Double Strand Breaks and Cell-Cycle Arrest Induced by the Cyanobacterial Toxin Cylindrospermopsin in HepG2 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
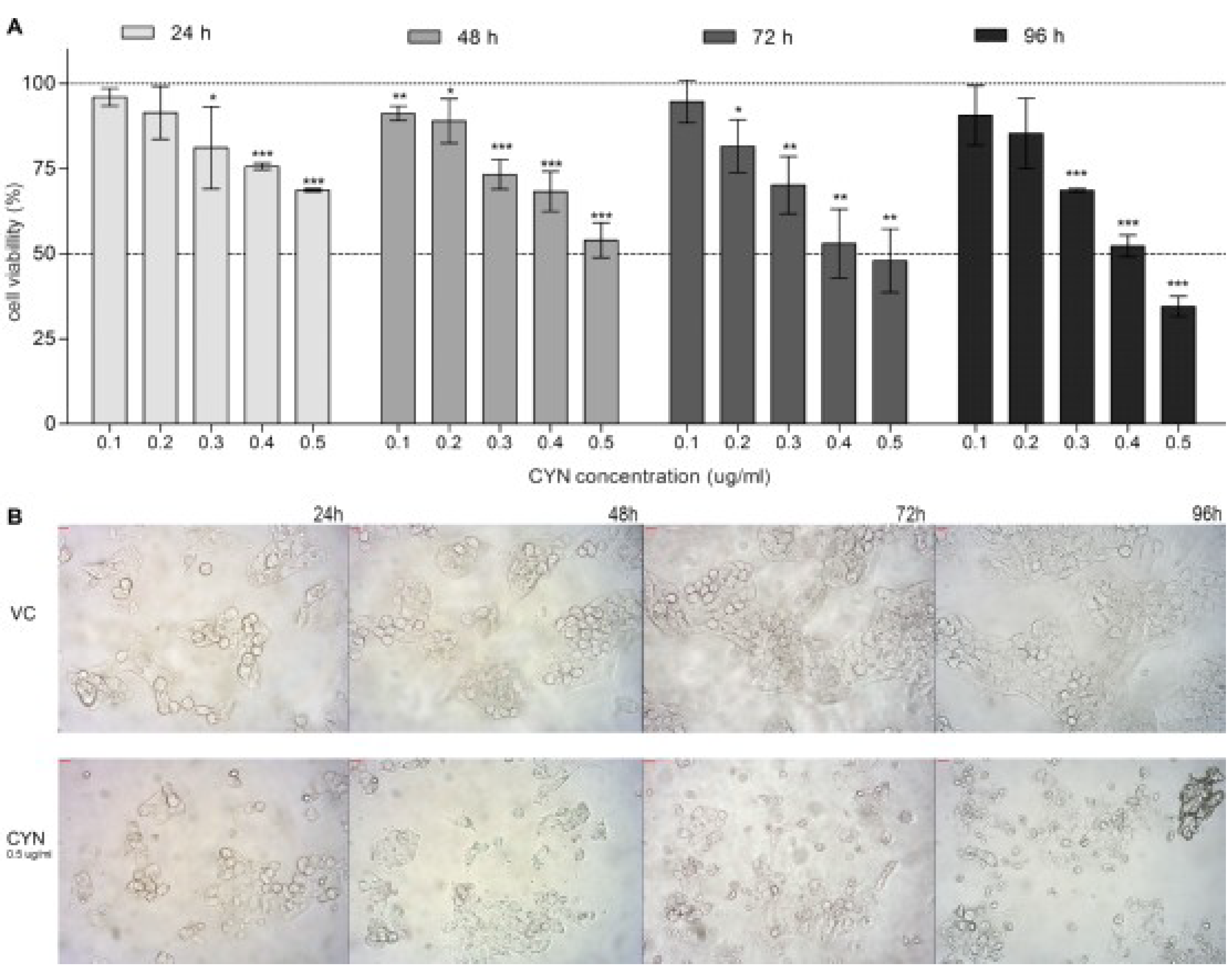
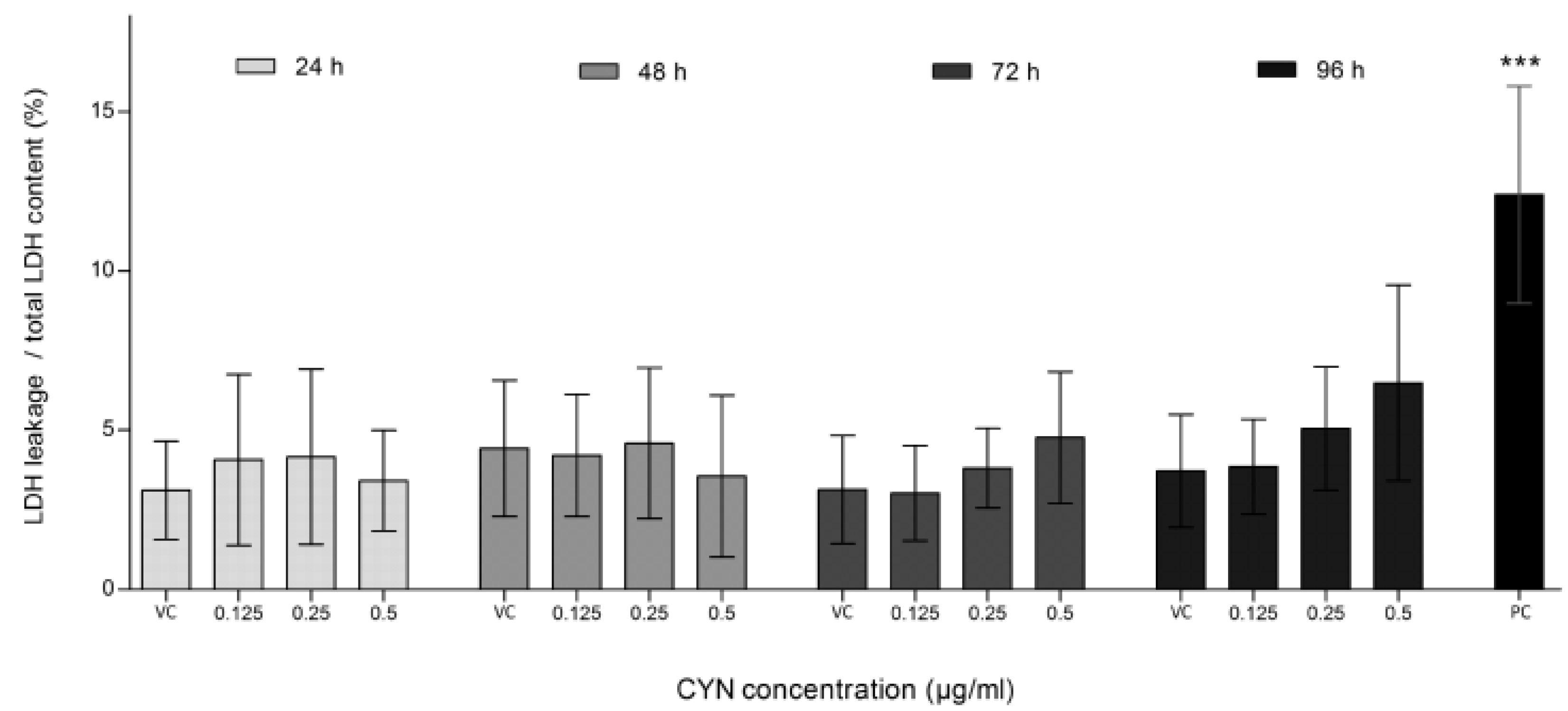
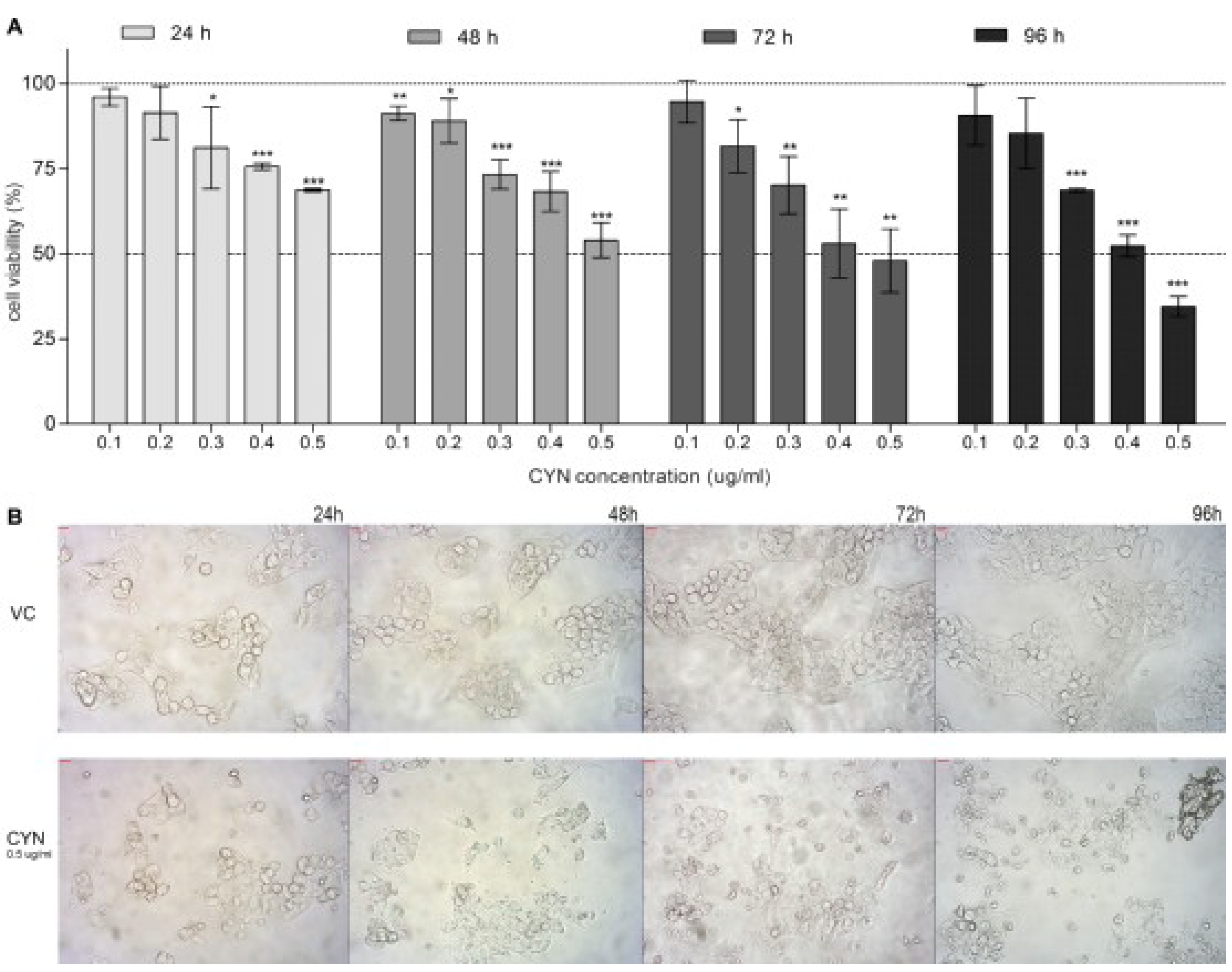
2.1. Viability of HepG2 Cells after CYN Exposure

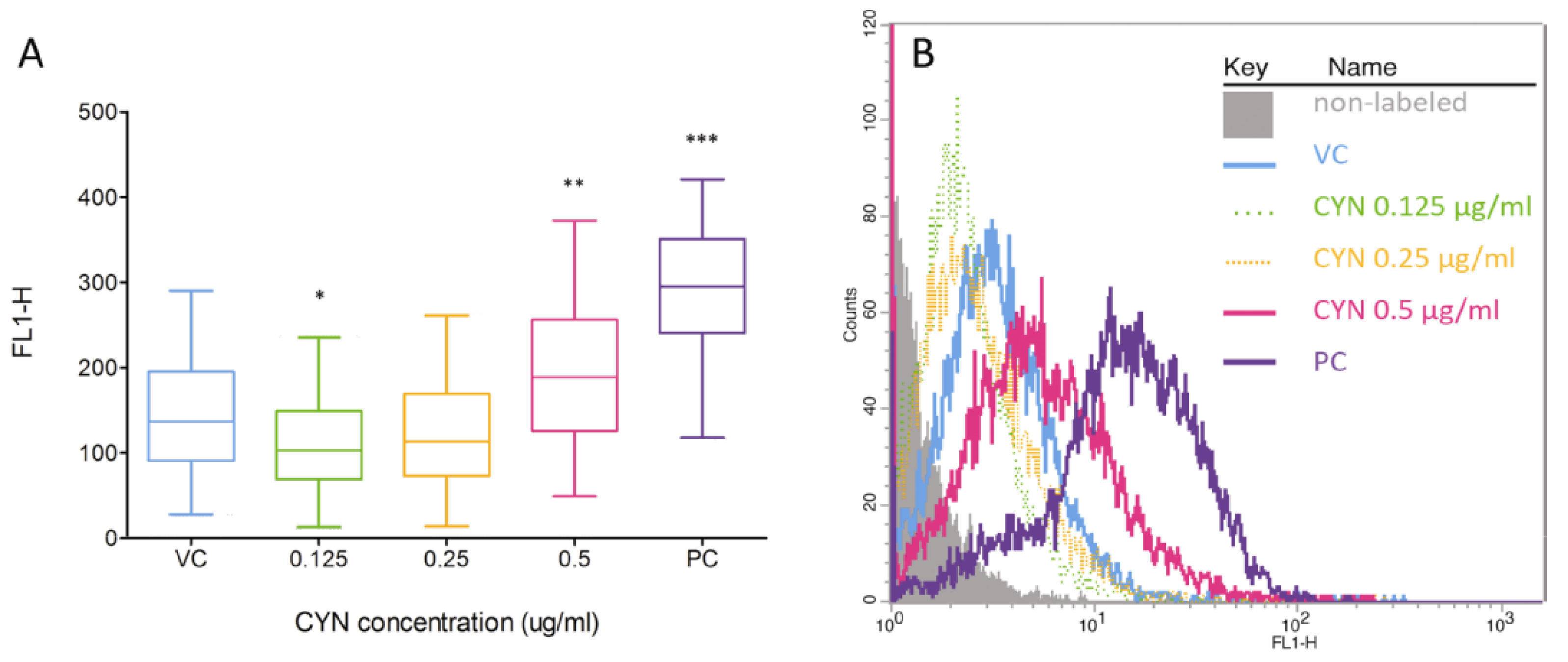
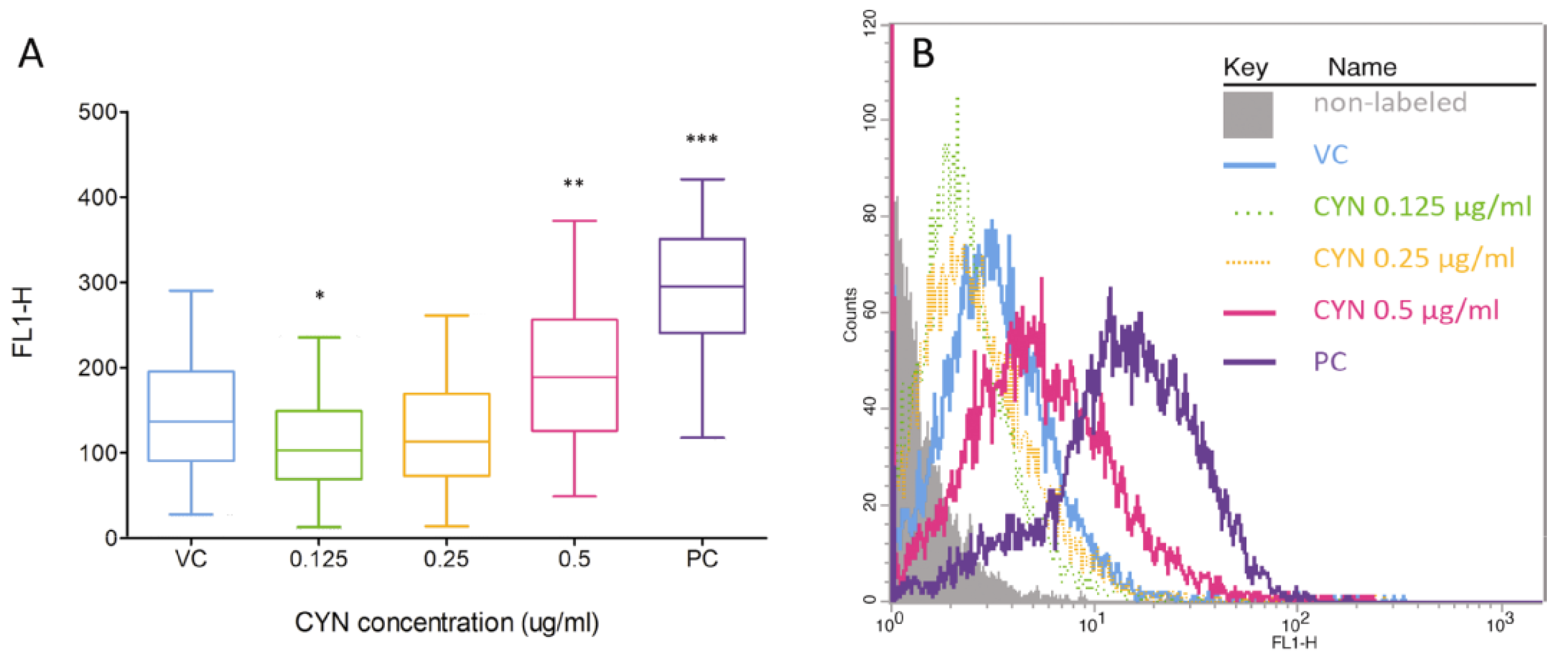
2.2. Formation of DNA Double Strand Beaks (DSBs) by CYN
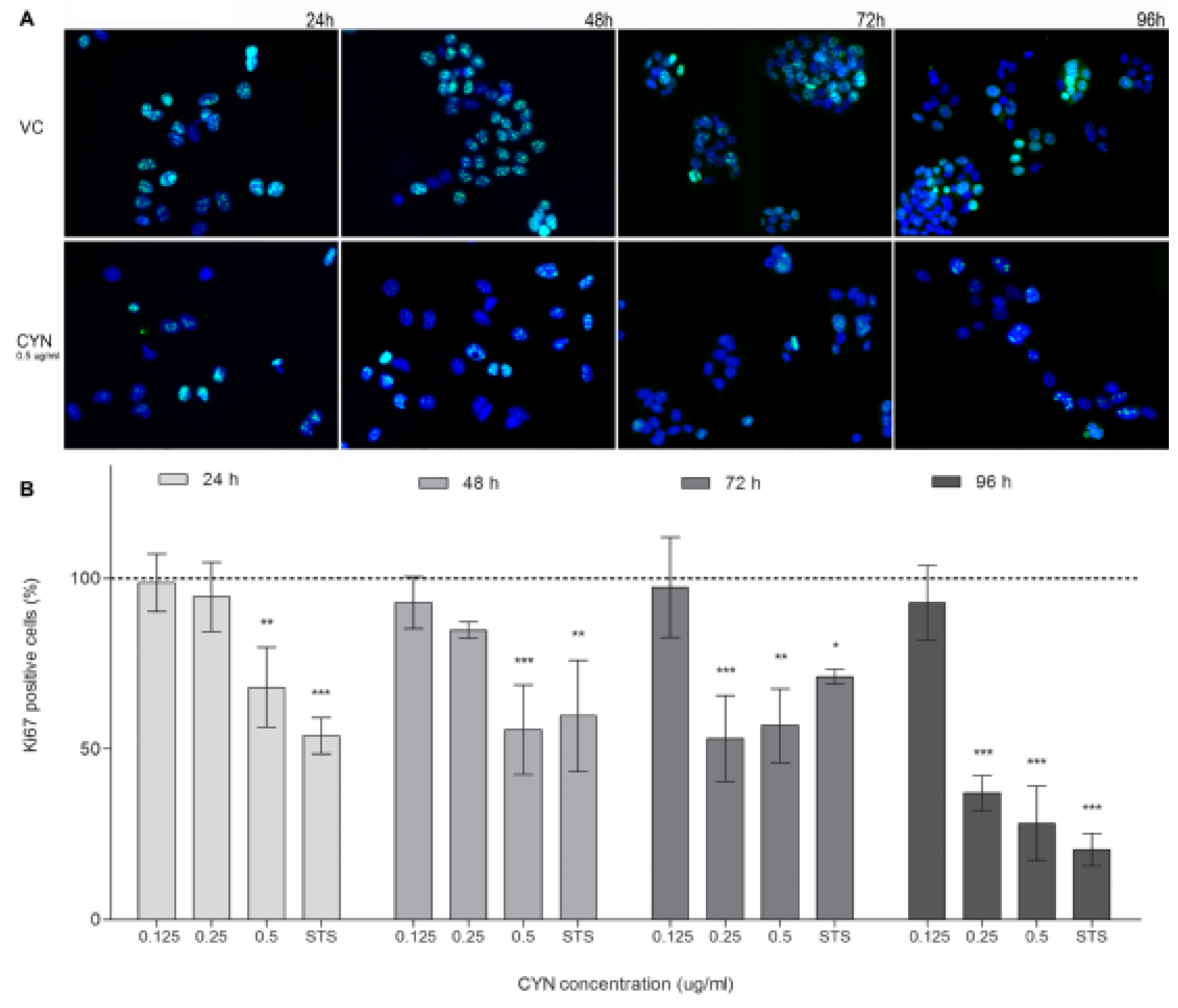
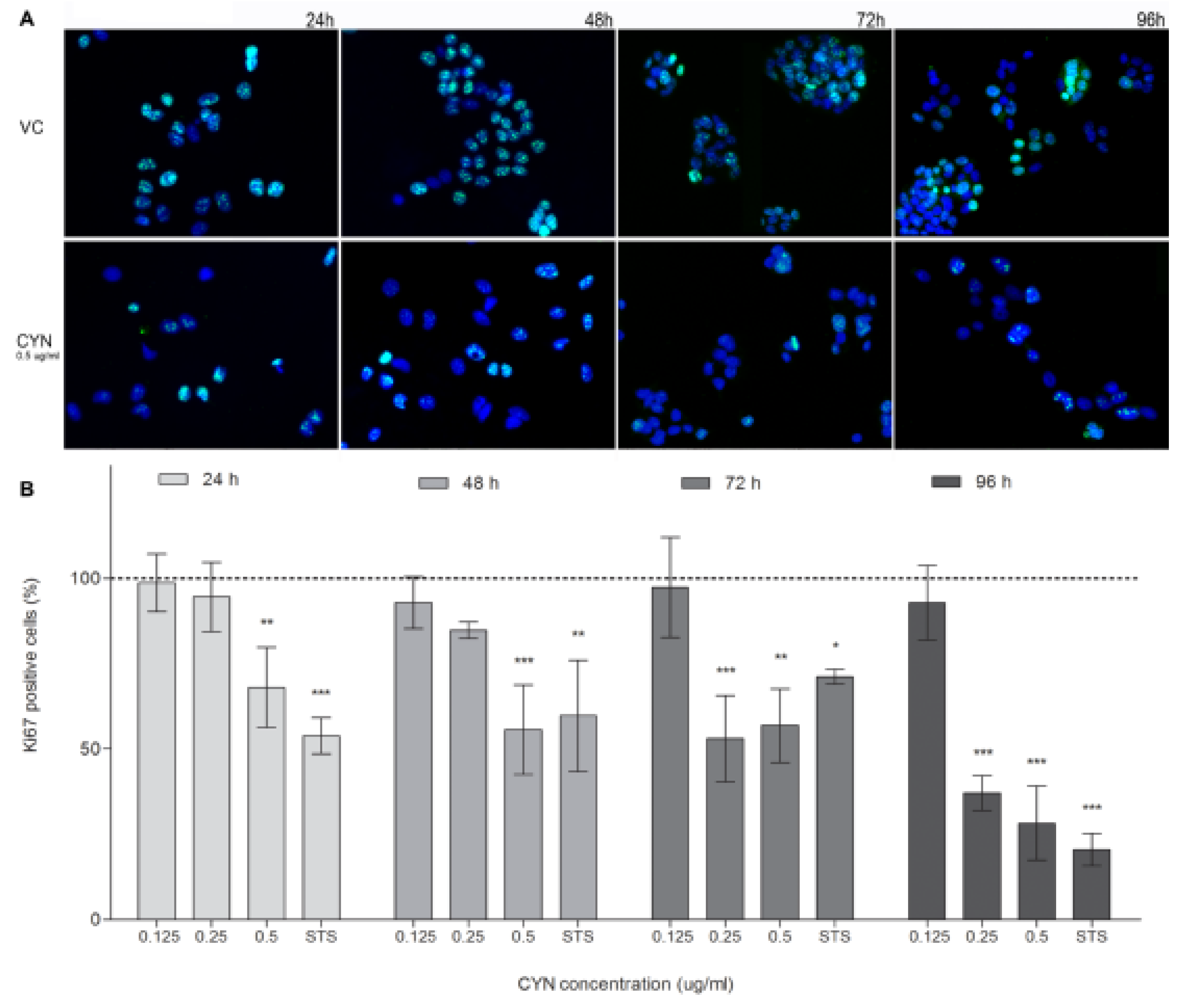
2.3. Influence of CYN on Cell Proliferation
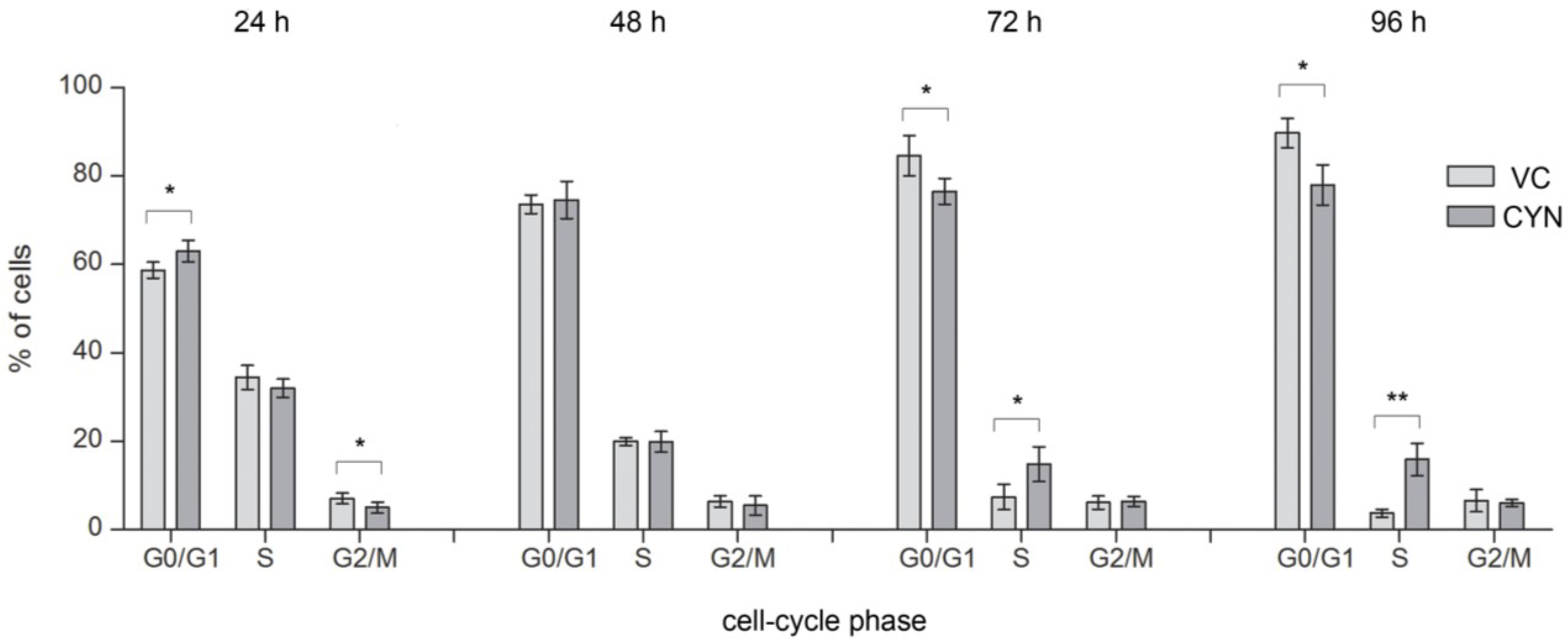
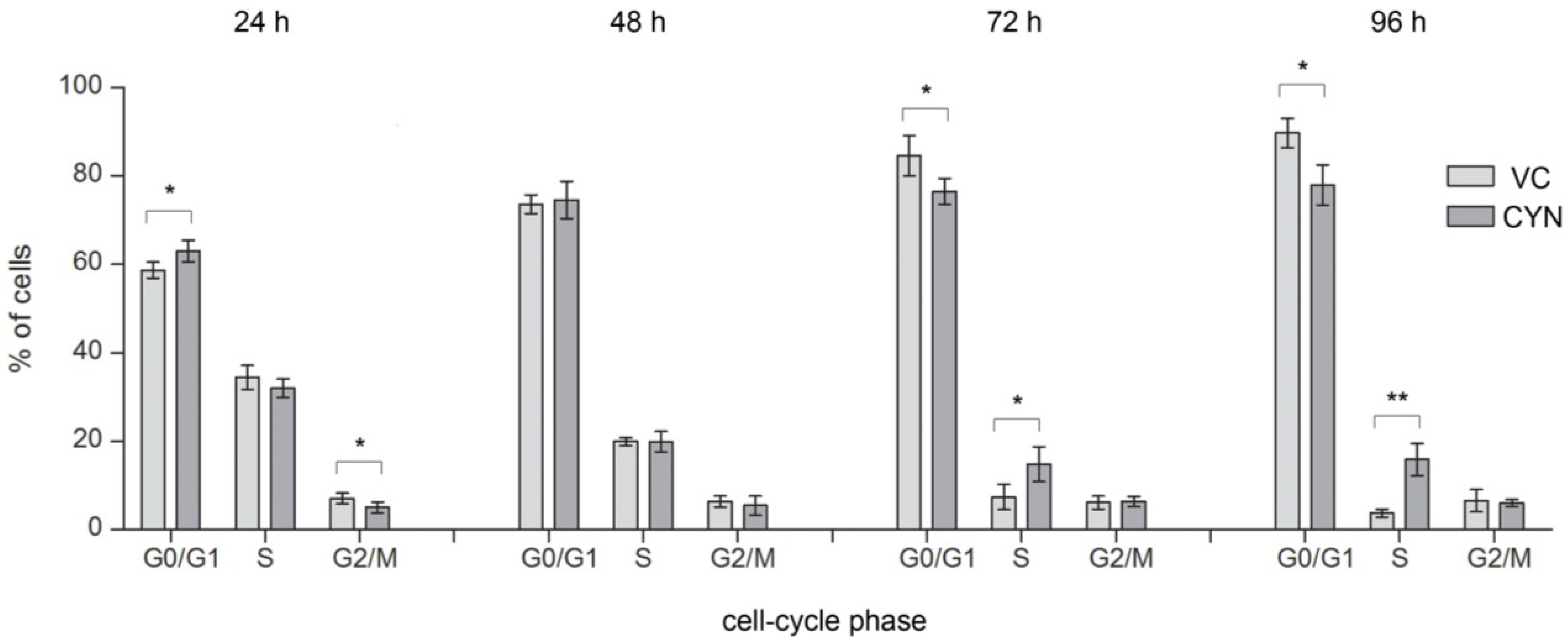
2.4. Influence on the Cell-Cycle Progression
3. Experimental Section
3.1. Chemicals
3.2. Cell Culture
3.3. Cell Viability—MTT Assay
3.4. Lactate Dehydrogenase (LDH) Leakage
3.5. DSB Detection—H2AX Foci Analysis
3.6. Cell-Proliferation—Immunocytochemical Staining of the Proliferation Marker Ki67
3.7. Cell-Cycle Analysis by Flow Cytometry
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Moreira, C.; Azevedo, J.; Antunes, A.; Vasconcelos, V. Cylindrospermopsin: Occurrence, methods of detection and toxicology. J. Appl. Microbiol. 2013, 114, 605–620. [Google Scholar] [CrossRef]
- Ohtani, I.; Moore, R.E.; Runnegar, M.T.C. Cylindrospermopsin: A potent hepatotoxin from the blue-green alga Cylindrospermopsis raciborskii. J. Am. Chem. Soc. 1992, 114, 7941–7942. [Google Scholar] [CrossRef]
- Krohn, A.J.; Wahlbrink, T.; Prehn, J.H.M. Mitochondrial depolarization is not required for neuronal apoptosis. J. Neurosci. 1999, 19, 7394–7404. [Google Scholar]
- Žegura, B.; Štraser, A.; Filipič, M. Genotoxicity and potential carcinogenicity of cyanobacterial toxins—A review. Mutat. Res. Rev. Mutat. Res. 2011, 727, 16–41. [Google Scholar] [CrossRef]
- Hawkins, P.R.; Runnegar, M.T.; Jackson, A.R.; Falconer, I.R. Severe hepatotoxicity caused by the tropical cyanobacterium (blue-green alga) Cylindrospermopsis raciborskii (Woloszynska) Seenaya and Subba Raju isolated from a domestic water supply reservoir. Appl. Environ. Microbiol. 1985, 50, 1292–1295. [Google Scholar]
- Froscio, S.M.; Humpage, A.R.; Burcham, P.C.; Falconer, I.R. Cylindrospermopsin-induced protein synthesis inhibition and its dissociation from acute toxicity in mouse hepatocytes. Environ. Toxicol. 2003, 18, 243–251. [Google Scholar] [CrossRef]
- Bazin, E.; Mourot, A.; Humpage, A.R.; Fessard, V. Genotoxicity of a freshwater cyanotoxin, cylindrospermopsin, in two human cell lines: Caco-2 and HepaRG. Environ. Mol. Mutagen. 2010, 51, 251–259. [Google Scholar]
- Humpage, A.; Fontaine, F.; Froscio, S.; Burcham, P.; Falconer, I. Cylindrospermopsin genotoxicity and cytotoxicity: Role of cytochrome P-450 and oxidative stress. J. Toxicol. Environ. Health A 2005, 68, 739–753. [Google Scholar] [CrossRef]
- Humpage, A.R.; Fenech, M.; Thomas, P.; Falconer, I.R. Micronucleus induction and chromosome loss in transformed human white cells indicate clastogenic and aneugenic action of the cyanobacterial toxin, cylindrospermopsin. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2000, 472, 155–161. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Žegura, B. Genotoxic effects of the cyanobacterial hepatotoxin cylindrospermopsin in the HepG2 cell line. Arch. Toxicol. 2011, 85, 1617–1626. [Google Scholar] [CrossRef]
- Žegura, B.; Gajski, G.; Štraser, A.; Garaj-Vrhovac, V. Cylindrospermopsin induced DNA damage and alteration in the expression of genes involved in the response to DNA damage, apoptosis and oxidative stress. Toxicon 2011, 58, 471–479. [Google Scholar] [CrossRef]
- Shaw, G.R.; Seawright, A.A.; Moore, M.R.; Lam, P.K.S. Cylindrospermopsin, a cyanobacterial alkaloid: Evaluation of its toxicologic activity. Ther. Drug Monit. 2000, 22, 89–92. [Google Scholar] [CrossRef]
- Shen, X.; Lam, P.K.S.; Shaw, G.R.; Wickramasinghe, W. Genotoxicity investigation of a cyanobacterial toxin, cylindrospermopsin. Toxicon 2002, 40, 1499–1501. [Google Scholar] [CrossRef]
- Falconer, I.R.; Humpage, A.R. Preliminary evidence for in vivo tumour initiation by oral administration of extracts of the blue-green alga Cylindrospermopsis raciborskii containing the toxin cylindrospermopsin. Environ. Toxicol. 2001, 16, 192–195. [Google Scholar] [CrossRef]
- US Environmental Protection Agency, Creating a Cyanotoxin Target List for the Unregulated Contaminant Monitoring Ruley; US Environmental Protection Agency, Technical Service Center: Cincinnati, OH, USA, 2010.
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef]
- Salaün, P.; le Breton, M.; Morales, J.; Bellé, R.; Boulben, S.; Mulner-Lorillon, O.; Cormier, P. Signal transduction pathways that contribute to CDK1/cyclin B activation during the first mitotic division in sea urchin embryos. Exp. Cell Res. 2004, 296, 347–357. [Google Scholar] [CrossRef]
- O’Farrell, P.H. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001, 11, 512–519. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Žegura, B. Cylindrospermopsin induced transcriptional responses in human hepatoma HepG2 cells. Toxicol. In Vitro 2013, 27, 1809–1819. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Gorenc, I.; Žegura, B. The influence of cylindrospermopsin on oxidative DNA damage and apoptosis induction in HepG2 cells. Chemosphere 2013, 92, 24–30. [Google Scholar] [CrossRef]
- Fessard, V.; Bernard, C. Cell alterations but no DNA strand breaks induced in vitro by cylindrospermopsin in CHO K1 cells. Environ. Toxicol. 2003, 18, 353–359. [Google Scholar] [CrossRef]
- Lankoff, A.; Wojcik, A.; Lisowska, H.; Bialczyk, J.; Dziga, D.; Carmichael, W.W. No induction of structural chromosomal aberrations in cylindrospermopsin-treated CHO-K1 cells without and with metabolic activation. Toxicon 2007, 50, 1105–1115. [Google Scholar] [CrossRef]
- Dasika, G.K.; Lin, S.C.; Zhao, S.; Sung, P.; Tomkinson, A.; Lee, E.Y. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 1999, 18, 7883–7899. [Google Scholar]
- Habraken, Y.; Piette, J. NF-κB activation by double-strand breaks. Biochem. Pharmacol. 2006, 72, 1132–1141. [Google Scholar] [CrossRef]
- Norbury, C.J.; Hickson, I.D. Cellular Responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 367–401. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar]
- Sedelnikova, O.A.; Rogakou, E.P.; Panyutin, I.G.; Bonner, W.M. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat. Res. 2002, 158, 486–492. [Google Scholar]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Michael, D.; Oren, M. The p53 and Mdm2 families in cancer. Curr. Opin. Genet. Dev. 2002, 12, 53–59. [Google Scholar]
- Bruno, S.; Darzynkiewicz, Z. Cell cycle dependent expression and stability of the nuclear protein detected by Ki-67 antibody in HL-60 cells. Cell Prolif. 1992, 25, 31–40. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Ko, L.J.; Prives, C. p53: Puzzle and paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef]
- Bain, P.; Shaw, G.; Patel, B. Induction of p53-regulated gene expression in human cell lines exposed to the cyanobacterial toxin cylindrospermopsin. J. Toxicol. Environ. Health A 2007, 70, 1687–1693. [Google Scholar] [CrossRef]
- Shimizu, N.; Itoh, N.; Utiyama, H.; Wahl, G.M. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J. Cell Biol. 1998, 140, 1307–1320. [Google Scholar] [CrossRef]
- Yankiwski, V.; Marciniak, R.A.; Guarente, L.; Neff, N.F. Nuclear structure in normal and bloom syndrome cells. Proc. Natl. Acad. Sci. USA 2000, 97, 5214–5219. [Google Scholar] [CrossRef]
- Fenech, M. The in vitro micronucleus technique. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2000, 455, 81–95. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Zegura, B.; Zajc, I.; Lah, T.T.; Filipic, M. Patterns of microcystin-LR induced alteration of the expression of genes involved in response to DNA damage and apoptosis. Toxicon 2008, 51, 615–623. [Google Scholar] [CrossRef]
- FCSExtract Utility for Flow Cytometry Standard (FCS) Data. Earl F Glynn Stowers Institute for Medical Research. Available online: http://research.stowers-institute.org/efg/ScientificSoftware/Utility/FCSExtract/index.htm (accessed on 18 March 2013).
- Hreljac, I.; Zajc, I.; Lah, T.; Filipič, M. Effects of model organophosphorous pesticides on DNA damage and proliferation of HepG2 cells. Environ. Mol. Mutagen. 2008, 49, 360–367. [Google Scholar] [CrossRef]
- R Development Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012.
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D. R Development Core Team (2012) nlme: Linear and Nonlinear Mixed Effects Models; R Package Version 3.1-103; R Foundation for Statistical Computing: Vienna, Australia, 2012. [Google Scholar]
- Klitzke, S.; Fastner, J. Cylindrospermopsin degradation in sediments—The role of temperature, redox conditions, and dissolved organic carbon. Water Res. 2012, 46, 1549–1555. [Google Scholar] [CrossRef]
- Saker, M.L.; Eaglesham, G.K. The accumulation of cylindrospermopsin from the cyanobacterium Cylindrospermopsis raciborskii in tissues of the Redclaw crayfish Cherax quadricarinatus. Toxicon 1999, 37, 1065–1077. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alja, Š.; Filipič, M.; Novak, M.; Žegura, B. Double Strand Breaks and Cell-Cycle Arrest Induced by the Cyanobacterial Toxin Cylindrospermopsin in HepG2 Cells. Mar. Drugs 2013, 11, 3077-3090. https://doi.org/10.3390/md11083077
Alja Š, Filipič M, Novak M, Žegura B. Double Strand Breaks and Cell-Cycle Arrest Induced by the Cyanobacterial Toxin Cylindrospermopsin in HepG2 Cells. Marine Drugs. 2013; 11(8):3077-3090. https://doi.org/10.3390/md11083077
Chicago/Turabian StyleAlja, Štraser, Metka Filipič, Matjaž Novak, and Bojana Žegura. 2013. "Double Strand Breaks and Cell-Cycle Arrest Induced by the Cyanobacterial Toxin Cylindrospermopsin in HepG2 Cells" Marine Drugs 11, no. 8: 3077-3090. https://doi.org/10.3390/md11083077