Sequencing and Comparative Analysis of the Chloroplast Genome of Angelica polymorpha and the Development of a Novel Indel Marker for Species Identification

 ,
, 
 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. CP Genome Organization of A. polymorpha
2.2. Analysis of Repeated Sequences in the CP Genomes of A. polymorpha and L. officinale
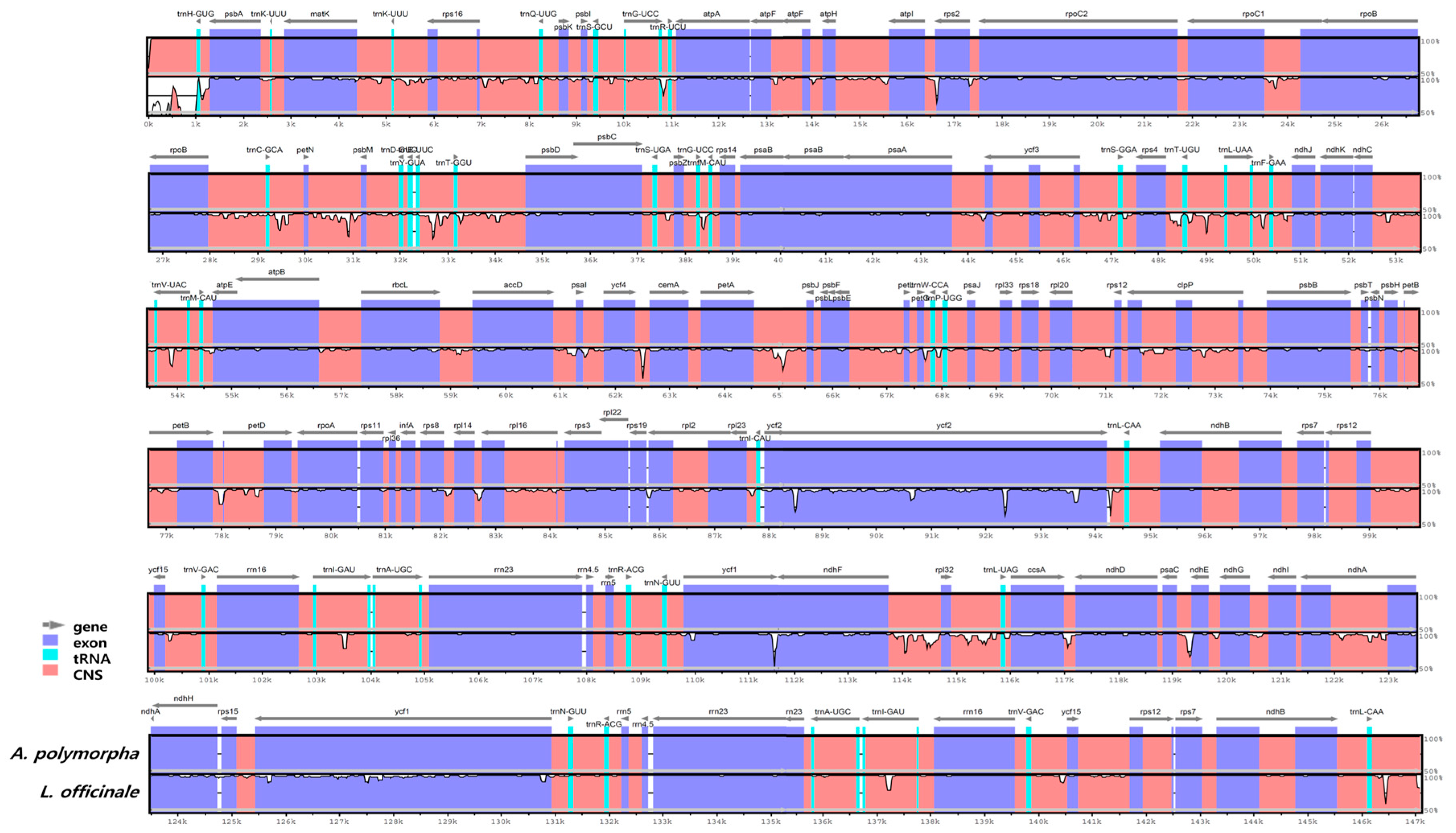
2.3. Comparative Analysis of the CP Genomes of A. polymorpha and L. officinale
2.4. Phylogenetic Relationship between A. polymorpha and L. officinale
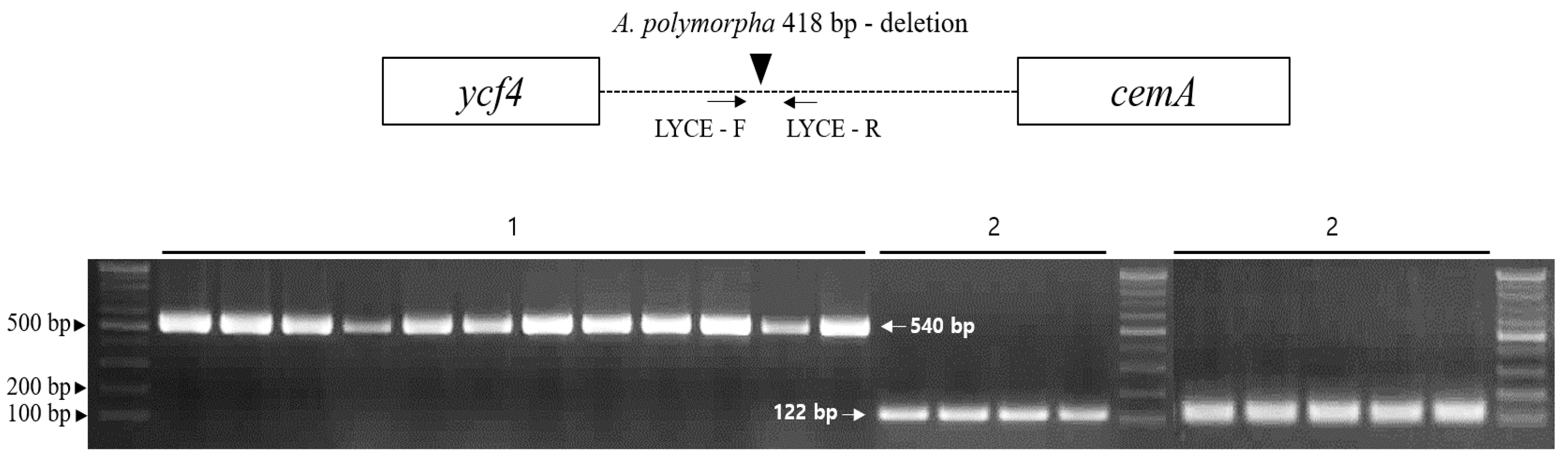
2.5. Development and Validation of an Indel Marker for Authentication of Cnidii Rhizoma
3. Materials and Methods
3.1. Plant Materials
3.2. Sequencing and Assembly of the CP Genome of A. polymorpha
3.3. Annotation and Comparative Analysis
3.4. Analysis of SSRs and Tandem and Palindromic Repeats in CP Genomes of A. polymorpha and L. officinale
3.5. Phylogenetic Analysis
3.6. Development and Validation of the LYCE Indel
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.; Al-Mssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Springer: Dordrecht, The Netherlands, 2012; pp. 103–126. [Google Scholar]
- Weng, M.-L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Cosner, M.E.; Jansen, R.K.; Palmer, J.D.; Downie, S.R. The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): Multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr. Genet. 1997, 31, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium x hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, J.J.; Huang, Y.T.; Chan, M.T.; Daniell, H.; Chang, W.J.; Hsu, C.T.; Liao, D.C.; Wu, F.H.; Lin, S.Y.; et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015, 5, 9040. [Google Scholar] [CrossRef] [PubMed]
- Frailey, D.C.; Chaluvadi, S.R.; Vaughn, J.N.; Coatney, C.G.; Bennetzen, J.L. Gene loss and genome rearrangement in the plastids of five Hemiparasites in the family Orobanchaceae. BMC Plant Biol. 2018, 18, 30. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, J.G.; Chen, X.L.; Cui, Y.X.; Xu, Z.C.; Li, Y.H.; Song, J.Y.; Duan, B.Z.; Yao, H. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci. Rep. 2017, 7, 12834. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Nayak, S.N.; May, G.D.; Jackson, S.A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Korea Institute of Oriental Medicine (KIOM). Defining Dictionary for Medicinal Herbs. 2019. Available online: http://boncho.kiom.re.kr/codex/ (accessed on 2 January 2019).
- Tian, E.; Liu, Q.; Chen, W.; Li, F.; Chen, A.; Li, C.; Chao, Z. Characterization of complete chloroplast genome of Angelica sinensis (Apiaceae), an endemic medical plant to China. Mitochondrial DNA B Resour. 2019, 4, 158–159. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.-F.; Cao, D.; Niu, J.-F.; Wang, Z.-Z. The complete chloroplast genome sequence of Angelica tsinlingensis (Apioideae). Mitochondrial DNA B Resour. 2018, 3, 480–481. [Google Scholar] [CrossRef]
- Deng, Y.-Q.; Wen, J.; Yu, Y.; He, X.-J. The complete chloroplast genome of Angelica nitida. Mitochondrial DNA B Resour. 2017, 2, 694–695. [Google Scholar] [CrossRef]
- Choi, S.A.; Kim, Y.J.; Lee, W.K.; Kim, K.Y.; Kim, J.H.; Seong, R.S. The complete chloroplast genome of the medicinal plant Angelica decursiva (Apiaceae) in Peucedani Radix. Mitochondrial DNA B Resour. 2016, 1, 210–211. [Google Scholar] [CrossRef]
- Choi, S.A.; Kim, Y.; Kim, K.-Y.; Kim, J.H.; Seong, R.S. The complete chloroplast genome sequence of the medicinal plant, Angelica gigas (Apiaceae). Mitochondrial DNA B Resour. 2016, 1, 280–281. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [PubMed]
- Kelchner, S.A. The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann. Mo. Bot. Gard. 2000, 87, 482–498. [Google Scholar] [CrossRef]
- Ogihara, Y.; Terachi, T.; Sasakuma, T. Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. USA 1988, 85, 8573–8577. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.B.; Braverman, J.M.; Soria-Hernanz, D.F. Patterns and relative rates of nucleotide and insertion/deletion evolution at six chloroplast intergenic regions in new world species of the Lecythidaceae. Mol. Biol. Evol. 2003, 20, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Huang, Y.M.; Kuo, L.Y.; Nguyen, Q.D.; Luu, H.T.; Callado, J.R.; Farrar, D.R.; Chiou, W.L. trnL-F is a powerful marker for DNA identification of field vittarioid gametophytes (Pteridaceae). Ann. Bot. 2013, 111, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoneberg Holt, S.D.; Horova, L.; Bures, P. Indel patterns of the plastid DNA trnL- trnF region within the genus Poa (Poaceae). J. Plant Res. 2004, 117, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Ingvarsson, P.K.; Ribstein, S.; Taylor, D.R. Molecular evolution of insertions and deletion in the chloroplast genome of silene. Mol. Biol. Evol. 2003, 20, 1737–1740. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Yang, S.; Choi, G.; Kim, W.J.; Moon, B.C. The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum Subgenus Lycoctonum. Molecules 2017, 22, 2012. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Kim, W.J.; Yang, S.; Yeo, S.M.; Li, H.; Moon, B.C. The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS ONE 2017, 12, e0184257. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Yun, B.K.; Yoon, Y.H.; Hong, S.Y.; Mekapogu, M.; Kim, K.H.; Yang, T.J. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 2015, 10, e0125332. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Yang, S.; Kim, W.J.; Noh, P.; Lee, H.O.; Moon, B.C. The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic Pharbitidis Semen (Seeds of I. nil or I. purpurea). Front. Plant Sci. 2018, 9, 965. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Downie, S.R.; Li, Q.; Yu, Y.; He, X.; Zhou, B. New insights into the phylogeny of Angelica and its allies (Apiaceae) with emphasis on East Asian species, inferred from nrDNA, cpDNA, and morphological evidence. Syst. Bot. 2013, 38, 266–281. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Yukawa, Y.; Miyamoto, T.; Obokata, J.; Sugiura, M. Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): Comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol. Biol. Evol. 2003, 20, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yi, X.; Yang, Y.X.; Su, Y.J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puerta, M.V.; Abbona, C.C. The chloroplast genome of Hyoscyamus niger and a phylogenetic study of the tribe Hyoscyameae (Solanaceae). PLoS ONE 2014, 9, e98353. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Yang, S.; Kim, W.J.; Noh, P.; Lee, H.O.; Moon, B.C. The complete chloroplast genome of Cnidium officinale Makino. Mitochondrial DNA B Resour. 2018, 3, 490–491. [Google Scholar] [CrossRef]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome biology 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Kim, W.J.; Yeo, S.M.; Choi, G.; Kang, Y.M.; Piao, R.; Moon, B.C. The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Liu, Q.; Hu, W.; Wang, T.; Xue, Q.; Messing, J. Dynamics of chloroplast genomes in green plants. Genomics 2015, 106, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Yang, S.; Kim, W.; Noh, P.; Lee, H.; Moon, B. Authentication of herbal medicines Dipsacus asper and Phlomoides umbrosa using DNA barcodes, chloroplast genome, and sequence characterized amplified region (SCAR) Marker. Molecules 2018, 23, 1748. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jung, J.-Y.; Choi, H.-I.; Kim, N.-H.; Park, J.Y.; Lee, Y.; Yang, T.-J. Diversity and evolution of major Panax species revealed by scanning the entire chloroplast intergenic spacer sequences. Genet. Resour. Crop Evol. 2013, 60, 413–425. [Google Scholar] [CrossRef]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Semon, M.; Perry, A.S.; Stefanovic, S.; Milbourne, D.; Barth, S.; Palmer, J.D.; et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.-J.; Downie, S.; van Wyk, B.-E.; Tilney, P. A molecular systematic investigation of Cymopterus and its allies (Apiaceae) based on phylogenetic analyses of nuclear (ITS) and plastid (rps16 intron) DNA sequences. S. Afr. J. Bot. 2004, 70, 407–416. [Google Scholar] [CrossRef]
- Sun, F.-J.; Downie, S.R. Phylogenetic relationships among the perennial, endemic Apiaceae subfamily Apioideae of western North America: Additional data from the cpDNA trnF-trnL-trnT region continue to support a highly polyphyletic Cymopterus. Plant Divers. Evol. 2010, 128, 151–172. [Google Scholar] [CrossRef]
- Pimenov, M.G.; Ostroumova, T.A.; Degtjareva, G.V.; Samigullin, T.H. Sillaphyton, a new genus of the Umbelliferae, endemic to the Korean peninsula. Botanica Pacifica 2016, 5, 31–41. [Google Scholar] [CrossRef]
- Wu, Z.; Raven, P.H.; Hong, D. Flora of China. Volume 14: Apiaceae through Ericaceae; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2005. [Google Scholar]
- Lee, Y.N. Flora of Korea; Kyo-Hak Publishing Co.: Seoul, Korea, 2002; 1265p. [Google Scholar]
- Makino, T. Observations on the Flora of Japan. (Continued from Vol. XXVII. p. 258.). Shokubutsugaku Zasshi 1914, 28, 20–30. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.C.; Lee, J.; Lee, H.O.; Joh, H.J.; Kim, N.H.; Park, H.S.; Yang, T.J. Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLoS ONE 2015, 10, e0117159. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Cheon, K.S.; Yoo, K.O.; Lee, H.O.; Cho, K.S.; Suh, J.T.; Kim, S.J.; Nam, J.H.; Sohn, H.B.; Kim, Y.H. Complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Front. Plant Sci. 2017, 8, 1696. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using mummer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics 2003, 00, 10.3.1–10.3.18. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Bohme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Warburton, P.E.; Giordano, J.; Cheung, F.; Gelfand, Y.; Benson, G. Inverted repeat structure of the human genome: The X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 2004, 14, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Gurusaran, M.; Ravella, D.; Sekar, K. Repex: Repeat extractor for biological sequences. Genomics 2013, 102, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T. MISA—Microsatellite Identification Tool. 2003. Available online: http://misaweb.ipk-gatersleben.de/ (accessed on 2 January 2019).
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of A. polymorpha and L. officinale are available from the authors and the herbarium of KIOM. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic 1 | A. polymorpha | L. officinale2 |
|---|---|---|
| Accession number | MH260705 | NC039760 [34] |
| Genome size | ||
| Total CP genome (bp) | 147,127 | 148,518 |
| Large single copy (LSC) region (bp) | 93,591 | 93,977 |
| Inverted repeat (IR) region (bp) | 17,870 | 18,467 |
| Small single copy (SSC) region (bp) | 17,796 | 17,607 |
| Number of unique genes | ||
| Total | 113 | 113 |
| Protein-coding genes | 79 | 79 |
| rRNA genes | 4 | 4 |
| tRNA genes | 30 | 30 |
| GC content (%) | ||
| Total genome | 37.5 | 37.6 |
| LSC region | 35.9 | 36.0 |
| IR regions | 45.0 | 44.8 |
| SSC region | 31.0 | 31.1 |
| Primer Name | Primer Sequence (5′→3′) | Position |
|---|---|---|
| LYCE-F | CGC TCA TTC TAG TCA AAG AAG ACG | ycf4-cemA |
| LYCE-R | CGC CAT CCA ATA TTT CTC TCA TGC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, I.; Yang, S.; Kim, W.J.; Song, J.-H.; Lee, H.-S.; Lee, H.O.; Lee, J.-H.; Ahn, S.-N.; Moon, B.C. Sequencing and Comparative Analysis of the Chloroplast Genome of Angelica polymorpha and the Development of a Novel Indel Marker for Species Identification. Molecules 2019, 24, 1038. https://doi.org/10.3390/molecules24061038
Park I, Yang S, Kim WJ, Song J-H, Lee H-S, Lee HO, Lee J-H, Ahn S-N, Moon BC. Sequencing and Comparative Analysis of the Chloroplast Genome of Angelica polymorpha and the Development of a Novel Indel Marker for Species Identification. Molecules. 2019; 24(6):1038. https://doi.org/10.3390/molecules24061038
Chicago/Turabian StylePark, Inkyu, Sungyu Yang, Wook Jin Kim, Jun-Ho Song, Hyun-Sook Lee, Hyun Oh Lee, Jung-Hyun Lee, Sang-Nag Ahn, and Byeong Cheol Moon. 2019. "Sequencing and Comparative Analysis of the Chloroplast Genome of Angelica polymorpha and the Development of a Novel Indel Marker for Species Identification" Molecules 24, no. 6: 1038. https://doi.org/10.3390/molecules24061038