A New Insight into Flowering Regulation: Molecular Basis of Flowering Initiation in Magnolia × soulangeana ‘Changchun’


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Observation of Flowering Initiation
2.3. Sample Collection
2.4. RNA Extraction and cDNA Library Construction
2.5. Illumina Sequencing, De novo Assembly, and Annotation
2.6. Gene Quantification and Differential Gene Expression Analysis
2.7. qRT-PCR Verification and Expression Analysis
2.8. Data Statistics and Analysis
3. Results
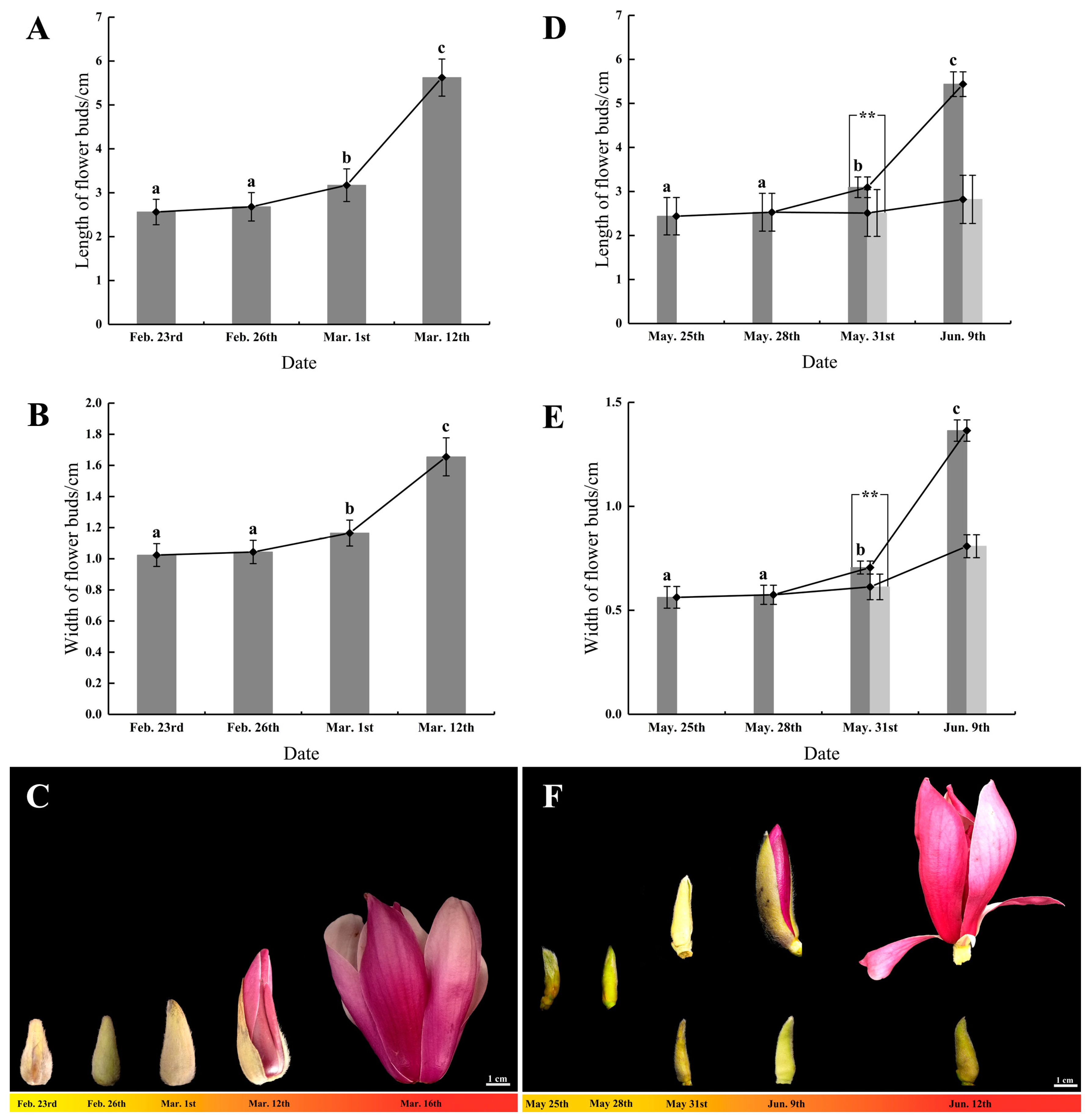
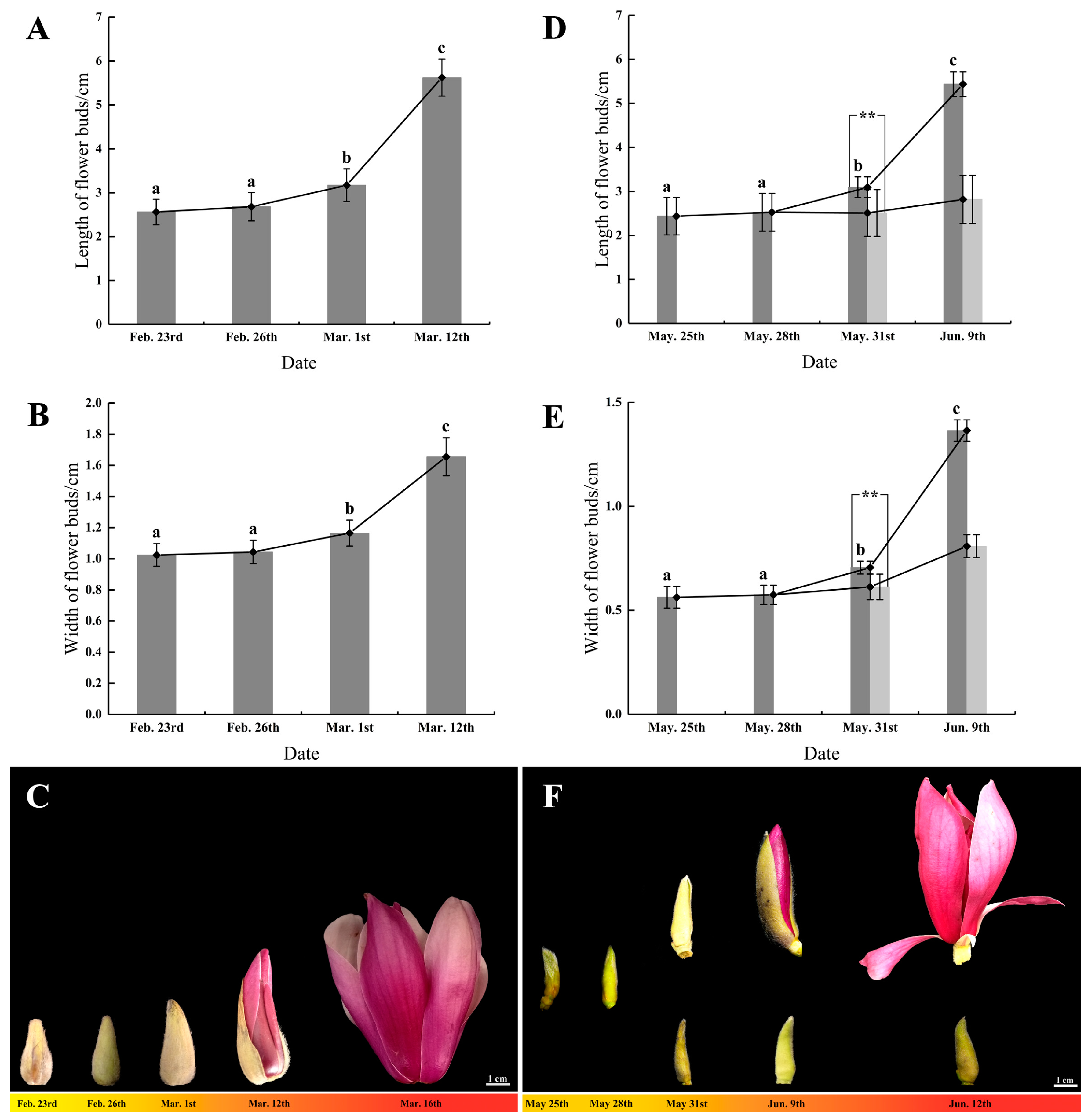
3.1. The Developmental Status of Flower Buds during Flowering Initiation
3.1.1. Flower Bud Growth and Morphological Changes in Spring Flowering
3.1.2. Flower Bud Growth and Morphological Changes in Summer Flowering
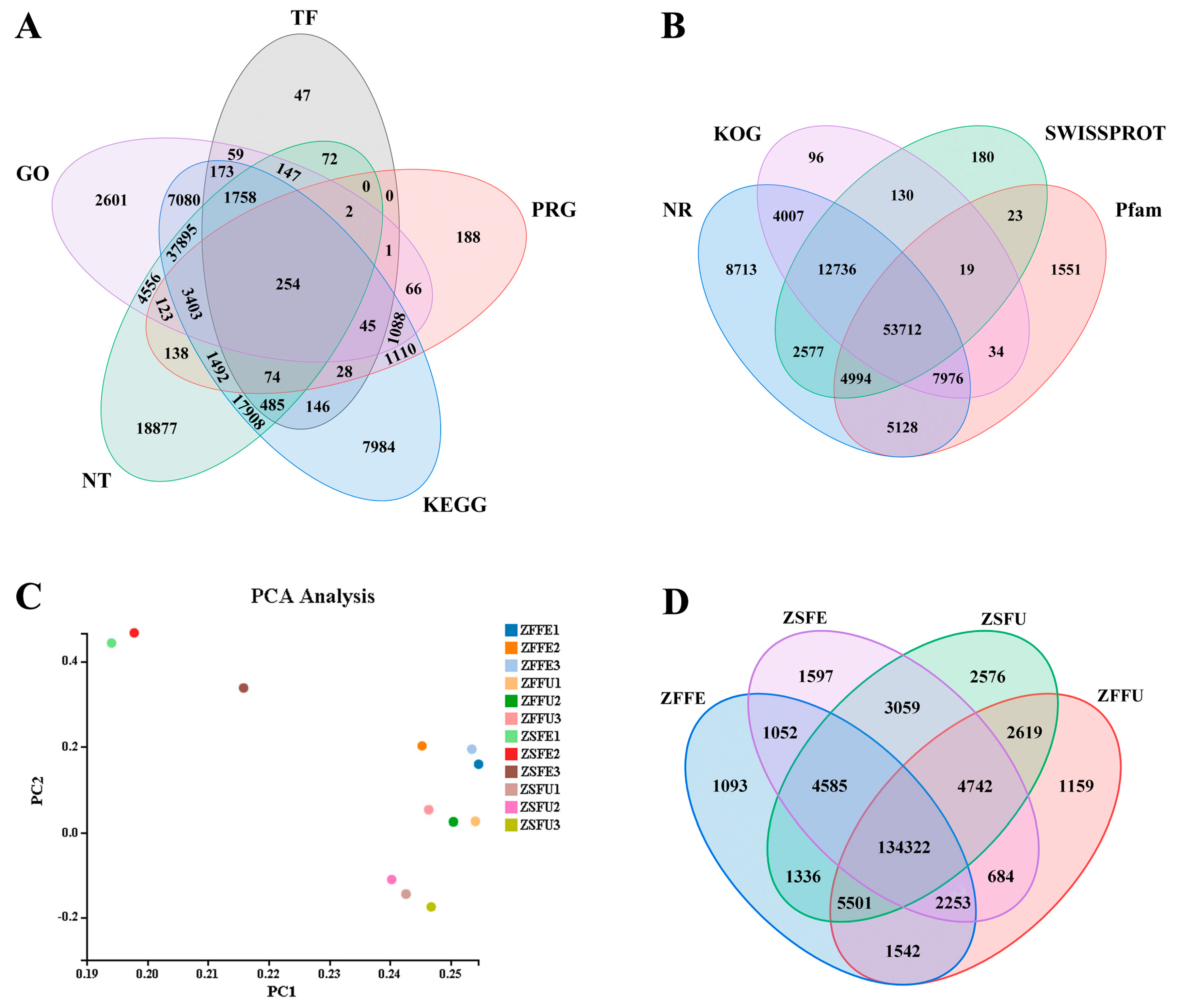
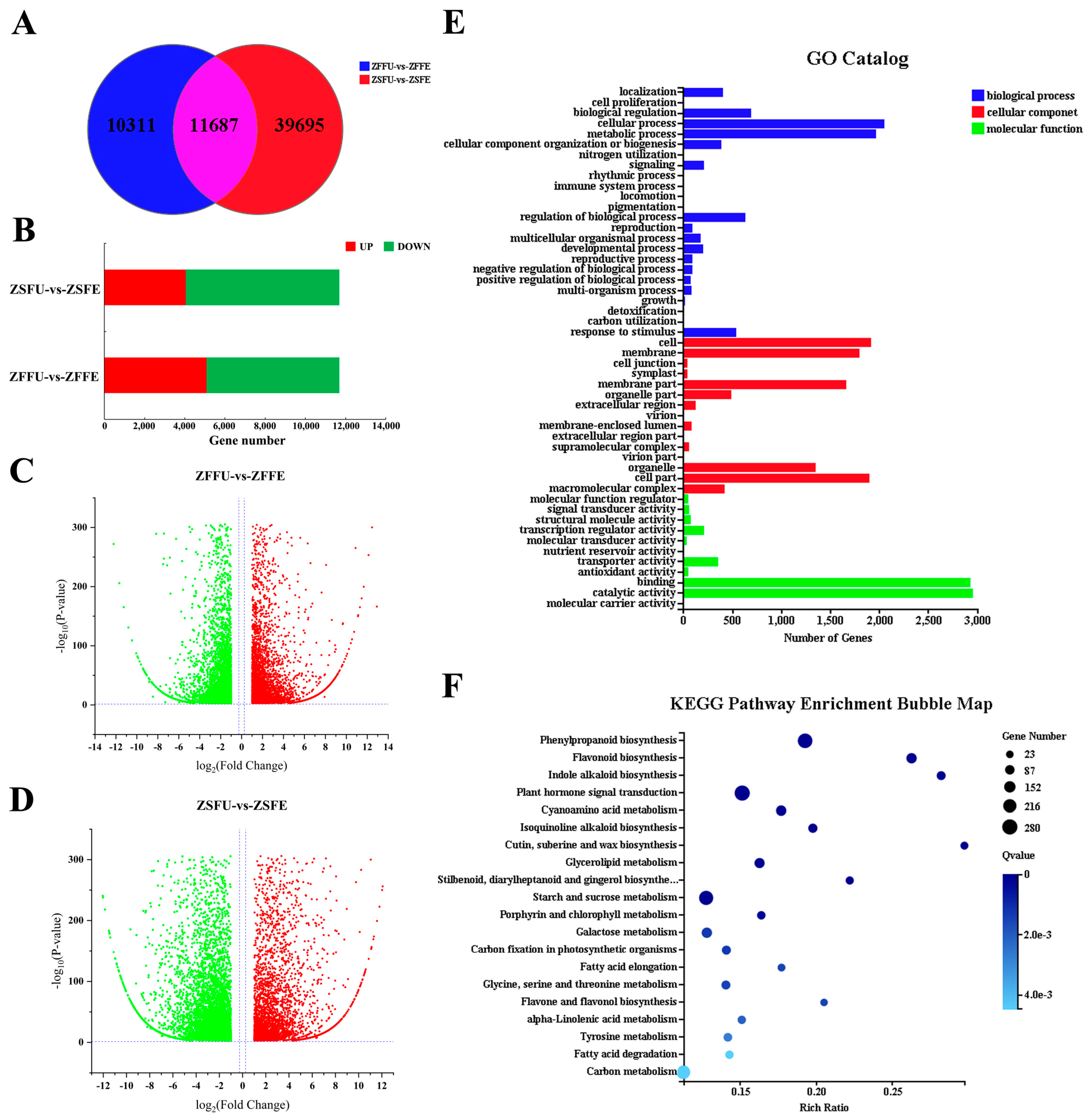
3.2. Transcriptome Sequencing and Function Annotation of Spring and Summer Flowering Initiation
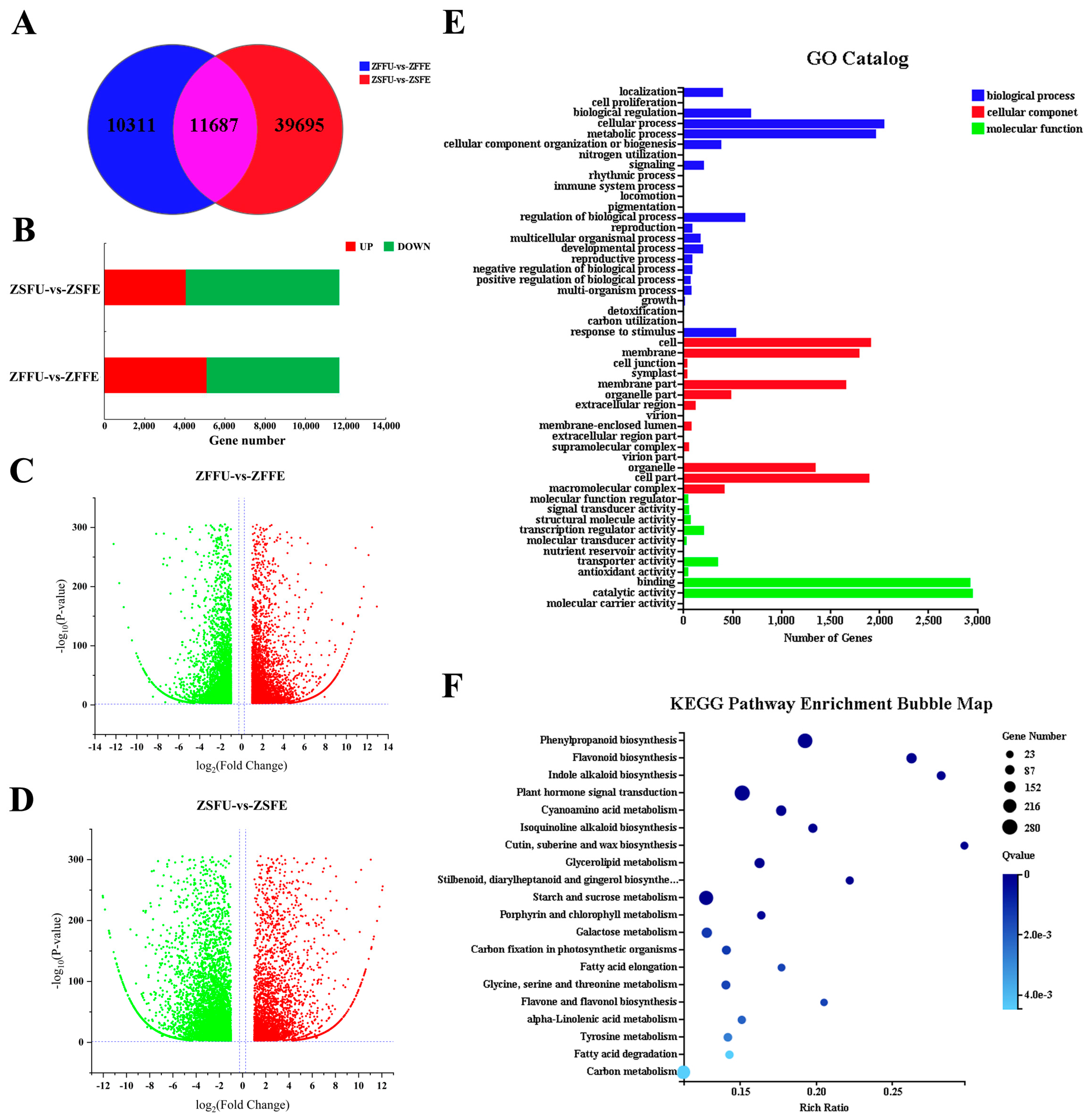
3.3. Gene Expression Analysis of Spring and Summer Flowering Initiation
3.4. Shared DEGs and Function Analysis of Spring and Summer Flowering Initiation
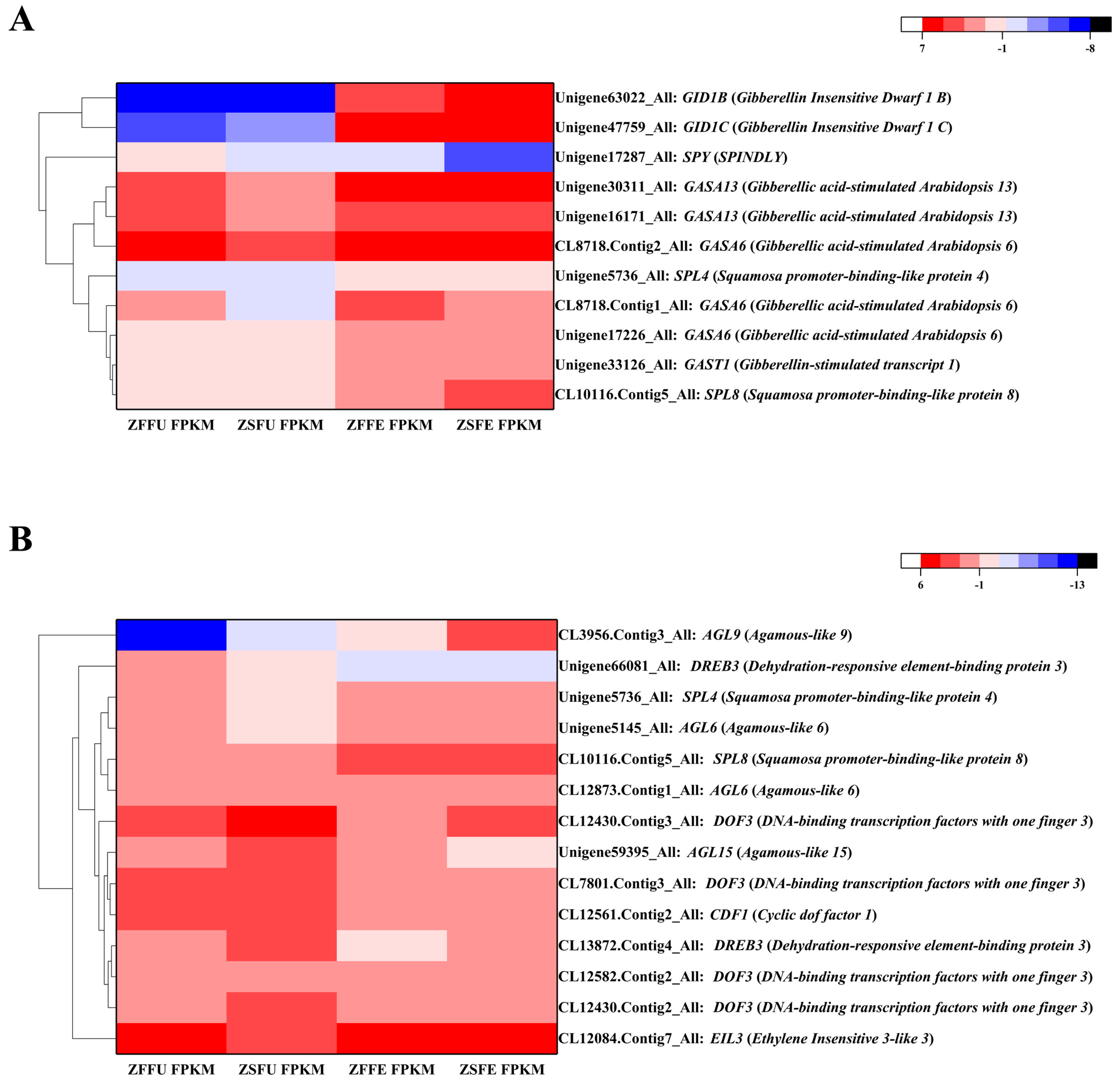
3.5. DEGs Related to Gibberellin Signaling in Spring and Summer Flowering Initiation
3.6. Related Transcription Factors in Spring and Summer Flowering Initiation
3.7. Expression Verification of Genes Related to Spring and Summer Flowering Initiation
4. Discussion
4.1. Flowering Initiation Is an Important Stage during ‘Changchun’ Flowering
4.2. Gibberellin Signaling Participates in Spring and Summer Flowering Initiation
4.3. Transcription Factors Participate in Spring and Summer Flowering Initiation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jung, C.; Pillen, K.; Staiger, D.; Coupland, G.; Korff, M. Editorial: Recent advances in flowering time control. Front. Plant Sci. 2017, 7, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.N.; Shi, S.Y.; Li, W.C.; Shu, B.; Liu, L.Q.; Xie, J.H. Transcriptome analysis of ‘Sijihua’ longan (Dimocarpus longan L.) based on next-generation sequencing technology. J. Hortic. Sci. Biotechnol. 2016, 91, 180–188. [Google Scholar] [CrossRef]
- Yu, G.; Zhou, Y.; Yu, J.J.; Hu, X.Q.; Teng, Y.; Yan, H.; Duan, J.A. Transcriptome and digital gene expression analysis unravels the novel mechanism of early fowering in Angelica sinensis. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, Y.; Zhang, C.L.; Qi, F.Y.; Li, X.P.; Mu, S.H.; Peng, Z.H. Characterization of the floral transcriptome of moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-Seq analysis. PLoS ONE 2014, 9, e98910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Qi, S.; Zhang, D.; Li, Y.; An, N.; Zhao, C.; Zhao, J.; Shah, K.; Han, M.; Xing, L. Comparative RNA-sequencing-based transcriptome profiling of buds from profusely flowering ‘Qinguan’ and weakly flowering ‘Nagafu no. 2’ apple varieties reveals novel insights into the regulatory mechanisms underlying floral induction. BMC Plant Biol. 2018, 18, 370. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Liu, L.; Huang, J.; Wang, Z.; Chen, F.; Zhang, Q.; Zheng, B.; Chen, M. Use of transcriptome sequencing to understand the pistillate flowering in hickory (Carya cathayensis Sarg.). BMC Genom. 2013, 14, 691. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.L.; Gao, J.; Xue, J.Q.; Xue, Y.Q.; Li, D.D.; Guan, Y.R.; Zhang, X.X. De novo sequencing of tree peony (Paeonia suffruticosa) transcriptome to identify critical genes involved in flowering and floral organ development. BMC Genom. 2019, 20, 572–593. [Google Scholar] [CrossRef]
- Brunner, A.M.; Evans, L.M.; Hsu, C.Y.; Shen, X. Vernalization and the chilling requirement to exit bud dormancy: Shared or separate regulation? Front. Plant Sci. 2014, 5, 732–738. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Li, Y.; Yang, Y.; Zhou, Y.; Zhao, K.; Zhang, Q. Isolation, functional characterization and evolutionary study of LFY1 gene in Prunus mume. Plant Cell Tissue Organ 2019, 136, 523–536. [Google Scholar] [CrossRef]
- Zhang, J.; Ai, X.; Sun, L.; Zhang, D.; Guo, W.; Deng, X.; Zhang, J. Transcriptome profile analysis of flowering molecular processes of early flowering trifoliate orange mutant and the wild-type [Poncirus trifoliata (L.) Raf.] by massively parallel signature sequencing. BMC Genom. 2011, 12, 63–82. [Google Scholar] [CrossRef] [Green Version]
- Horvath, D. Common mechanisms regulate flowering and dormancy. Plant Sci. 2009, 177, 523–531. [Google Scholar] [CrossRef]
- Sakamoto, D.; Nakamura, Y.; Sugiura, H.; Sugiura, T.; Asakura, T. Effect of 9-hydroxy-10-oxo-12(Z), 15(Z)-octadecadienoic acid (KODA) on endodormancy breaking in flower buds of Japanese pear. Hortscience 2010, 45, 1470–1474. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Tomes, S.; Karunairetnam, S.; Tustin, S.; Hellens, R.; Allan, A.; Macknight, R.; Varkonyi-Gasic, E. SVP-like MADS Box genes control dormancy and budbreak in apple. Front. Plant Sci. 2017, 8, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.T.; Bai, S.; Saito, T.; Imai, T.; Ito, A.; Moriguchi, T. Involvement of EARLY BUD-BREAK, an AP2/ERF transcription factor gene, in bud break in Japanese pear (Pyrus pyrifolia Nakai) lateral flower buds: Expression, histone modifications and possible target genes. Plant Cell Physiol. 2016, 57, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Chen, Q. Comparative transcriptome analysis of nonchilled, chilled, and late-pink bud reveals flowering pathway genes involved in chilling-mediated flowering in blueberry. BMC Plant Biol. 2018, 18, 98–110. [Google Scholar] [CrossRef]
- Yu, C.; Guo, X.; Luo, L.; Pan, H.; Zhang, Q. Species-specific genes account for the differences in floral transition between continuous-flowering and once-flowering roses. J. Plant Biochem. Biot. 2019, 28, 312–319. [Google Scholar] [CrossRef]
- Rivera, G.; Borchert, R. Induction of flowering in tropical trees by a 30-min reduction in photoperiod: Evidence from field observations and herbarium specimens. Tree Physiol. 2001, 21, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Sun, L.; Liu, X.; Liu, C.; Yin, Z. Nutritional effect and rhythm of spring and summer flowering in Magnolia soulangeana ‘Changchun’. Bull. Bot. Res. 2019, 39, 192–199. [Google Scholar] [CrossRef]
- Liu, W.; Hilu, K.; Wang, Y. From leaf and branch into a flower: Magnolia tells the story. Bot. Stud. 2014, 55, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, J.; Zheng, W. The complete chloroplast genome of threatened Magnolia laevifolia, a rare ornamental shrub with strong aromatic flowers. Conserv. Genet. Resour. 2018, 10, 339–342. [Google Scholar] [CrossRef]
- Porter, E.A.; Kite, G.C.; Veitch, N.C.; Geoghegan, I.A.; Larsson, S.; Simmonds, M.S.J. Phenylethanoid glycosides in tepals of Magnolia salicifolia and their occurrence in flowers of Magnoliaceae. Phytochemistry 2015, 117, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Morshedloo, M.R.; Quassinti, L.; Bramucci, M.; Lupidi, G.; Maggi, F. Chemical composition, antioxidant activity and cytotoxicity on tumour cells of the essential oil from flowers of Magnolia grandiflora cultivated in Iran. Nat. Prod. Res. 2017, 31, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chen, F.; Jing, D.; Liu, Z.; Ma, L. Isolation and characterization of an AGAMOUS-Like gene from Magnolia wufengensis (Magnoliaceae). Plant Mol. Biol. Rep. 2012, 30, 690–698. [Google Scholar] [CrossRef]
- Jing, D.; Liu, Z.; Zhang, B.; Ma, J.; Han, Y.; Chen, F. Two ancestral APETALA3 homologs from the basal angiosperm Magnolia wufengensis (Magnoliaceae) can affect flower development of Arabidopsis. Gene 2014, 537, 100–107. [Google Scholar] [CrossRef]
- Fan, L.; Chen, M.; Dong, B.; Wang, N.; Yu, Q.; Wang, X.; Xuan, L.; Wang, Y.; Zhang, S.; Shen, Y. Transcriptomic analysis of flower bud differentiation in Magnolia sinostellata. Genes 2018, 9, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Villacreses, J.; Blanc, N.; Espinoza, L.; Martinez, C.; Pastor, G.; Manque, P.; Undurraga, S.F.; Polanco, V. High quality RNA extraction from Maqui berry for its application in next-generation sequencing. SpringerPlus 2016, 5, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−△△CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, L.P.; Chen, L.G.; Yu, D.Q. Transcription factor WRKY75 interacts with DELLA proteins to affect flowering. Plant Physiol. 2018, 176, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warwell, M.V.; Shaw, R.G. Phenotypic selection on growth rhythm in whitebark pine under climatic conditions warmer than seed origins. J. Evol. Biol. 2018, 31, 1284–1299. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Li, J.; Li, X.; Wu, B.; Wang, J.; Liu, Z.; Yin, H. Genome-wide transcriptome profiling provides insights into floral bud development of summer-flowering Camellia azalea. Sci. Rep. 2015, 5, 9729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Tuan, P.A.; Katsumi-Horigane, A.; Bai, S.; Ito, A.; Sekiyama, Y.; Ono, H.; Moriguchi, T. Development of flower buds in the Japanese pear (Pyrus pyrifolia) from late autumn to early spring. Tree Physiol. 2015, 35, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, H.; Ooka, T.; Jotatsu, H.; Sasaki, R.; Tao, R. Expression analysis of PpDAM5 and PpDAM6 during flower bud development in peach (Prunus persica). Sci. Hortic. 2011, 129, 844–848. [Google Scholar] [CrossRef]
- Alburquerque, N.; Burgos, L.; Egea, J. Apricot flower bud development and abscission related to chilling, irrigation and type of shoots. Sci. Hortic. 2003, 98, 265–276. [Google Scholar] [CrossRef]
- Galvao, V.C.; Schmid, M. Regulation of flowering by endogenous signals. In Advances in Botanical Research; Fornara, F., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 72, pp. 63–102. [Google Scholar] [CrossRef]
- Nelson, S.K.; Steber, C.M. Gibberellin hormone signal perception: Down-regulating DELLA repressors of plant growth and development. In Annual Plant Reviews; Hedden, P., Thomas, S.G., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2016; Volume 49, pp. 153–188. [Google Scholar] [CrossRef]
- Griffiths, J.; Murase, K.; Rieu, I.; Zentella, R.; Zhang, Z.; Powers, S.J.; Gong, F.; Phillips, A.L.; Hedden, P.; Sun, T.; et al. Genetic characterization and functional analysis of the GID1 gibberellin receptors in Arabidopsis. Plant Cell 2006, 18, 3399–3414. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Cui, J.; Fu, X.Q.; Yan, T.X.; Tang, K.X. Cloning and characterization of DELLA genes in Artemisia annua. Genet Mol. Res. 2015, 14, 10037–10049. [Google Scholar] [CrossRef]
- Galvao, V.C.; Horrer, D.; Kuettner, F.; Schmid, M. Spatial control of flowering by DELLA proteins in Arabidopsis thaliana. Development 2012, 139, 4072–4082. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Kodaira, K.; Maruyama, K.; Mizoi, Z.; Tran, L.; Fujita, Y.; Morimoto, K.; Shinozaki, K.; Yamaguchi-Shinozaki, K. SPINDLY, a negative regulator of gibberellic acid signaling, is involved in the plant abiotic stress response. Plant Physiol. 2011, 157, 1900–1913. [Google Scholar] [CrossRef] [Green Version]
- Zentella, R.; Sui, N.; Barnhill, B.; Hsieh, W.; Hu, J.; Shabanowitz, J.; Boyce, M.; Olszewski, N.E.; Zhou, P.; Hunt, D.F.; et al. The Arabidopsis O-fucosyltransferase SPINDLY activates nuclear growth repressor DELLA. Nat. Chem. Biol. 2017, 13, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Galvão, V.C.; Zhang, Y.; Horrer, D.; Zhang, T.; Hao, Y.; Feng, Y.; Wang, S.; Schmid, M.; Wang, J. Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA PROMOTER BINDING–LIKE transcription factors. Plant Cell 2012, 24, 3320–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Schwarz, S.; Saedler, H.; Huijser, P. SPL8, a local regulator in a subset of gibberellin-mediated developmental processes in Arabidopsis. Plant Mol. Biol. 2007, 63, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Wang, H.; Yu, H.; Zhong, C.; Zhang, X.; Peng, J.; Wang, X. GASA14 regulates leaf expansion and abiotic stress resistance by modulating reactive oxygen species accumulation. J. Exp. Bot. 2013, 64, 1637–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinovich, L.; Weiss, D. The Arabidopsis cysteinerich protein GASA4 promotes GA responses and exhibits redox activity in bacteria and in planta. Plant J. 2010, 64, 1018–1027. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Wang, X. Expression pattern of GASA, downstream genes of DELLA, in Arabidopsis. Chin. Sci. Bull. 2008, 53, 3839–3846. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Kang, S.G.; Hah, C.; Jang, J.C. Molecular and cellular characterization of GA-Stimulated transcripts GASA4 and GASA6 in Arabidopsis thaliana. Plant Sci. 2016, 246, 1–10. [Google Scholar] [CrossRef]
- Shi, L.; Gast, R.T.; Gopalraj, M.; Olszewski, N.E. Characterization of a shoot-specific, GA3- and ABA regulated gene from tomato. Plant J. 1992, 2, 153–159. [Google Scholar] [CrossRef]
- Kovalchuk, N.; Jia, W.; Eini, O.; Morran, S.; Pyvovarenko, T.; Fletcher, S.; Bazanova, N.; Harris, J.; Beck-Oldach, K.; Shavrukov, Y.; et al. Optimization of TaDREB3 gene expression in transgenic barley using cold-inducible promoters. Plant Biotechnol. J. 2013, 11, 659–670. [Google Scholar] [CrossRef]
- Lin, P.; Shen, C.; Chen, H.; Yao, X.; Lin, J. Improving tobacco freezing tolerance by co-transfer of stress-inducible CbCBF and CbICE53 genes. Biol. Plant. 2017, 61, 520–528. [Google Scholar] [CrossRef]
- Fornara, F.; Panigrahi, K.C.S.; Gissot, L.; Sauerbrunn, N.; Rühl, M.; Jarillo, J.A.; Coupland, G. Arabidopsis DOF transcription factors act redundantly to reduce CONSTANS expression and are essential for a photoperiodic flowering response. Dev. Cell 2009, 17, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaizumi, T. Arabidopsis circadian clock and photoperiodism: Time to think about location. Curr. Opin. Plant Biol. 2010, 13, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yang, M.; Zhang, W.; Chen, F.; Shen, S. A putative flowering-time-related Dof transcription factor gene, JcDof3, is controlled by the circadian clock in Jatropha curcas. Plant Sci. 2011, 181, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.K.; Biswas, B.; Gresshoff, P.M. Classical ethylene insensitive mutants of the Arabidopsis EIN2 orthologue lack the expected ‘hypernodulation’ response in Lotus japonicus. J. Integr. Plant Biol. 2013, 55, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hsu, Y.; Bartholomew, D.P.; Maruthasalam, S.; Lin, C. Delaying natural flowering in pineapple through foliar application of aviglycine, an inhihitor of ethylene biosynthesis. Hortscience 2007, 42, 1188–1191. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.R.; Allan, A.C.; Chen, K.S.; Ferguson, I.B. Kiwifruit EIL and ERF genes involved in regulating fruit ripening. Plant Physiol. 2010, 153, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Hong, M.; Chung, Y.; An, G. Ectopic expression of tobacco MADS genes modulates flowering time and plant architecture. Mol. Cells 2000, 9, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Müssig, C.; Altmann, T. Changes in gene expression in response to altered SHL transcript levels. Plant Mol. Biol. 2003, 53, 805–820. [Google Scholar] [CrossRef]
- Li, H.; Liang, W.; Jia, R.; Yin, C.; Zong, J.; Kong, H.; Zhang, D. The AGL6-like gene OsMADS6 regulates floral organ and meristem identities in rice. Cell Res. 2010, 20, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhang, Q.; Wang, L.; Duan, K.; Pan, A.; Tang, X.; Sui, S.; Li, M. The AGL6-like gene CpAGL6, a potential regulator of floral time and organ identity in wintersweet (Chimonanthus praecox). J. Plant Growth Regul. 2011, 30, 343–352. [Google Scholar] [CrossRef]
- Katahata, S.; Futamura, N.; Igasaki, T.; Shinohara, K. Functional analysis of SOC1-like and AGL6-like MADS-box genes of the gymnosperm Cryptomeria japonica. Tree Genet. Genomes 2014, 10, 317–327. [Google Scholar] [CrossRef]
- Fernandez, D.E.; Wang, C.; Zheng, Y.; Adamczyk, B.J.; Singhal, R.; Hall, P.K.; Perry, S.E. The MADS-domain factors AGAMOUS-LIKE15 and AGAMOUS-LIKE18, along with SHORT VEGETATIVE PHASE and AGAMOUS-LIKE24, are necessary to block floral gene expression during the vegetative phase. Plant Physiol. 2014, 165, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.J.; Lehti-Shiu, M.D.; Fernandez, D.E. The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant J. 2007, 50, 1007–1019. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads (M) | Clean Reads (M) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| ZFFU | 77.20 | 71.00 | 97.62 | 89.75 |
| ZFFE | 74.71 | 68.64 | 97.57 | 89.36 |
| ZSFU | 74.71 | 67.70 | 97.98 | 90.46 |
| ZSFE | 76.37 | 69.09 | 97.84 | 89.99 |
| Sample | Total Number | Mean Length | 200–2000 nt | N50 | GC (%) |
|---|---|---|---|---|---|
| ZFFU | 55786.33 | 1094.33 | 84.90% | 1690.00 | 44.13 |
| ZFFE | 53792.00 | 1087.67 | 85.18% | 1683.00 | 44.19 |
| ZSFU | 63000.67 | 986.33 | 87.28% | 1627.67 | 43.89 |
| ZSFE | 54682.00 | 1045.67 | 85.86% | 1692.33 | 44.17 |
| Values | Total | NR 1 | NT 2 | Swissprot 3 | KEGG 4 |
|---|---|---|---|---|---|
| Number | 168120 | 99843 | 87184 | 74371 | 80923 |
| Percentage | 100% | 59.39% | 51.86% | 44.24% | 48.13% |
| Values | KOG 5 | Pfam 6 | GO 7 | TF 8 | PRG 9 |
| Number | 78710 | 73437 | 59251 | 3291 | 8012 |
| Percentage | 46.82% | 43.68% | 35.24% | 1.96% | 4.77% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Z.; Sun, L.; Wei, Q.; Ju, Y.; Zou, X.; Wan, X.; Liu, X.; Yin, Z. A New Insight into Flowering Regulation: Molecular Basis of Flowering Initiation in Magnolia × soulangeana ‘Changchun’. Genes 2020, 11, 15. https://doi.org/10.3390/genes11010015
Jiang Z, Sun L, Wei Q, Ju Y, Zou X, Wan X, Liu X, Yin Z. A New Insight into Flowering Regulation: Molecular Basis of Flowering Initiation in Magnolia × soulangeana ‘Changchun’. Genes. 2020; 11(1):15. https://doi.org/10.3390/genes11010015
Chicago/Turabian StyleJiang, Zheng, Liyong Sun, Qiang Wei, Ye Ju, Xuan Zou, Xiaoxia Wan, Xu Liu, and Zengfang Yin. 2020. "A New Insight into Flowering Regulation: Molecular Basis of Flowering Initiation in Magnolia × soulangeana ‘Changchun’" Genes 11, no. 1: 15. https://doi.org/10.3390/genes11010015