Genes 2021, 12(9), 1415; https://doi.org/10.3390/genes12091415 - 15 Sep 2021
Cited by 10 | Viewed by 5228
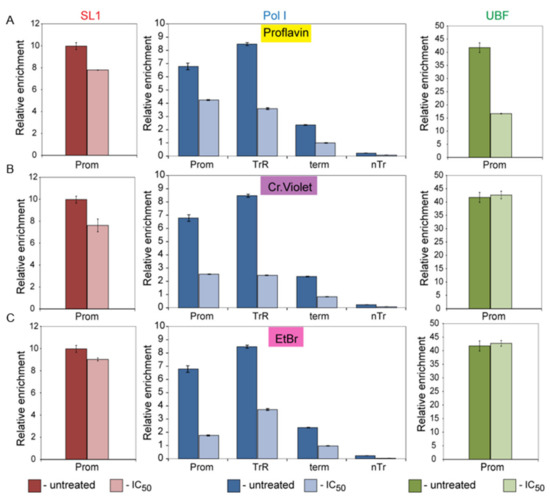
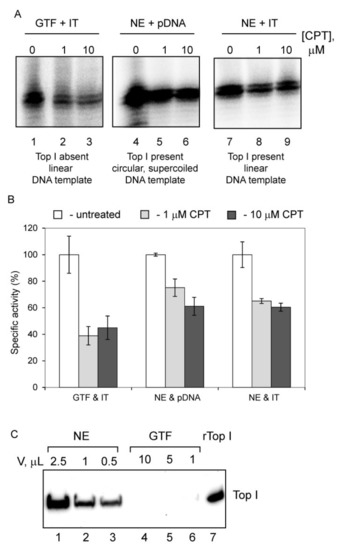
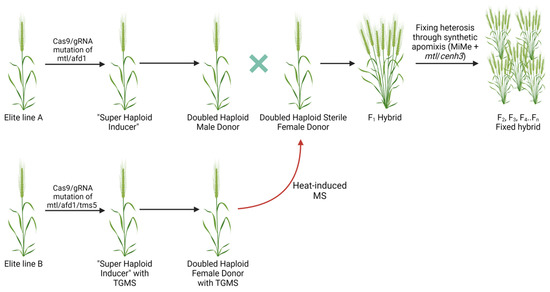
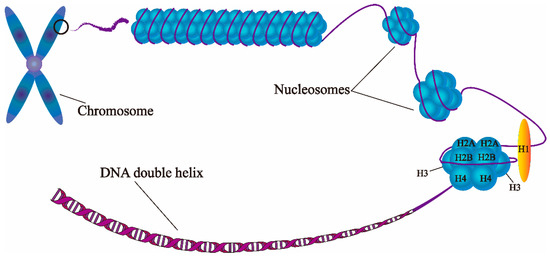
Abstract
The eukaryotic nucleus is continuously being exposed to endogenous and exogenous sources that cause DNA breaks, whose faithful repair requires the activity of dedicated nuclear machineries. DNA is packaged into a variety of chromatin domains, each characterized by specific molecular properties that regulate
[...] Read more.
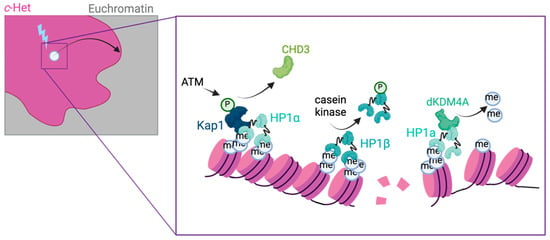
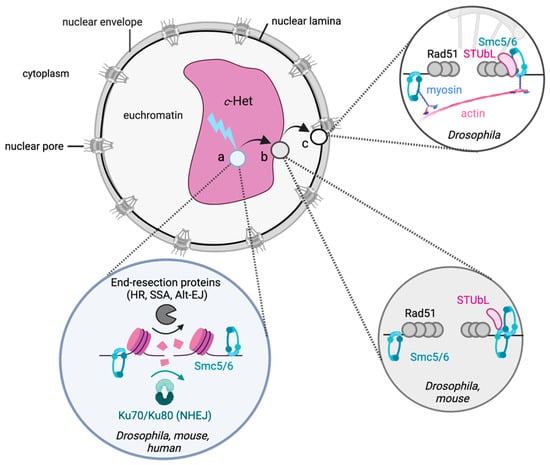
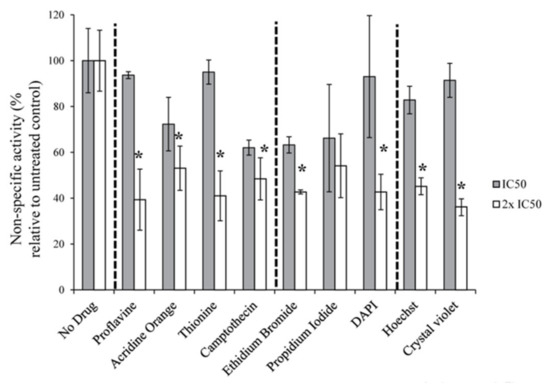
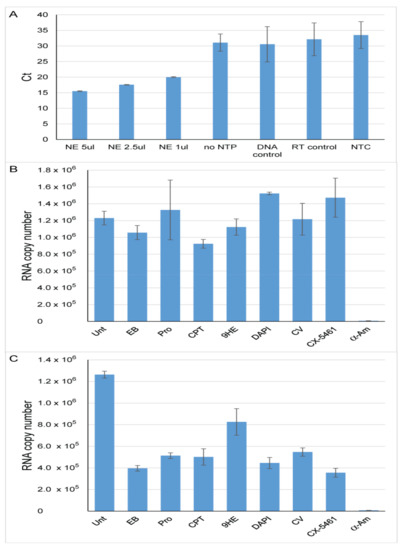
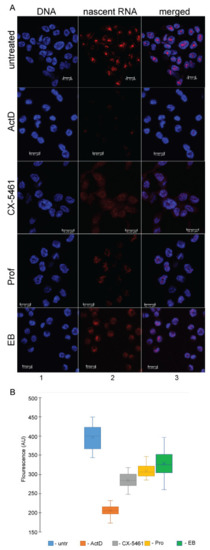
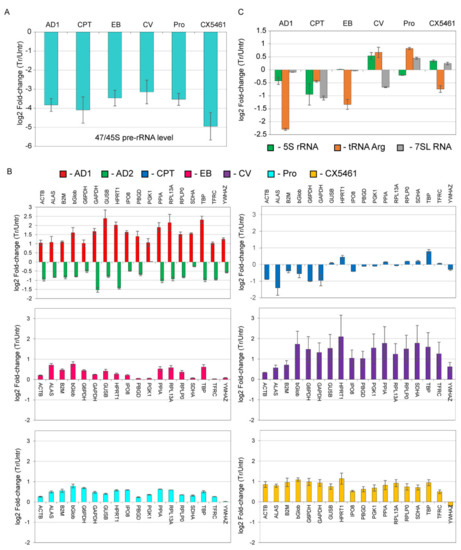
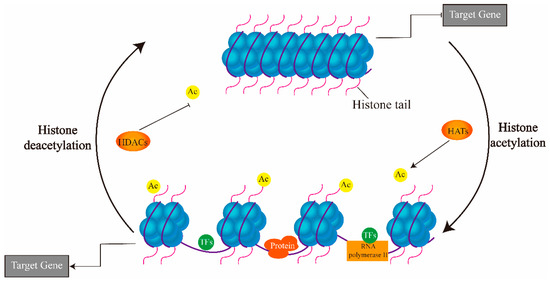
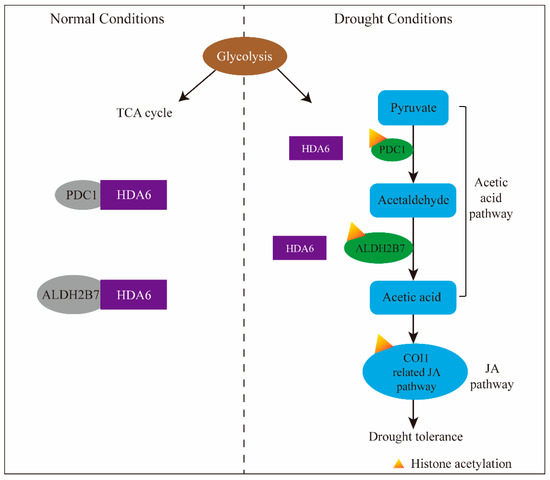
The eukaryotic nucleus is continuously being exposed to endogenous and exogenous sources that cause DNA breaks, whose faithful repair requires the activity of dedicated nuclear machineries. DNA is packaged into a variety of chromatin domains, each characterized by specific molecular properties that regulate gene expression and help maintain nuclear structure. These different chromatin environments each demand a tailored response to DNA damage. Silenced chromatin domains in particular present a major challenge to the cell’s DNA repair machinery due to their specific biophysical properties and distinct, often repetitive, DNA content. To this end, we here discuss the interplay between silenced chromatin domains and DNA damage repair, specifically double-strand breaks, and how these processes help maintain genome stability.
Full article
(This article belongs to the Special Issue DNA Damage Response Mechanisms in Model Systems)
►
Show Figures
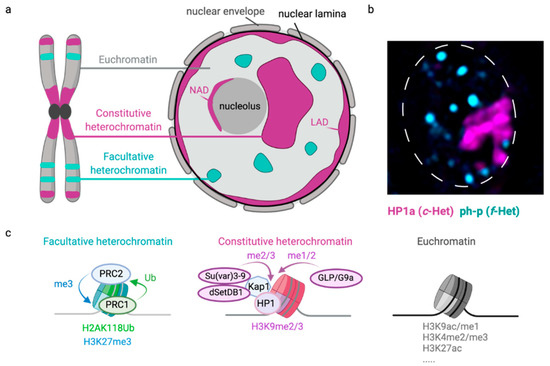
Figure 1





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}