
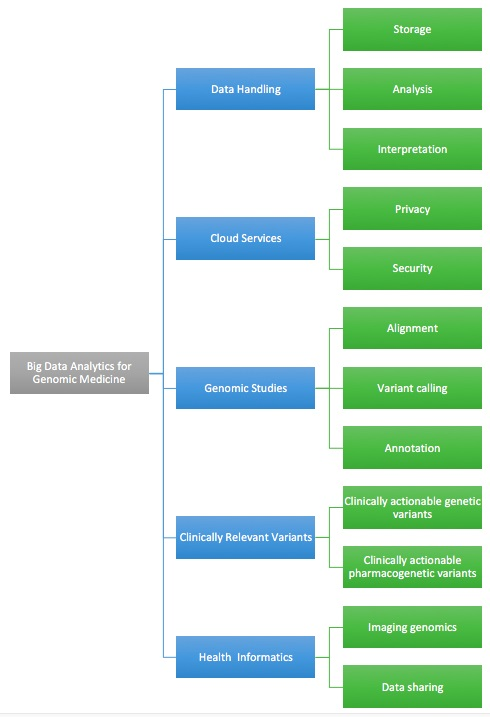
Big Data Analytics for Genomic Medicine

Abstract
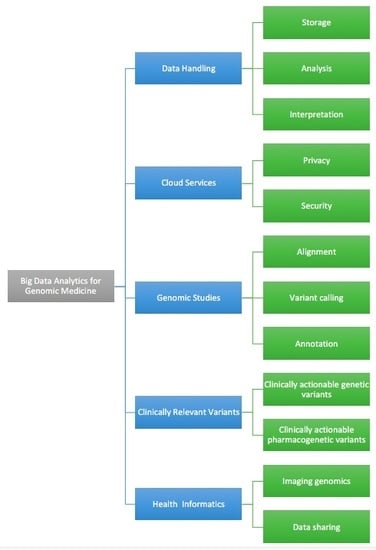
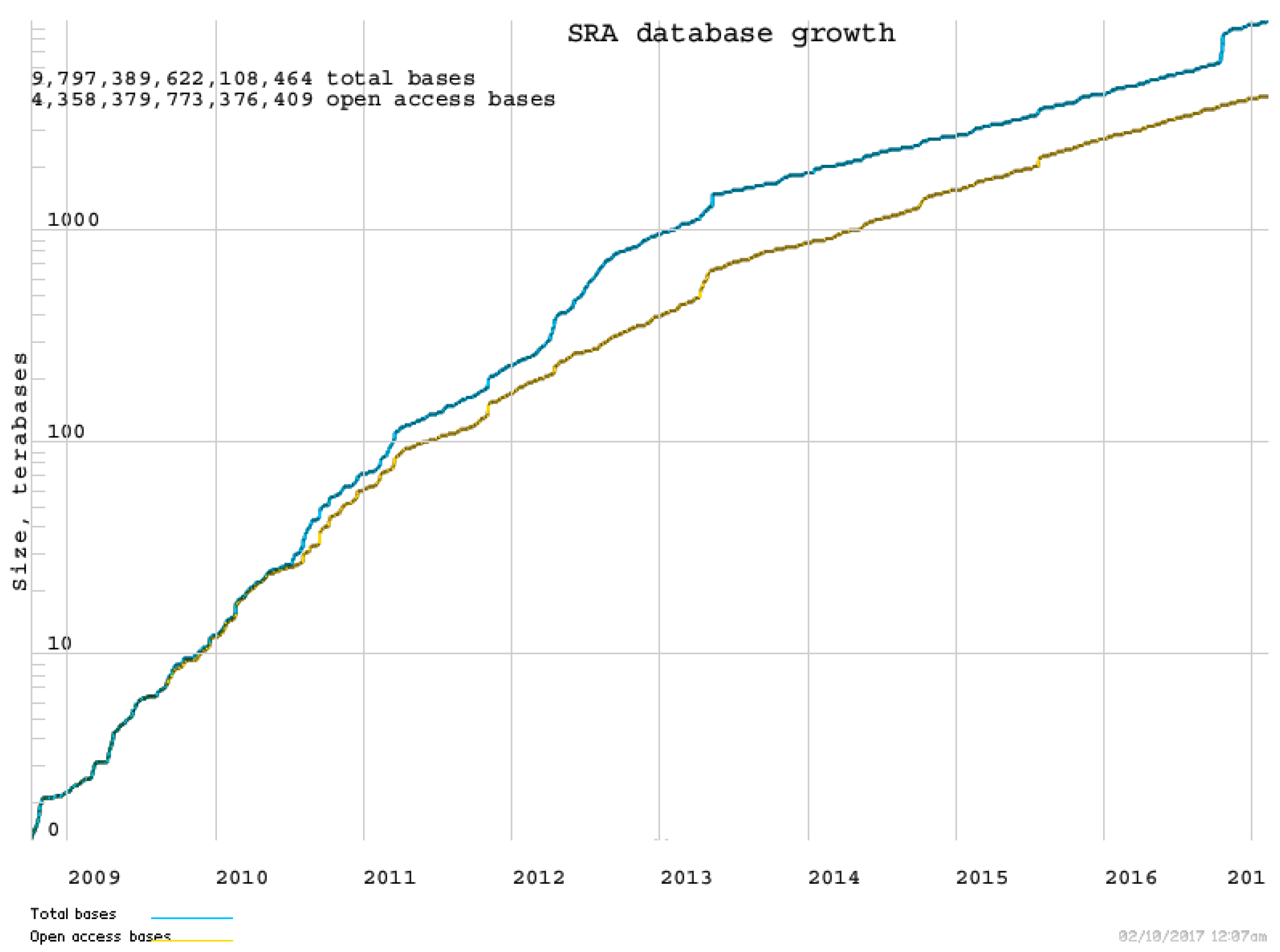
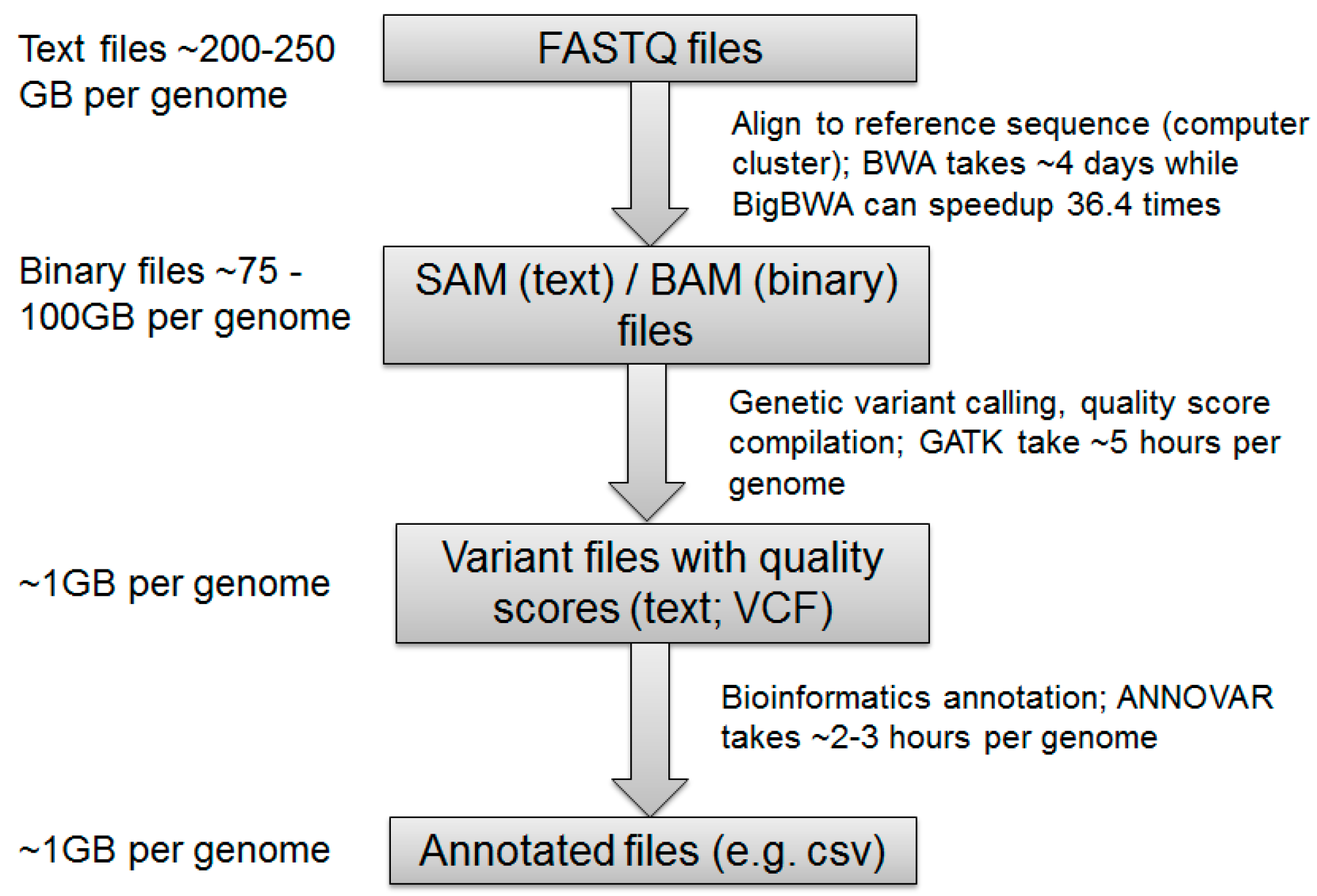
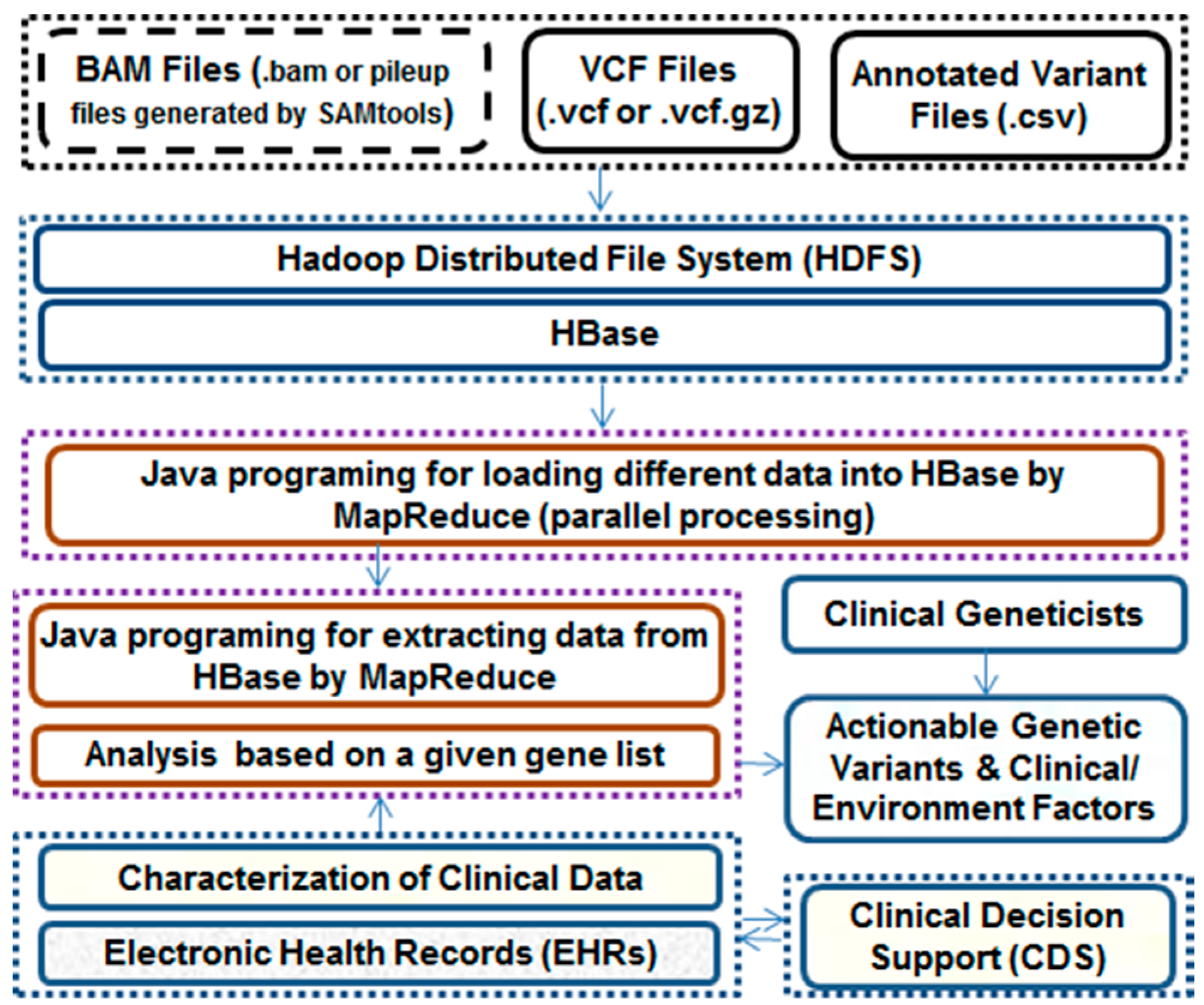
:
1. Introduction
2. Challenges of Handling Genomic and Clinical Data
2.1. Challenges in Manipulating Genomic Data
2.2. Challenges in Manipulating Clinical Data
3. Big Data on the Cloud
3.1. Cloud Computing
3.2. Privacy and Security Challenges of Cloud Computing
4. Big Data Analytics in Genomic Studies
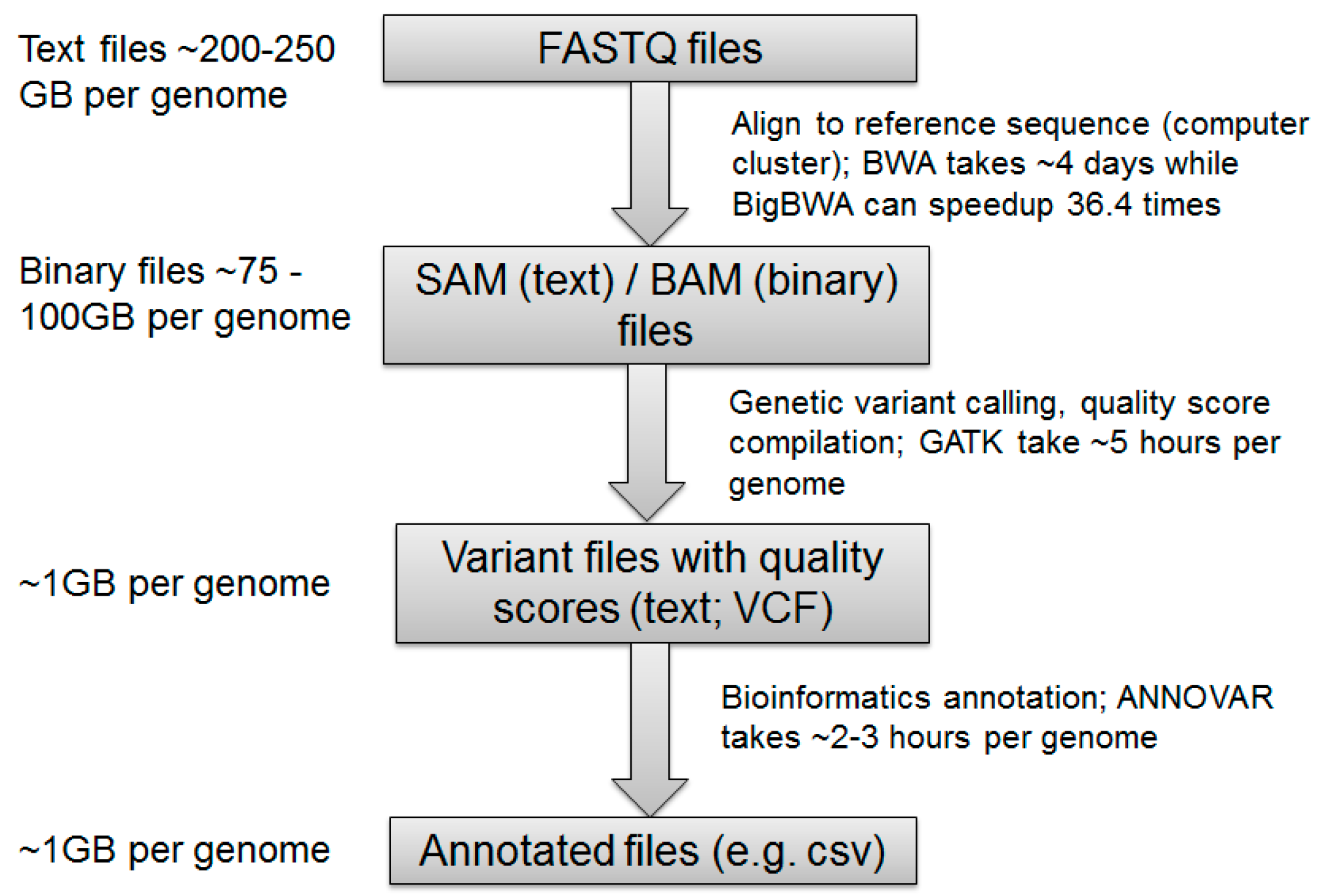
4.1. NGS Read Alignment
4.2. Calling Variants
4.3. Variant Annotation
4.4. Statistical Analysis of Genomic Data
4.5. Security of Genomic Data
5. Analysis of Genomic and Clinical Data
5.1. Clinically Actionable Genetic Variants
5.2. Clinically Actionable Pharmacogenetic Variants
6. Big Data Analytics in Health Research
6.1. Health Informatics
6.2. Medical Imaging Analysis
6.3. Data Sharing
7. Discussion
8. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.C.; He, M.M. Challenges of identifying clinically actionable genetic variants for precision medicine. J. Healthc. Eng. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Vassy, J.L.; Korf, B.R.; Green, R.C. How to know when physicians are ready for genomic medicine. Sci. Transl. Med. 2015, 7, 287fs219. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Hayden, M.R. Hunting human disease genes: Lessons from the past, challenges for the future. Hum. Genet. 2013, 132, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, O.; Kuivaniemi, H.; Tromp, G.; Faucett, W.A.; Li, R.; Manolio, T.A.; Sanderson, S.C.; Kannry, J.; Zinberg, R.; Basford, M.A.; et al. The Electronic Medical Records and Genomics (eMERGE) Network: Past, present, and future. Genet. Med. 2013, 15, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, R.R.; Lyons-Weiler, M.; Petrosko, P.; Dhir, R.; Becich, M.J.; LaFramboise, W.A. Clinical integration of next-generation sequencing technology. Clin. Lab. Med. 2012, 32, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Baro, E.; Degoul, S.; Beuscart, R.; Chazard, E. Toward a literature-driven definition of big data in healthcare. BioMed Res. Int. 2015, 2015, 639021. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Jing, S.; Yi, J.; Zhen, W. Innovative Testing and Measurement Solutions for Smart Grid; John Wiley & Sons: Singapore, 2015. [Google Scholar]
- Raghupathi, W.; Raghupathi, V. Big data analytics in healthcare: Promise and potential. Health Inf. Sci. Syst. 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Chute, C.G.; Ullman-Cullere, M.; Wood, G.M.; Lin, S.M.; He, M.; Pathak, J. Some experiences and opportunities for big data in translational research. Genet. Med. 2013, 15, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Gulcher, J.R.; Jonsson, P.; Kong, A.; Kristjansson, K.; Frigge, M.L.; Karason, A.; Einarsdottir, I.E.; Stefansson, H.; Einarsdottir, A.S.; Sigurthoardottir, S.; et al. Mapping of a familial essential tremor gene, FET1, to chromosome 3q13. Nat. Genet. 1997, 17, 84–87. [Google Scholar] [CrossRef] [PubMed]
- McCarty, C.A.; Nair, A.; Austin, D.M.; Giampietro, P.F. Informed consent and subject motivation to participate in a large, population-based genomics study: The marshfield clinic personalized medicine research project. Community Genet. 2007, 10, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Butte, A.J.; Kohane, I.S. Creation and implications of a phenome-genome network. Nat. Biotechnol. 2006, 24, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lee, L.; Chen, J.; Collins, R.; Wu, F.; Guo, Y.; Linksted, P.; Peto, R. Cohort profile: The Kadoorie Study of Chronic Disease in China (KSCDC). Int. J. Epidemiol. 2005, 34, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen-Torvik, L.J.; Stallings, S.C.; Gordon, A.S.; Almoguera, B.; Basford, M.A.; Bielinski, S.J.; Brautbar, A.; Brilliant, M.H.; Carrell, D.S.; Connolly, J.J.; et al. Design and anticipated outcomes of the eMERGE-PGx project: A multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin. Pharmacol. Ther. 2014, 96, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Pong-Wong, R.; Canela-Xandri, O.; Rawlik, K.; Haley, C.S.; Tenesa, A. Evaluating the contribution of genetics and familial shared environment to common disease using the UK biobank. Nat. Genet. 2016, 48, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Grabe, H.J.; Assel, H.; Bahls, T.; Dorr, M.; Endlich, K.; Endlich, N.; Erdmann, P.; Ewert, R.; Felix, S.B.; Fiene, B.; et al. Cohort profile: Greifswald approach to individualized medicine (GANI_MED). J. Transl. Med. 2014, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Kvale, M.N.; Hesselson, S.E.; Zhan, Y.; Aquino, C.; Cao, Y.; Cawley, S.; Chung, E.; Connell, S.; Eshragh, J.; et al. Next generation genome-wide association tool: Design and coverage of a high-throughput European-optimized SNP array. Genomics 2011, 98, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Saal, L.H.; Vallon-Christersson, J.; Hakkinen, J.; Hegardt, C.; Grabau, D.; Winter, C.; Brueffer, C.; Tang, M.H.; Reutersward, C.; Schulz, R.; et al. The Sweden Cancerome Analysis Network—Breast (SCAN-B) initiative: A large-scale multicenter infrastructure towards implementation of breast cancer genomic analyses in the clinical routine. Genome Med. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Postmus, I.; Trompet, S.; Deshmukh, H.A.; Barnes, M.R.; Li, X.; Warren, H.R.; Chasman, D.I.; Zhou, K.; Arsenault, B.J.; Donnelly, L.A.; et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat. Commun. 2014, 5, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiber, G.E.; LaCroix, A.Z. Older women veterans in the women’s health initiative. Gerontologist 2016, 56 (Suppl. 1), S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Fellowes, A.; Doig, K.; Ellul, J.; Bosma, T.J.; Irwin, D.; Vedururu, R.; Tan, A.Y.; Weiss, J.; Chan, K.S.; et al. Assessing the clinical value of targeted massively parallel sequencing in a longitudinal, prospective population-based study of cancer patients. Br. J. Cancer 2015, 112, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. Clingen—The clinical genome resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Abuin, J.M.; Pichel, J.C.; Pena, T.F.; Amigo, J. Bigbwa: Approaching the burrows-wheeler aligner to big data technologies. Bioinformatics 2015, 31, 4003–4005. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Person, T.N.; Hebbring, S.J.; Heinzen, E.; Ye, Z.; Schrodi, S.J.; McPherson, E.W.; Lin, S.M.; Peissig, P.L.; Brilliant, M.H.; et al. Seqhbase: A big data toolset for family based sequencing data analysis. J. Med. Genet. 2015, 52, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Veltman, J.A.; Gilissen, C. Novel bioinformatic developments for exome sequencing. Hum. Genet. 2016, 135, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Jamoom, E.W.; Yang, N.; Hing, E. Adoption of certified electronic health record systems and electronic information sharing in physician offices: United states, 2013 and 2014. NCHS Data Brief 2016, 236, 1–8. [Google Scholar]
- Slee, V.N. The international classification of diseases: Ninth revision (ICD-9). Ann. Intern. Med. 1978, 88, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Wojczynski, M.K.; Tiwari, H.K. Definition of phenotype. Adv. Genet. 2008, 60, 75–105. [Google Scholar] [PubMed]
- Rice, J.P.; Saccone, N.L.; Rasmussen, E. Definition of the phenotype. Adv. Genet. 2001, 42, 69–76. [Google Scholar] [PubMed]
- Gurwitz, D.; Pirmohamed, M. Pharmacogenomics: The importance of accurate phenotypes. Pharmacogenomics 2010, 11, 469–470. [Google Scholar] [CrossRef] [PubMed]
- Samuels, D.C.; Burn, D.J.; Chinnery, P.F. Detecting new neurodegenerative disease genes: Does phenotype accuracy limit the horizon? Trends Genet. 2009, 25, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Richesson, R.L.; Sun, J.; Pathak, J.; Kho, A.N.; Denny, J.C. Clinical phenotyping in selected national networks: Demonstrating the need for high-throughput, portable, and computational methods. Artif. Intell. Med. 2016, 71, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.N.; Pacheco, J.A.; Peissig, P.L.; Rasmussen, L.; Newton, K.M.; Weston, N.; Crane, P.K.; Pathak, J.; Chute, C.G.; Bielinski, S.J.; et al. Electronic medical records for genetic research: Results of the emerge consortium. Sci. Transl. Med. 2011, 3, 79re71. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Tafti, A.P.; He, K.Y.; Wang, K.; He, M.M. Sparktext: Biomedical text mining on big data framework. PLoS ONE 2016, 11, e0162721. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, A.; Daugelaite, J.; Sleator, R.D. ‘Big data’, Hadoop and cloud computing in genomics. J. Biomed. Inf. 2013, 46, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.J.; de la Torre, I.; Fernandez, G.; Lopez-Coronado, M. Analysis of the security and privacy requirements of cloud-based electronic health records systems. J. Med. Internet Res. 2013, 15, e186. [Google Scholar] [CrossRef] [PubMed]
- Takabi, H.; Joshi, J.B.D.; Ahn, G.-J. Security and privacy challenges in cloud computing environments. IEEE Secur. Priv. 2010, 8, 24–31. [Google Scholar] [CrossRef]
- Calder, B.; Wang, J.; Ogus, A.; Nilakantan, N.; Skjolsvold, A.; McKelvie, S.; Xu, Y.; Srivastav, S.; Wu, J.; Simitci, H.; et al. Windows azure storage: A highly available cloud storage service with strong consistency. In Proceedings of the Twenty-Third ACM Symposium on Operating Systems Principles, Cascais, Portugal, 23–26 October 2011; pp. 143–157.
- Fusaro, V.A.; Patil, P.; Gafni, E.; Wall, D.P.; Tonellato, P.J. Biomedical cloud computing with Amazon Web Services. PLoS Comput. Biol. 2011, 7, e1002147. [Google Scholar] [CrossRef] [PubMed]
- Kong, J. A practical approach to improve the data privacy of virtual machines. In Proceedings of the 2010 10th IEEE International Conference on Computer and Information Technology, Bradford, UK, 29 June–1 July 2010; pp. 936–941.
- Aziz, A.; Kawamoto, K.; Eilbeck, K.; Williams, M.S.; Freimuth, R.R.; Hoffman, M.A.; Rasmussen, L.V.; Overby, C.L.; Shirts, B.H.; Hoffman, J.M.; et al. The genomic CDS sandbox: An assessment among domain experts. J. Biomed. Inf. 2016, 60, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Waterman, M.S. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics 1988, 2, 231–239. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Church, D.M.; Schneider, V.A.; Steinberg, K.M.; Schatz, M.C.; Quinlan, A.R.; Chin, C.S.; Kitts, P.A.; Aken, B.; Marth, G.T.; Hoffman, M.M.; et al. Extending reference assembly models. Genome Biol. 2015, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.C. Cloudburst: Highly sensitive read mapping with mapreduce. Bioinformatics 2009, 25, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Schatz, M.C.; Lin, J.; Pop, M.; Salzberg, S.L. Searching for snps with cloud computing. Genome Biol. 2009, 10, R134. [Google Scholar] [CrossRef] [PubMed]
- Pireddu, L.; Leo, S.; Zanetti, G. Seal: A distributed short read mapping and duplicate removal tool. Bioinformatics 2011, 27, 2159–2160. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.J.; Chen, R.; Lam, H.Y.; Karczewski, K.J.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011, 29, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Ajay, S.S.; Parker, S.C.; Abaan, H.O.; Fajardo, K.V.; Margulies, E.H. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011, 21, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wu, Y.; Narzisi, G.; O’Rawe, J.A.; Barron, L.T.; Rosenbaum, J.; Ronemus, M.; Iossifov, I.; Schatz, M.C.; Lyon, G.J. Reducing indel calling errors in whole genome and exome sequencing data. Genome Med. 2014, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. Available online: http://arxiv.org/abs/1207.3907 (accessed on 15 October 2016).
- Evani, U.S.; Challis, D.; Yu, J.; Jackson, A.R.; Paithankar, S.; Bainbridge, M.N.; Jakkamsetti, A.; Pham, P.; Coarfa, C.; Milosavljevic, A.; et al. Atlas2 Cloud: A framework for personal genome analysis in the cloud. BMC Genom. 2012, 13 (Suppl. 6), S19. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. Genomics 2012. [Google Scholar]
- Bao, R.; Huang, L.; Andrade, J.; Tan, W.; Kibbe, W.A.; Jiang, H.; Feng, G. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inf. 2014, 13, 67–82. [Google Scholar]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 15 October 2016).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Mark, A.; Afrasiabi, C.; Tsueng, G.; Juchler, M.; Gopal, N.; Stupp, G.S.; Putman, T.E.; Ainscough, B.J.; Griffith, O.L.; et al. High-performance web services for querying gene and variant annotation. Genome Biol. 2016, 17, 91. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. Clinvar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Stephens, Z.D.; Lee, S.Y.; Faghri, F.; Campbell, R.H.; Zhai, C.; Efron, M.J.; Iyer, R.; Schatz, M.C.; Sinha, S.; Robinson, G.E. Big data: Astronomical or genomical? PLoS Biol. 2015, 13, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Hazin, R.; Brothers, K.B.; Malin, B.A.; Koenig, B.A.; Sanderson, S.C.; Rothstein, M.A.; Williams, M.S.; Clayton, E.W.; Kullo, I.J. Ethical, legal, and social implications of incorporating genomic information into electronic health records. Genet. Med. 2013, 15, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.B.; Kaye, J.; Terry, S.F. Governance through privacy, fairness, and respect for individuals. EGEMS 2016, 4, 1207. [Google Scholar] [PubMed]
- The Workgroup for Electronic Data Interchange. Issues and Trends in Electronic Genomic Data Exchange; The Workgroup for Electronic Data Interchange: Washington, DC, USA, 2015. [Google Scholar]
- Department of Health and Human Services Office of the Secretary. Modifications to the HIPAA Privacy, Security, Enforcement, and Breach Notification Rules under the Health Information Technology for Economic and Clinical Health Act and the Genetic Information Nondiscrimination Act; Department of Health and Human Services, Ed.; Federal Register: Washington, DC, USA, 2013.
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Daneshjou, R.; Zappala, Z.; Kukurba, K.; Boyle, S.M.; Ormond, K.E.; Klein, T.E.; Snyder, M.; Bustamante, C.D.; Altman, R.B.; Montgomery, S.B. Path-scan: A reporting tool for identifying clinically actionable variants. Pac. Symp. Biocomput. 2014, 229–240. [Google Scholar]
- Zhou, W.; Zhao, H.; Chong, Z.; Mark, R.J.; Eterovic, A.K.; Meric-Bernstam, F.; Chen, K. Clinsek: A targeted variant characterization framework for clinical sequencing. Genome Med. 2015, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Wells, Q.S.; Stallings, S.; Bush, W.S.; Gordon, A.; Nickerson, D.A.; Kim, J.H.; Crosslin, D.R.; Jarvik, G.P.; Carrell, D.S.; et al. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. J. Am. Med. Assoc. 2016, 315, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. Long QT syndrome and potentially pathogenic genetic variants. J. Am. Med. Assoc. 2016, 315, 2467. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- He, K.Y.; Zhao, Y.; McPherson, E.W.; Li, Q.; Xia, F.; Weng, C.; Wang, K.; He, M.M. Pathogenic mutations in cancer-predisposing genes: A survey of 300 patients with whole-genome sequencing and lifetime electronic health records. PLoS ONE 2016, 11, e0167847. [Google Scholar] [CrossRef] [PubMed]
- Gharani, N.; Keller, M.A.; Stack, C.B.; Hodges, L.M.; Schmidlen, T.J.; Lynch, D.E.; Gordon, E.S.; Christman, M.F. The coriell personalized medicine collaborative pharmacogenomics appraisal, evidence scoring and interpretation system. Genome Med. 2013, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Klein, T.E. CPIC: Clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Teutsch, S.M.; Bradley, L.A.; Palomaki, G.E.; Haddow, J.E.; Piper, M.; Calonge, N.; Dotson, W.D.; Douglas, M.P.; Berg, A.O. The evaluation of genomic applications in practice and prevention (EGAPP) initiative: Methods of the EGAPP working group. Genet. Med. 2009, 11, 3–14. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. Sift web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Baucom, A.; Mukhyala, K.; Manning, G.; Zhang, Z. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genom. 2013, 14 (Suppl. 3), S7. [Google Scholar]
- Flanagan, S.E.; Patch, A.M.; Ellard, S. Using sift and polyphen to predict loss-of-function and gain-of-function mutations. Genet. Test. Mol. Biomark. 2010, 14, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Mazza, T. Congruency in the prediction of pathogenic missense mutations: State-of-the-art web-based tools. Brief. Bioinform. 2013, 14, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm (accessed on 30 July 2016).
- The Clinical Pharmacogenetics Implementation Consortium (CPIC). Available online: https://www.pharmgkb.org/ (accessed on 30 July 2016).
- Peters, S.G.; Buntrock, J.D. Big data and the electronic health record. J. Ambul. Care Manag. 2014, 37, 206–210. [Google Scholar] [CrossRef] [PubMed]
- DeFrances, C. Electronic Health Records and “Big Data” for Health Care. Available online: http://www.cdc.gov/nchs/data/bsc/bscpres_defrances_may_2016.pdf (accessed on 15 October 2016).
- Kitchen, R.R.; Rozowsky, J.S.; Gerstein, M.B.; Nairn, A.C. Decoding neuroproteomics: Integrating the genome, translatome and functional anatomy. Nat. Neurosci. 2014, 17, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Laird, A.R.; Eickhoff, S.B.; Fox, P.M.; Uecker, A.M.; Ray, K.L.; Saenz, J.J., Jr.; McKay, D.R.; Bzdok, D.; Laird, R.W.; Robinson, J.L.; et al. The brainmap strategy for standardization, sharing, and meta-analysis of neuroimaging data. BMC Res. Notes 2011, 4, 349. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E. Functional brain imaging and human brain function. J. Neurosci. 2003, 23, 3959–3962. [Google Scholar] [PubMed]
- Albrecht, J.; Kopietz, R.; Frasnelli, J.; Wiesmann, M.; Hummel, T.; Lundstrom, J.N. The neuronal correlates of intranasal trigeminal function—An ALE meta-analysis of human functional brain imaging data. Brain Res. Rev. 2010, 62, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Glasser, M.F.; Coalson, T.S.; Robinson, E.C.; Hacker, C.D.; Harwell, J.; Yacoub, E.; Ugurbil, K.; Andersson, J.; Beckmann, C.F.; Jenkinson, M.; et al. A multi-modal parcellation of human cerebral cortex. Nature 2016, 536, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Dinov, I.; Lozev, K.; Petrosyan, P.; Liu, Z.; Eggert, P.; Pierce, J.; Zamanyan, A.; Chakrapani, S.; van Horn, J.; Parker, D.S.; et al. Neuroimaging study designs, computational analyses and data provenance using the LONI pipeline. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Alyass, A.; Turcotte, M.; Meyre, D. From big data analysis to personalized medicine for all: Challenges and opportunities. BMC Med. Genom. 2015, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Dinov, I.D. Methodological challenges and analytic opportunities for modeling and interpreting big healthcare data. GigaScience 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Stephens, K.A.; Baldwin, L.M.; Keppel, G.A.; Whitener, R.J.; Echo-Hawk, A.; Korngiebel, D. Developing governance for federated community-based EHR data sharing. AMIA Jt. Summits Transl. Sci. Proc. 2014, 2014, 71–76. [Google Scholar] [PubMed]
- Tarczy-Hornoch, P.; Amendola, L.; Aronson, S.J.; Garraway, L.; Gray, S.; Grundmeier, R.W.; Hindorff, L.A.; Jarvik, G.; Karavite, D.; Lebo, M.; et al. A survey of informatics approaches to whole-exome and whole-genome clinical reporting in the electronic health record. Genet. Med. 2013, 15, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Peissig, P.L.; Nikolai, A.; Brilliant, M. Personalized medicine. In Drug Discovery and Evaluation: Pharmacological Assays; Hock, F.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–16. [Google Scholar]
- Peterson, J.F.; Bowton, E.; Field, J.R.; Beller, M.; Mitchell, J.; Schildcrout, J.; Gregg, W.; Johnson, K.; Jirjis, J.N.; Roden, D.M.; et al. Electronic health record design and implementation for pharmacogenomics: A local perspective. Genet. Med. 2013, 15, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.M.; et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the discovehr study. Science 2016, 354. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.L.; Jain, S.K.; Levy, M.A. Integrating cancer genomic data into electronic health records. Genome Med. 2016, 8, 113. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Project | Start Year | Aims | Website | Country |
|---|---|---|---|---|
| deCODE genetics | 1996 | To utilize population-based genomic data and EHRs to investigate inherited causes of common diseases | http://www.decode.com/ [13] | USA |
| PMRP | 2002 | To enroll >20,000 participants to form a resource enabling researchers to study which genes cause diseases, which genes predict reactions to drugs, and how environment and genes work together to cause diseases | http://www.marshfieldresearch.org/chg/pmrp/ [14] | USA |
| I2B2 | 2004 | To enable clinical researchers to use existing clinical data and genomic data for discovery research; to facilitate the design of targeted therapies for individual patients with diseases having genetic origins | http://www.i2b2.org/ [15] | USA |
| CKB | 2004 | To identify the complex interplay between genes and environmental factors on the risks of common chronic diseases | http://www.ckbiobank.org/ [16] | China |
| eMERGE | 2007 | To develop methods and best strategies for utilizing EHRs for genomic research in support of implementing genomic medicine | http://emerge-network.org/ [17] | USA |
| UK Biobank | 2007 | To improve the prevention, diagnosis, and treatment of a wide range of serious and life-threatening illnesses through a collection of 500,000 volunteers' biosamples and medical records | http://www.ukbiobank.ac.uk/ [18] | UK |
| GANI_MED | 2009 | To develop targeted strategies for the prevention, diagnosis, and therapy of diseases, tailored to the specific characteristics of an individual patient or a well-defined patient group. Specifically, these strategies should improve prediction models for health and disease outcomes and also avoid inefficient therapy strategies and adverse side effects | http://www2.medizin.uni-greifswald.de/gani_med/index.php?L=1&id=603 [19] | Germany |
| KP RPGEH | 2009 | To explore the genetic and environmental factors that influence common disease | http://www.rpgeh.kaiser.org/ [20] | USA |
| SCAN-B Initiative | 2010 | To improve survival and quality of life for breast cancer patients through the introduction of gene expression and genomic tumor profiling into the clinical routine for breast cancer | http://scan.bmc.lu.se/index.php/Main_Page [21] | Sweden |
| PGPop | 2010 | To understand how a person’s genetic make-up affects his or her response to medications | http://pgpop.mc.vanderbilt.edu/ [22] | USA |
| MVP | 2011 | To enroll one million volunteers and use their clinical and genetic data to improve health care for veterans | http://www.research.va.gov/mvp/ [23] | USA |
| Cancer 2015 Study | 2015 | To classify cancers molecularly using MPS to promote more targeted treatment of cancer patients and improve patient survival and outcomes | [24] | Australia |
| Precision Medicine Initiative | 2016 | To gain better insights into the biological, environmental, and behavioral influences for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle by using the genomic and clinical data of a million Americans | https://www.nih.gov/precision-medicine-initiative-cohort-program [1] | USA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, K.Y.; Ge, D.; He, M.M. Big Data Analytics for Genomic Medicine. Int. J. Mol. Sci. 2017, 18, 412. https://doi.org/10.3390/ijms18020412
He KY, Ge D, He MM. Big Data Analytics for Genomic Medicine. International Journal of Molecular Sciences. 2017; 18(2):412. https://doi.org/10.3390/ijms18020412
Chicago/Turabian StyleHe, Karen Y., Dongliang Ge, and Max M. He. 2017. "Big Data Analytics for Genomic Medicine" International Journal of Molecular Sciences 18, no. 2: 412. https://doi.org/10.3390/ijms18020412
APA StyleHe, K. Y., Ge, D., & He, M. M. (2017). Big Data Analytics for Genomic Medicine. International Journal of Molecular Sciences, 18(2), 412. https://doi.org/10.3390/ijms18020412