School of Resources and Environmental Engineering, Ludong University, Yantai 264025, China
Genes 2023, 14(2), 293; https://doi.org/10.3390/genes14020293 - 22 Jan 2023
Cited by 3 | Viewed by 4154
Abstract
To address the plant adaptability of sorghum (Sorghum bicolor) in salinity, the research focus should shift from only selecting tolerant varieties to understanding the precise whole-plant genetic coping mechanisms with long-term influence on various phenotypes of interest to expanding salinity, improving
[...] Read more.
To address the plant adaptability of sorghum (Sorghum bicolor) in salinity, the research focus should shift from only selecting tolerant varieties to understanding the precise whole-plant genetic coping mechanisms with long-term influence on various phenotypes of interest to expanding salinity, improving water use, and ensuring nutrient use efficiency. In this review, we discovered that multiple genes may play pleiotropic regulatory roles in sorghum germination, growth, and development, salt stress response, forage value, and the web of signaling networks. The conserved domain and gene family analysis reveals a remarkable functional overlap among members of the bHLH (basic helix loop helix), WRKY (WRKY DNA-binding domain), and NAC (NAM, ATAF1/2, and CUC2) superfamilies. Shoot water and carbon partitioning, for example, are dominated by genes from the aquaporins and SWEET families, respectively. The gibberellin (GA) family of genes is prevalent during pre-saline exposure seed dormancy breaking and early embryo development at post-saline exposure. To improve the precision of the conventional method of determining silage harvest maturity time, we propose three phenotypes and their underlying genetic mechanisms: (i) the precise timing of transcriptional repression of cytokinin biosynthesis (IPT) and stay green (stg1 and stg2) genes; (ii) the transcriptional upregulation of the SbY1 gene and (iii) the transcriptional upregulation of the HSP90-6 gene responsible for grain filling with nutritive biochemicals. This work presents a potential resource for sorghum salt tolerance and genetic studies for forage and breeding.
Full article
(This article belongs to the Section Plant Genetics and Genomics)
▼
Show Figures
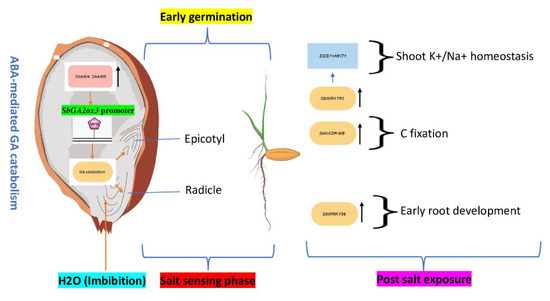
Figure 1







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}