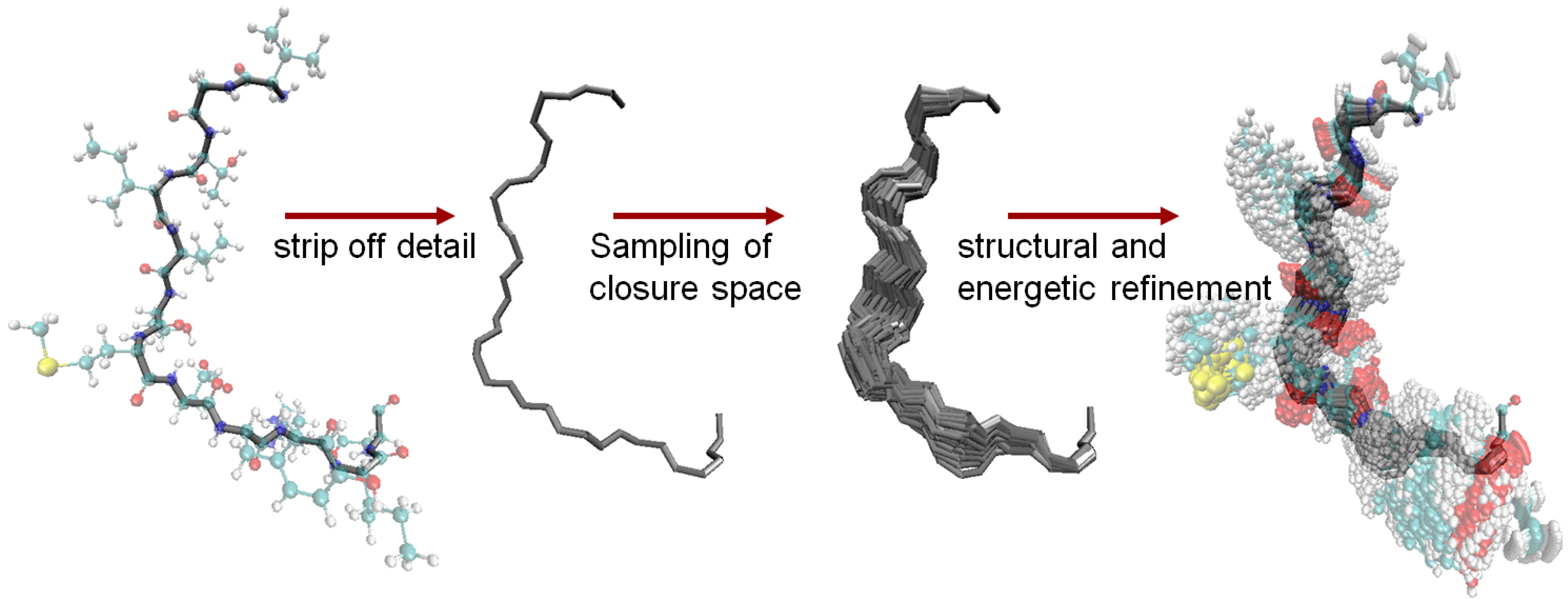
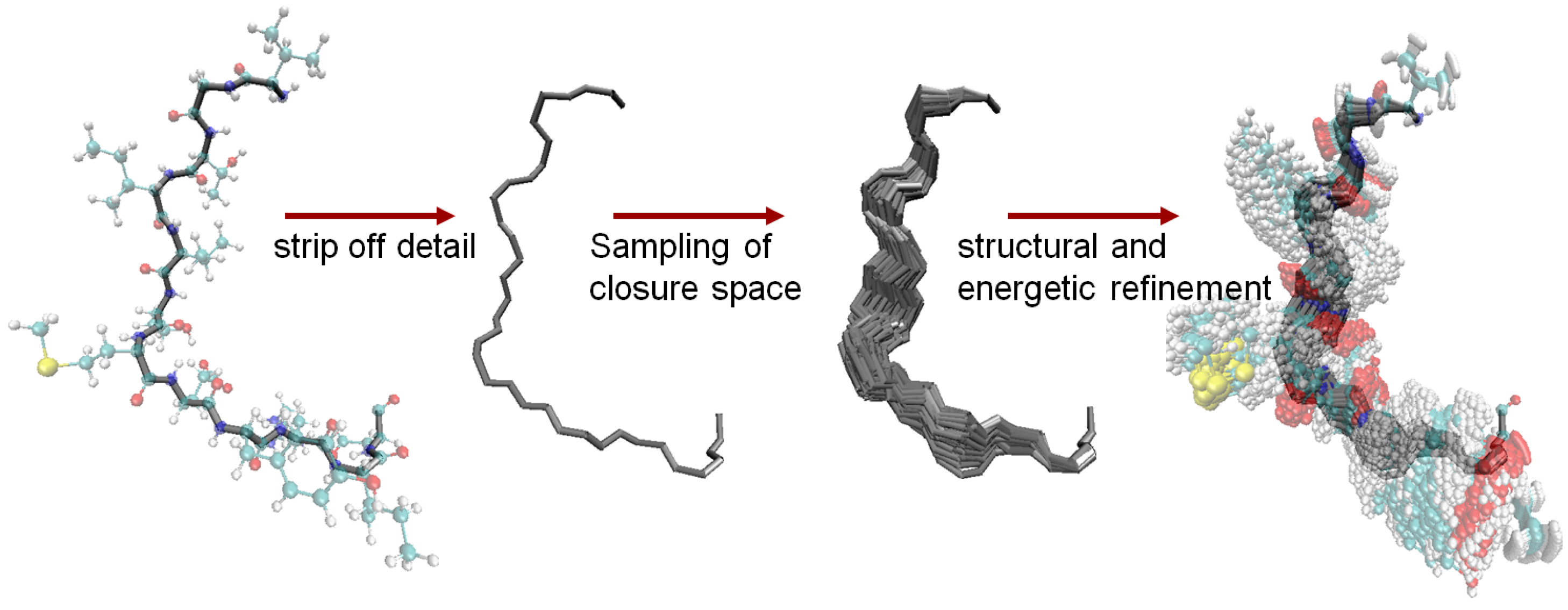
6.4. The Fragment Ensemble Method
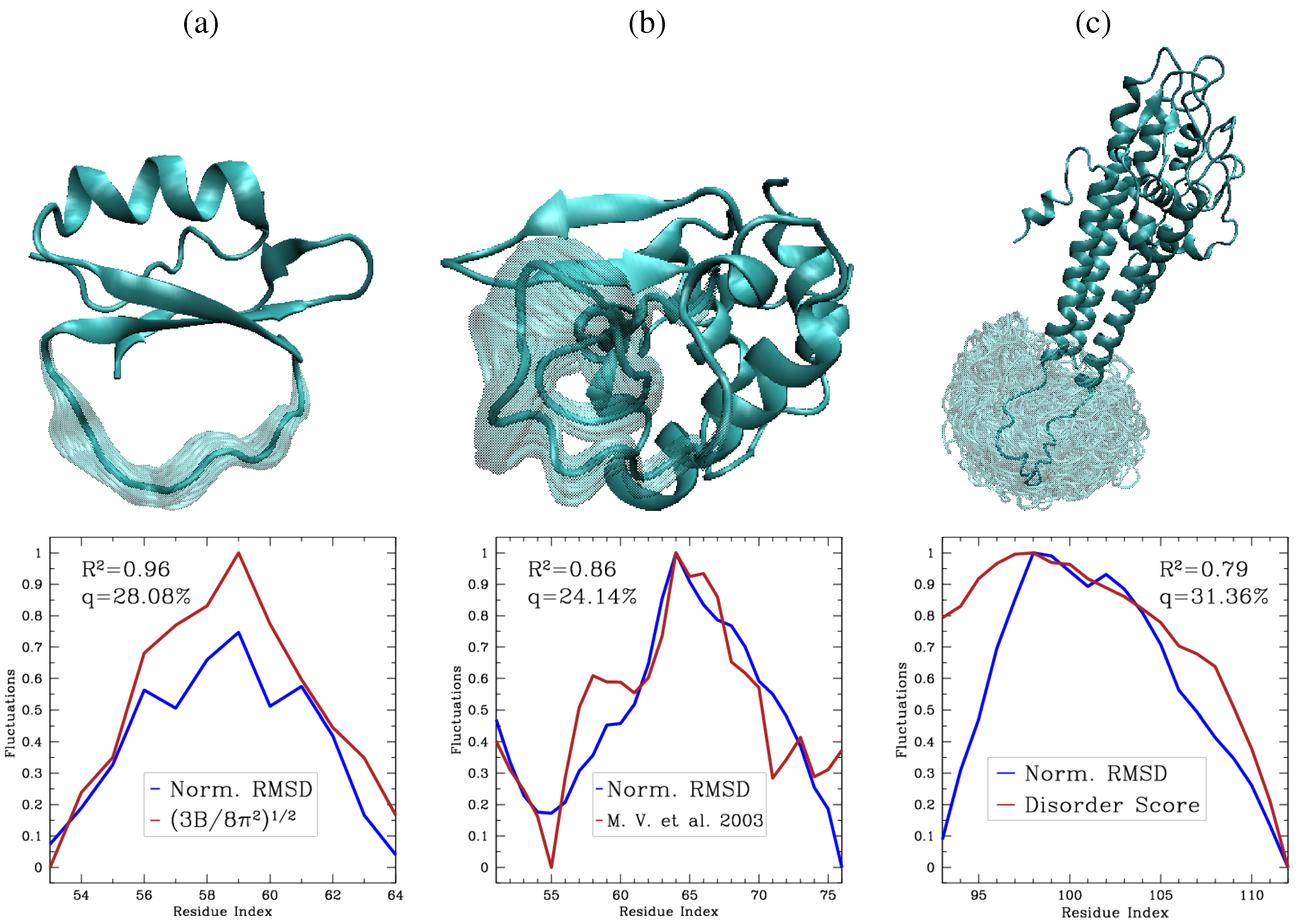
7. Conclusions
Acknowledgements
References
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Kuriyan, J. Molecular Dynamics and protein function. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 6679–6685. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Koga, N.; Takada, S.; Onuchic, J.N.; Wolynes, P.G. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11844–11849. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; de Groot, B.L. Detection of functional modes in protein dynamics. PLoS Comp. Biol. 2009, 5, e1000480. [Google Scholar] [CrossRef] [PubMed]
- Jenzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 6 February 2012). [Google Scholar] [CrossRef]
- Decanniere, K.; Muyldermans, S.; Wyns, L. Canonical antigen-binding loop structures in immunoglobulins: more structures, more canonical classes? J. Mol. Biol. 2000, 300, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Likitvivatanavong, S.; Aimanova, K.G.; Gill, S.S. Loop residues of the receptor binding domain of Bacillus thuringiensis Cry11Ba toxin are important for mosquitocidal activity. FEBS Lett. 2007, 583, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Lepsik, M.; Field, M.J. Binding of calcium and other metal ions to the EF-hand loops of calmodulin studied by quantum chemical calculations and molecular dynamics simulations. J. Phys. Chem. 2007, 111, 10012–10022. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.M.; Marintcheva, B.; Cook, T.; Lee, S.J.; Tabor, S.; Richardson, C.C. A unique loop in T7 DNA polymerase mediates the binding of helicase-primase, DNA binding protein, and processivity factor. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 5096–5101. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; de Veer, S.J.; Sit, K.C.; Reboul, C.F.; Buckle, A.M.; Harris, J.M. Mastering the canonical loop of serine protease inhibitors: Enhancing potency by optimising the internal hydrogen bond network. PLoS One 2011, 6, e19302. [Google Scholar] [CrossRef] [PubMed]
- Thanki, N.; Zeelen, J.P.; Mathieu, M.; Laenicke, R.; Abagyan, R.A.; Wierenga, R.K.; Schliebs, W. Protein engineering with monomeric triosephosphate isomerase (monoTIM): The modelling and structure verification of a seven residue loop. Protein Eng. 1997, 10, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliand, G.; Bhat, T.N.; Weissig, H.; N., S.I.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Eicken, C.; Sharma, V.; Klabunde, T.; Lawrenz, M.B.; Hardham, J.M.; Norris, S.J.; Sacchettini, J.C. Crystal structure of Lyme disease variable surface antigen VlsE of Borrelia Burgdorferi. J. Biol. Chem. 2002, 277, 21691–21696. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomolec. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.J.; Rader, A.J.; Kuhn, L.A.; Thorpe, M.F. Protein flexibility predictions using graph theory. Protein. Struct. Funct. Bioinf. 2001, 44, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Mamonova, T.; Hespenheide, B.; Straub, R.; Thorpe, M.F.; Kurnikova, M. Protein flexibility using constraints from molecular dynamics simulations. J. Phys. Biol. 2005, 2, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.; Jagodzinski, F.; Li, Y.; Streinu, I. KINARI-Web: A server for protein rigidity analysis. Nucleic Acids Res. 2011, 39, W177–W183. [Google Scholar] [CrossRef] [PubMed]
- Dunbrack Jr., R.L. Comparative modeling of CASP3 targets using PSI-BLAST and SCWRL. Protein. Struct. Funct. Bioinf. 1999, 37, 81–87. [Google Scholar] [CrossRef]
- Bradley, P.; Misura, K.M.S.; Baker, D. Toward high-resolution de novo structure prediction for dmall proteins. Science 2005, 309, 1868–1871. [Google Scholar] [CrossRef] [PubMed]
- Craig, J. Introduction to Robotics: Mechanics and Control, 2nd ed.; Addison-Wesley: Boston, MA, USA, 1989; p. 450. [Google Scholar]
- Zhang, M.; Kavraki, L.E. A new method for fast and accurate derivation of molecular conformations. Chem. Inf. Comput. Sci. 2002, 42, 64–70. [Google Scholar] [CrossRef]
- Zhang, M.; Kavraki, L.E. Finding solutions of the inverse kinematics problem in computer-aided drug design. In Currents in Computational Molecular Biology; Florea, L., Walenz, B., Hannenhalli, S., Eds.; ACM Press: Washington, DC, USA, 2002; Number TR02-385; pp. 214–215. [Google Scholar]
- Engh, R.A.; Huber, R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. 1991, A47, 392–400. [Google Scholar] [CrossRef]
- Abayagan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Manocha, D.; Zhu, Y. Kinematic manipulation of molecular chains subject to rigid constraints. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 285–293. [Google Scholar] [PubMed]
- Singh, A.P.; Latombe, J.C.; Brutlag, D.L. A motion planning approach to flexible ligand binding. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 7, 252–261. [Google Scholar]
- Apaydin, M.S.; Singh, A.P.; Brutlag, D.L.; Latombe, J.C. Capturing molecular energy landscapes with probabilistic conformational roadmaps. Proc. IEEE Int. Conf. Robot. Autom. 2001, 1, 932–939. [Google Scholar]
- Amato, N.M.; Dill, K.A.; Song, G. Using motion planning to map protein folding landscapes and analyze folding kinetics of known native structures. J. Comp. Biol. 2002, 10, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Apaydin, M.S.; Brutlag, D.L.; Guestrin, C.; Hsu, D.; Latombe, J.C. Stochastic roadmap simulation: An efficient representation and algorithm for analyzing molecular motion. J. Comp. Biol. 2003, 10, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Amato, N.M. A Motion planning approach to folding: From paper craft to protein folding. IEEE Trans. Robot. Autom. 2004, 20, 60–71. [Google Scholar] [CrossRef]
- Cortes, J.; Simeon, T.; de Angulo, R.; Guieysse, D.; Remaud-Simeon, M.; Tran, V. A path planning approach for computing large-amplitude motions of flexible molecules. Bioinformatics 2005, 21, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Streinu, I.; Brock, O. A methodology for efficiently sampling the conformation space of molecular structures. J. Phys. Biol. 2005, 2, S108–S115. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Jernigan, R.L.; Chirikjian, G.S. Efficient generation of feasible pathways for protein conformationa transitions. Biophys. J. 2002, 83, 1620–1630. [Google Scholar] [CrossRef]
- Georgiev, I.; Donald, B.R. Dead-end elimintation with backbone flexibility. Bioinformatics 2007, 23, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.H.; Apaydin, M.S.; Brutlag, D.L.; Hsu, D.; Latombe, J.C. Using stochastic roadmap simulation to predict experimental quantities in protein folding kinetics: Folding rates and phi-values. J. Comp. Biol. 2007, 14, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, S.; Cortes, J.; Stefaniu, A.; Simeon, T. An NMA-guided path planning approach for computing large-amplitude conformational changes in proteins. Protein. Struct. Funct. Bioinf. 2008, 70, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Dunbrack, R.L., Jr.; Cohen, F.E. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997, 6, 1661–1681. [Google Scholar] [CrossRef] [PubMed]
- Clementi, C. Coarse-grained models of protein folding: Toy-models or predictive tools? Curr. Opinion Struct. Biol. 2008, 18, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Chan, H.S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Socci, N.D.; Onuchic, J.N.; Wolynes, P.G. Protein folding mechanisms and the multidimensional folding funnel. Protein. Struct. Funct. Bioinf. 1998, 32, 136–158. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Wolynes, P.G. Theory of protein folding. Curr. Opinion Struct. Biol. 1997, 14, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, J.N.; Luthey-Schulten, Z.; Wolynes, P.G. Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem. 1997, 48, 545–600. [Google Scholar] [CrossRef] [PubMed]
- Ejtehadi, M.R.; Avall, S.P.; Plotkin, S.S. Three-body interactions improve the prediction of rate and mechanism in protein folding models. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15088–15093. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, P.J.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Protein. Struct. Funct. Bioinf. 2004, 55, 351–367. Available online: http://www.francisco.compchem.ucsf.edu/~jacobson/Software:PLOP (accessed on 6 February 2012). [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. Available online: http://modbase.compbio.ucsf.edu/modloop/ (accessed on 6 February 2012). [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Soto, C.S.; Honig, B. Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7432–7437. Available online: http://wiki.c2b2.columbia.edu/honiglab public/index.php/Software:Loopy (accessed on 6 February 2012). [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Honig, B. Evaluation of the conformational free energies of loops in proteins. Proteins 1994, 18, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kolodny, R.; Guibas, L.; Levitt, M.; Koehl, P. Inverse kinematics in biology: The protein loop closure problem. Int. J. Robot. Res. 2005, 24, 151–163. [Google Scholar] [CrossRef]
- Primrose, E.J.F. On the input-output equation of the general 7R- mechanism. Mech. Mach. Theory 1986, 21, 509–510. [Google Scholar] [CrossRef]
- Manocha, D.; Canny, J. Efficient inverse kinematics for general 6R manipulator. IEEE Trans. Robot. Autom. 1994, 10, 648–657. [Google Scholar] [CrossRef]
- Manocha, D.; Zhu, Y.; Wright, W. Conformational analysis of molecular chains using nano-kinematics. Comput. Appl. Biosci. 1995, 11, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Go, N.; Scheraga, H.J. Ring closure and local conformational deformations of chain molecules. Macromolecules 1970, 3, 178–187. [Google Scholar] [CrossRef]
- Bruccoleri, R.E.; Karplus, M. Chain closure with bond angle variations. Macromolecules 1985, 18, 2676–2773. [Google Scholar] [CrossRef]
- Palmer, K.A.; Scheraga, H.A. Standard-geometry chains fitted to x-ray derived structures: Validation of the rigid-geometry approximation. I. chain closure through a limited search of “loop” conformations. J. Comput. Chem. 1991, 12, 505–526. [Google Scholar] [CrossRef]
- Wedemeyer, W.J.; Scheraga, H.J. Exact analytical loop closure in proteins using polynomial equations. J. Comput. Chem. 1999, 20, 819–844. [Google Scholar] [CrossRef]
- Coutsias, E.A.; Seok, C.; Jacobson, M.; Dill, K. A kinematic view of loop closure. J. Comput. Chem. 2004, 25, 510–528. Available online: http://dillgroup. ucsf.edu/loop closure/ (accessed on 6 February 2012). [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; White, R.A.; Wang, L.; Goldman, R.; Kavraki, L.E.; Hasset, B. Improving conformational searches by geometric screening. Bioinformatics 2005, 21, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Chirikjian, G.S. General methods for computing hyper-redundant manipulator inverse kinematics. In Proceedings of the 1993 IEEE/RSJ International Conference on Intelligent Robots and Systems ’93, IROS ’93, Yokohama, Japan, 26–30 July 1993; Volume 2, pp. 1067–1073.
- Zhang, M.; Kavraki, L.E. Solving molecular inverse kinematics problems for protein folding and drug design. In Currents in Computational Molecular Biology; ACM Press: New York, NY, USA, 2002; pp. 214–215. [Google Scholar]
- Fine, R.M.; Wang, H.J.; Shenkin, P.; Yarmush, D.; Levinthal, C. Predicting antibody hypervariable loop conformations. II: Minimization and molecular dynamics studies of MCPC603 from many randomly generated loop conformations. Proteins 1986, 1, 342–362. [Google Scholar] [CrossRef] [PubMed]
- Shenkin, P.S.; Yarmush, D.L.; Fine, R.; Wang, H.J.; Levinthal, C. Predicting antibody hypervariable loop conformations. I: Ensembles of random conformations for ring-like structures. Biopolymers 1987, 26, 2053–2085. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.T.; Chen, C.C. A combined optimization method for solving the inverse kinematics problem of mechanical manipulators. IEEE Trans. Robot. Autom. 1991, 7, 489–499. [Google Scholar] [CrossRef]
- Ring, C.S.; Kneller, D.G.; Langridge, R.; Cohen, F.E. Taxonomy and conformational analysis of loops in proteins. J. Mol. Biol. 1992, 224, 685–699. [Google Scholar] [CrossRef]
- Canutescu, A.A.; Dunbrack, R.L. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003, 12, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Lotan, I. Algorithms exploiting the chain structure of proteins. Ph.D. Thesis, Stanford University, Stanford, CA, USA, 2004. [Google Scholar]
- Lotan, I.; van den Bedem, H.; Deacon, A.M.; Latombe, J.C. Computing protein structures from electron density maps: The mising loop problem. In Algorithmic Foundations of Robotics VI; Erdman, M., Hsu, D., Overmars, M., van der Stappen, F., Eds.; Springer: Berlin, Germany, 2004; pp. 153–168. [Google Scholar]
- van den Bedem, H.; Lotan, I.; Latombe, J.C.; Deacon, A.M. Real-space protein-model completion: An inverse-kinematics approach. Acta Crystallogr. 2005, D61, 2–13. Avaiable online: https://simtk.org/home/looptk/Software:LoopTK (accessed on 6 February 2012). [Google Scholar] [CrossRef] [PubMed]
- Shehu, A.; Clementi, C.; Kavraki, L.E. Modeling protein conformational ensembles: From missing loops to equilibrium fluctuations. Protein. Struct. Funct. Bioinf. 2006, 65, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A.; Clementi, C.; Kavraki, L.E. Sampling conformation space to model equilibrium fluctuations in proteins. Algorithmica 2007, 48, 303–327. [Google Scholar] [CrossRef]
- Zhu, K.; Pincus, D.L.; Zhao, S.; Friesner, R.A. Long loop prediction using the protein local optimization program. Protein. Struct. Funct. Bioinf. 2006, 65, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Sellers, B.D.; Zhu, K.; Zhao, S.; Friesner, R.A.; Jacobson, M.P. Toward better refinement of comparative models: Predicting loops in inexact environments. Protein. Struct. Funct. Bioinf. 2008, 72, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M.; Prilusky, J.; Manning, N.O. Canonical structures for the hypervariable loops of immunoglobulins. J. Mol. Biol. 1987, 196, 901–917. [Google Scholar] [CrossRef]
- Claessens, M.; Cutsem, E.; Lasters, I.; Wodak, S. Modeling the polypeptide backbone with ‘spare parts’ from known protein structures. Protein Eng. 1989, 4, 335–345. [Google Scholar] [CrossRef]
- Summers, N.L.; Karplus, M. Modeling of globular proteins. A distance-based data search procedure for the construction of insertion/deletion regions and Pro- non-Pro mutations. J. Mol. Biol. 1990, 216, 991–1016. [Google Scholar] [CrossRef]
- Tramontano, A.; Lesk, A. Common features of the conformations of antigen-binding loops in immunoglobulins and application to modeling loop conformations. Proteins 1992, 13, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol. 1992, 226, 507–533. [Google Scholar] [CrossRef]
- Topham, C.M.; McLeod, A.; Eisenmenger, F.; Overington, J.P.; Johnson, M.; Blundell, T. Fragment ranking in modeling of protein structure: Conformationally constrained environmental amino acid substitution tables. J. Mol. Biol. 1993, 229, 194–220. [Google Scholar] [CrossRef] [PubMed]
- Lessel, U.; Schomburg, D. Similarities between protein structures. Protein Eng. 1994, 7, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.C.R.; Thornton, J.M. Structural families in loops of homologous proteins—Automatic classification, modeling and application to antibodies. J. Mol. Biol. 1996, 263, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, Z.; Lai, L. Protein loops on structurally similar scaffolds: Database and conformational analysis. Biopolymers 1999, 49, 481–495. [Google Scholar] [CrossRef]
- Jones, T.A.; Thirup, S. Using known substructures in protein model building and crystallography. EMBO J. 1986, 5, 819–822. [Google Scholar] [PubMed]
- Chothia, C.; Lesk, A.; Tramontano, A.; Levitt, M.S.; Gill, S.J.; Air, G.; Sheriff, S.; Padla, E.; Davies, D.; Tulip, W.R.; et al. Conformation of immunoglobulin hypervariable regions. Nature 1989, 342, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Morea, V.; Tramontano, A.; Rustici, M.; Chothia, C.; Lesk, A.M. Conformations of the third hypervariable region in the VH domain of immunoglobulins. J. Mol. Biol. 1998, 275, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Fidelis, K.; Stern, P.S.; Bacon, D.; Moult, J. Comparison of systematic search and database methods for constructing segments of protein structure. Protein Eng. 1994, 7, 953–960. [Google Scholar] [CrossRef] [PubMed]
- van Vlijmen, H.W.T.; Karplus, M. PDB-based protein loop prediction: Parameters for selection and methods for optimization. J. Mol. Biol. 1997, 267, 975–1001. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; James, M.N.G. An algorithm for determining the conformation of polypeptide segments in proteins by systematic search. Proteins 1986, 1, 146–163. [Google Scholar] [CrossRef] [PubMed]
- Du, P.C.; Andrec, M.; Levy, R.M. Have we seen all structures corresponding to short protein fragments in the Protein Data Bank? An update. Protein Eng. 2003, 16, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Tossato, C.E.; Bindewald, E.; Hesser, J.; Maenner, R. A divide and conquer approach to fast loop modeling. Protein Eng. 2002, 15, 279–286. [Google Scholar] [CrossRef]
- Lee, J.; Lee, D.; Park, H.; Coutsias, E.A.; Seok, C. Protein loop modeling by using fragment assembly and analytical loop closure. Protein. Struct. Funct. Bioinf. 2010, 78, 3428–3436. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W.F. Molecular dynamics simulations. Curr. Opinion Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glättli, A.; Hünenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. Engl. 2006, 45, 4064–4092. [Google Scholar] [CrossRef] [PubMed]
- Bruccoleri, R.E.; Haber, E.; Novotny, J. Structure of antibody hypervariable loops reproduced by a conformational search algorithm. Nature 1988, 335, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Bruccoleri, R.E.; Karplus, M. Prediction of the folding of short poly-peptide segments by uniform conformational sampling. Biopolymers 1987, 26, 137–168. [Google Scholar] [CrossRef] [PubMed]
- Bruccoleri, R.E. Application of systematic conformational search to protein modeling. Mol. Simulat. 1993, 10, 151–174. [Google Scholar] [CrossRef]
- Brower, R.C.; Vasmatzis, G.; Silverman, M.; DeLisi, C. Exhaustive conformational search and simulated annealing for models of lattice peptides. Biopolymers 1993, 33, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Deane, C.M.; Blundell, T.L. A novel exhaustive search algorithm for predicting the conformation of polypeptide segments in proteins. Proteins 2000, 40, 135–144. [Google Scholar] [CrossRef]
- DePristo, M.A.; de Bakker, P.I.; Lovell, S.C.; Blundell, T.L. Ab initio construction of polypeptide fragments: Efficient generation of accurate, representative ensembles. Protein. Struct. Funct. Bioinf. 2003, 51, 41–55. Available online: http://mordred.bioc.cam.ac.uk/~rapper/Software:RAPPER (accessed on 6 February 2012). [Google Scholar] [CrossRef] [PubMed]
- Bruccoleri, R.E.; Karplus, M. Conformational Sampling using high temperature molecular dynamics. Biopolymers 1990, 29, 1847–1862. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.H.; Scheraga, H.A. Pattern recognition in the prediction of protein structure. I. Tripeptide conformational probabilities calculated from the amino acid sequence. J. Comput. Chem. 1989, 10, 770–797. [Google Scholar] [CrossRef]
- Lambert, M.H.; Scheraga, H.A. Pattern recognition in the prediction of protein structure. II. Chain conformation from a probability-directed search procedure. J. Comput. Chem. 1989, 10, 798–816. [Google Scholar] [CrossRef]
- Lambert, M.H.; Scheraga, H.A. Pattern recognition in the prediction of protein structure. III. An importance-sampling minimization procedure. J. Comput. Chem. 1989, 10, 817–831. [Google Scholar] [CrossRef]
- Dudek, M.; Scheraga, H.J. Protein structure prediction using a combination of sequence homology and global energy minimization. I. Global energy minimization of surface loops. J. Comput. Chem. 1990, 11, 121–151. [Google Scholar] [CrossRef]
- Dudek, M.J.; Ramnarayan, K.; Ponder, J.W. Protein structure prediction using a combination of sequence homology and global energy minimization II. Energy functions. J. Comput. Chem. 1998, 19, 548–573. [Google Scholar] [CrossRef]
- Tanner, J.J.; Nell, L.J.; McCammon, J.A. Anti-insulin antibody structure and conformation. II. Molecular Dynamics with explicit solvent. Biopolymers 1992, 32, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Rao, U.; Teeter, M.M. Improvement of turn prediction by molecular dynamics: A case study of a1-Purothionin. Protein Eng. 1993, 6, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, N.; Higo, J.; Kidera, A. Free energy landscapes of short peptides by enhanced conformational sampling. J. Mol. Biol. 2000, 296, 197–216. [Google Scholar] [CrossRef] [PubMed]
- McGarrah, D.B.; Judson, R.S. Analysis of the genetic algorithm method of molecular conformation determination. J. Comput. Chem. 1993, 14, 1385–1395. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Moult, J. Ab initio structure prediction for small polypeptides and protein fragments using genetic algorithms. Proteins 1995, 23, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; DeLisi, C. Determining minimum energy conformations of polypeptides by dynamic programming. Biopolymers 1990, 29, 1755–1772. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.V.; Reva, B.A. Search for the stable state of a short chain in a molecular field. Protein Eng. 1992, 5, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.; Mathiowetz, A.M.; Chan, S.I.; Goddard, W.A. De novo prediction of polypeptide conformations using dihedral probability grid Monte Carlo methodology. Protein Sci. 1995, 4, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Rapp, C.S.; Friesner, R.A. Prediction of loop geometries using a generalized born model of solvation effects. Proteins 1999, 35, 173–183. [Google Scholar] [CrossRef]
- Higo, J.; Collura, V.; Garnier, J. Development of an extended simulated annealing method: Application to the modeling of complementary determining regions of immunoglobulins. Biopolymers 1992, 32, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Collura, V.; Higo, J.; Garnier, J. Modelling of protein loops by simulated annealing. Protein Sci. 1993, 2, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Carlacci, L.; Englander, L. The loop problem in proteins: A Monte Carlo simulated annealing approach. Biopolymers 1993, 33, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Carlacci, L.; Englander, S.W. Loop problem in proteins: Developments on the Monte Carlo simulated annealing approach. J. Comput. Chem. 1996, 17, 1002–1012. [Google Scholar] [CrossRef]
- Vasmatzis, G.; C.Brower, R.; DeLisi, C. Predicting immunoglobulin-like hypervariable loops. Biopolymers 1994, 34, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.; Zheng, Q.; Vajda, S.; DeLisi, C. Computing the structure of bound peptides. Application to antigen recognition by class I major histocompatibility complex receptors. J. Mol. Biol. 1993, 234, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Rosenfeld, R.; DeLisi, C.; Kyle, D.J. Multiple copy sampling in protein loop modelling: Computational efficiency and sensitivity to dihedral perturbations. Protein Sci. 1994, 3, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.; Rosenfeld, R. Simultaneous modeling of multiple loops in proteins. Protein Sci. 1995, 4, 496–505. [Google Scholar]
- Kidera, A. Enhanced conformational sampling in Monte Carlo simulations of proteins: Application to a constrained peptide. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 9886–9889. [Google Scholar] [CrossRef] [PubMed]
- Koehl, P.; Delarue, M. New mean field self-consistent formalism providing simultaneously both gap closure and side chain positioning in protein homology modelling. Nat. Struct. Biol. 1995, 2, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Samudrala, R.; Moult, J. A graph-theoretic algorithm for comparative modeling of protein structure. J. Mol. Biol. 1998, 279, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.S.; Fasnacht, M.; Zhu, J.; Forrest, L.; Honig, B. Loop modeling: Sampling, filtering, and scoring. Protein. Struct. Funct. Bioinf. 2008, 70, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Yakey, J.; LaValle, S.M.; Kavraki, L.E. Randomized path planning for linkages with closed kinematic chains. IEEE Trans. Robot. Autom. 2001, 17, 951–959. [Google Scholar] [CrossRef]
- Han, L.; Amato, N.M. A kinematics-based probabilistic roadmap method for closed chain systems. In Algorithmic and Computational Robotics: New Directions; Donald, B.R., Lynch, K.M., Rus, D., Eds.; AK Peters: MA, USA, 2001; pp. 233–246. [Google Scholar]
- Xie, D.; Amato, N.M. A kinematics-based probabilistic roadmap method for high DOF closed chain systems. In Proceedings of the 2004 IEEE International Conference on Robotics and Automation, 26 April–1 May 2004; Volume 1, pp. 473–478.
- Cortes, J.; Simeon, T.; Laumond, J.P. A Random Loop Generator for planning the motions of closed kinematic chains using PRM methods. In Proceedings of the 2002 IEEE International Conference on Robotics and Automation, 2002; Volume 2, pp. 2141–2146.
- Cortes, J.; Simeon, T. Probabilistic motion planning for parallel mechanisms. In Proceedings of the 2003 IEEE International Conference on Robotics and Automation, 14–19 September 2003; Volume 3, pp. 4354–4359.
- Cortes, J.; Simeon, T.; Remauld-Simeon, M.; Tran, V. Geometric algorithms for the conformational analysis of long protein loops. J. Comput. Chem. 2004, 25, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Milgram, R.J.; Liu, G.; Latombe, J.C. On the structure of the inverse kinematics map of a fragment of protein backbone. J. Comput. Chem. 2008, 29, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Dhanik, A.; Marz, N.; Propper, R.; Kou, C.; Liu, G.; van den Bedem, H.; Latombe, J.C.; Halperin-Landsberg, I.; Altman, R.B. Efficient algorithms to explore conformation spaces of flexible protein loops. In Proceedings of the IEEE/ACM Transactions on Computational Biology and Bioinformatics, October–December 2008; pp. 534–545.
- Ko, J.; Lee, D.; Park, H.; Coutsias, E.A.; Lee, J.; Seok, C. The FALC-Loop web server for protein loop modeling. Nucleic Acids Res. 2011, 39, W210–W214. Available online: http://falc-loop.seoklab.org/ (accessed on 6 February 2012). [Google Scholar] [CrossRef] [PubMed]
- Mandell, D.J.; Coutsias, E.A.; Kortemme, T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 2009, 6, 510–528. [Google Scholar] [CrossRef] [PubMed]
- Coutsias, E.A.; Seok, C.; Wester, M.J.; Dill, K.A. Resultants and loop closure. Int. J. Quantum Chem. 2006, 106, 176–189. [Google Scholar] [CrossRef]
- Nilmeier, J.; Hua, L.; Coutsias, E.; Jacobson, M.P. Assessing loop flexibility by hierarchical Monte Carlo sampling. J. Chem. Theory Comput. 2011, 7, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Rosenfeld, R.; Vajda, S.; DeLisi, C. Loop closure via bond scaling and relaxation. J. Comput. Chem. 1992, 14, 556–565. [Google Scholar] [CrossRef]
- Zheng, Q.; Rosenfeld, R.; Vajda, S.; DeLisi, C. Determining protein loop conformation using scaling-relaxation techniques. Protein Sci. 1993, 2, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhu, F.; Rassokhin, D.N.; Agrafiotis, D.K. A Self-organizing algorithm for modeling protein loops. PLoS Comp. Biol. 2009, 5, e1000478. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rata, I.; Jakobsson, E. Sampling multiple scoring functions can improve protein loop structure prediction accuracy. J. Chem. Inf. Model. 2011, 51, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Zhou, Y. Accurate and efficient loop selections by the DFIRE-based all-atom statistical potential. Protein Sci. 2004, 13, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Fitzkee, N.C.; Fleming, P.J.; Rose, G.D. The protein coil library: A structural database of nonhelix, nonstrand fragments derived from the PDB. Protein. Struct. Funct. Bioinf. 2005, 58, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Storn, R.; Price, K. Differential evolution—A simple and efficient heuristic for global optimization over continuous spaces. J. Global Optim. 2004, 11, 341–359. [Google Scholar] [CrossRef]
- LaValle, S.M.; Kuffner, J.J. Randomized kinodynamic planning. Int. J. Robot. Res. 2001, 20, 378–400. [Google Scholar] [CrossRef]
- Kavraki, L.E.; Svetska, P.; Latombe, J.C.; Overmars, M. Probabilistic roadmaps for path planning in high-dimensional configuration spaces. IEEE Trans. Robot. Autom. 1996, 12, 566–580. [Google Scholar] [CrossRef]
- Debeire-Gosselin, M.; Loonis, M.; Samain, E.; Debeire, P. Purification and properties of a 22kDa endoxylanase excreted by a new strain of thermophilic bacterium. Xylans and Xylanases; Elsevier Science Publishers: Amsterdam, The Netherlands, 1997; pp. 463–466. [Google Scholar]
- Harris, G.W.; Pickersgill, R.; Connerton, I.; Debeire, P.; Touzel, J.P.; Breton, C.; Pérez, S. Structural basis of the properties of an industrially relevant thermophilic xylanase. Protein. Struct. Funct. Bioinf. 1997, 29, 77–86. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Pacci, E.; Dobson, C.; Karplus, M. Rare fluctuations of native proteins sampled by equilibrium hydrogen exchange. J. Am. Chem. Soc. 2003, 125, 15686–15687. [Google Scholar] [CrossRef]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Sequence complexity of disordered protein. Protein. Struct. Funct. Bioinf. 2001, 42, 38–48. [Google Scholar]
- Shehu, A.; Kavraki, L.E.; Clementi, C. On the characterization of protein native state ensembles. Biophys. J. 2007, 92, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.; Chavan, A.G.; Day, R.; Lennox, K.P.; Sukhanov, P.; Dahl, D.B.; Vannucci, M.; Tsai, J. Near-native protein loop sampling using nonparametric density estimation accommodating sparcity. PLoS Comp. Biol. 2011, 7, e1002234. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, M.F.; Ming, L. Macromolecular flexibility. Phil. Mag. 2004, 84, 1323–31137. [Google Scholar] [CrossRef]
- Wells, S.; Menor, S.; Hespenheide, B.; Thorpe, M.F. Constrained geometric simulation of diffusive motion in proteins. J. Phys. Biol. 2005, 2, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.W.; Speranskiy, K.; Thorpe, M.F. Generating stereochemically acceptable protein pathways. Protein. Struct. Funct. Bioinf. 2010, 78, 2908–2921. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Zhang, L.; Latombe, J.C. Sampling-based exploration of folded state of a protein under kinematic and geometric constraints. Protein. Struct. Funct. Bioinf. 2011, 80, 25–43. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/.)
Share and Cite
Shehu, A.; Kavraki, L.E. Modeling Structures and Motions of Loops in Protein Molecules. Entropy 2012, 14, 252-290. https://doi.org/10.3390/e14020252
Shehu A, Kavraki LE. Modeling Structures and Motions of Loops in Protein Molecules. Entropy. 2012; 14(2):252-290. https://doi.org/10.3390/e14020252
Chicago/Turabian StyleShehu, Amarda, and Lydia E. Kavraki. 2012. "Modeling Structures and Motions of Loops in Protein Molecules" Entropy 14, no. 2: 252-290. https://doi.org/10.3390/e14020252