by
Nikoletta Psatha 1, Kiriaki Paschoudi 1,2, Anastasia Papadopoulou 2 and Evangelia Yannaki 2,3,*
and Evangelia Yannaki 2,3,*
and Evangelia Yannaki 2,3,*
1
Department of Genetics, Development and Molecular Biology, School of Biology, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece
2
Gene and Cell Therapy Center, Hematology Clinic, George Papanikolaou Hospital, Exokhi, 57010 Thessaloniki, Greece
3
Department of Hematology, School of Medicine, University of Washington, Seattle, WA 98195, USA
Genes 2022, 13(12), 2222; https://doi.org/10.3390/genes13122222 - 27 Nov 2022
Cited by 19 | Viewed by 7182
Abstract
The tremendous evolution of genome-editing tools in the last two decades has provided innovative and effective approaches for gene therapy of congenital and acquired diseases. Zinc-finger nucleases (ZFNs), transcription activator- like effector nucleases (TALENs) and CRISPR-Cas9 have been already applied by ex vivo
[...] Read more.
The tremendous evolution of genome-editing tools in the last two decades has provided innovative and effective approaches for gene therapy of congenital and acquired diseases. Zinc-finger nucleases (ZFNs), transcription activator- like effector nucleases (TALENs) and CRISPR-Cas9 have been already applied by ex vivo hematopoietic stem cell (HSC) gene therapy in genetic diseases (i.e., Hemoglobinopathies, Fanconi anemia and hereditary Immunodeficiencies) as well as infectious diseases (i.e., HIV), and the recent development of CRISPR-Cas9-based systems using base and prime editors as well as epigenome editors has provided safer tools for gene therapy. The ex vivo approach for gene addition or editing of HSCs, however, is complex, invasive, technically challenging, costly and not free of toxicity. In vivo gene addition or editing promise to transform gene therapy from a highly sophisticated strategy to a “user-friendly’ approach to eventually become a broadly available, highly accessible and potentially affordable treatment modality. In the present review article, based on the lessons gained by more than 3 decades of ex vivo HSC gene therapy, we discuss the concept, the tools, the progress made and the challenges to clinical translation of in vivo HSC gene editing.
Full article
(This article belongs to the Special Issue Gene Editing for Therapy and Reverse Genetics of Blood Diseases)
▼
Show Figures
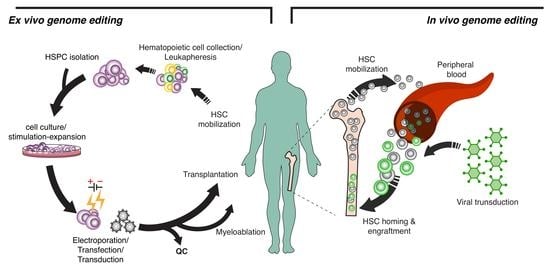
Graphical abstract








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}