HDX and Native Mass Spectrometry Reveals the Different Structural Basis for Interaction of the Staphylococcal Pathogenicity Island Repressor Stl with Dimeric and Trimeric Phage dUTPases

 , ,
, ,
Abstract
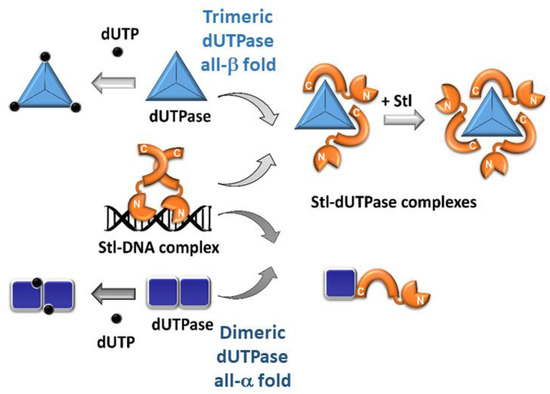
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cloning, Expression, and Purification of Proteins
2.2. dUTPase Enzyme Activity Assay
2.3. Native Gel Electrophoresis
2.4. Chemical Crosslinking
2.5. Native Mass Spectrometry
2.6. HDX-MS
2.7. Homology Models
3. Results and Discussion
3.1. Stl Inhibits the Enzymatic Activity of Homodimeric and Homotrimeric dUTPases with Comparable Inhibition Constant
3.2. Mechanism of Interaction with Stl is Markedly Different between Homodimeric and Homotrimeric Phage dUTPases
3.3. dUTPase Active Sites are Directly Involved in the Complex Formation with Stl
3.4. Different Regions of Stl Mediate the Promiscuity of this Protein for dUTPase Binding
5. Conclusions
Supplementary Materials
Author Contributions
Note Added in Proof
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzgerald, J.R. Human origin for livestock-associated methicillin-resistant Staphylococcus aureus. MBio 2012, 3, e00082-12. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M. Horizontal gene transfer in human pathogens. Crit. Rev. Microbiol. 2013, 7828, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Christie, G.E.; Penadés, J.R. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 2010, 8, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.A.; Ruzin, A.; Ross, H.F.; Kurepina, N.; Novick, R.P. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 1998, 29, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Tormo-Más, M.A.; Mir, I.; Shrestha, A.; Tallent, S.M.; Campoy, S.; Lasa, I.; Barbé, J.; Novick, R.P.; Christie, G.E.; Penadés, J.R. Moonlighting bacteriophage proteins derepress staphylococcal pathogenicity islands. Nature 2010, 465, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, J.E.; Németh, V.; Papp-Kádár, V.; Nyíri, K.; Leveles, I.; Bendes, Á.Á.; Zagyva, I.; Róna, G.; Pálinkás, H.L.; Besztercei, B.; et al. Highly potent dUTPase inhibition by a bacterial repressor protein reveals a novel mechanism for gene expression control. Nucleic Acids Res. 2014, 42, 11912–11920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiques, E.; Quiles-Puchalt, N.; Donderis, J.; Ciges-Tomas, J.R.; Alite, C.; Bowring, J.Z.; Humphrey, S.; Penadés, J.R.; Marina, A. Another look at the mechanism involving trimeric dUTPases in Staphylococcus aureus pathogenicity island induction involves novel players in the party. Nucleic Acids Res. 2016, 44, 5457–5469. [Google Scholar] [CrossRef]
- Nyíri, K.; Papp-Kádár, V.; Szabó, J.E.; Németh, V.; Vértessy, B.G. Exploring the role of the phage-specific insert of bacteriophage Φ11 dUTPase. Struct. Chem. 2015, 26, 1425–1432. [Google Scholar] [CrossRef]
- Hirmondó, R.; Szabó, J.E.; Nyíri, K.; Tarjányi, S.; Dobrotka, P.; Tóth, J.; Vértessy, B.G. Cross-species inhibition of dUTPase via the Staphylococcal Stl protein perturbs dNTP pool and colony formation in Mycobacterium. DNA Repair 2015, 30, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Frígols, B.; Quiles-Puchalt, N.; Mir-Sanchis, I.; Donderis, J.; Elena, S.F.; Buckling, A.; Novick, R.P.; Marina, A.; Penadés, J.R. Virus satellites drive viral evolution and ecology. PLoS Genet. 2015, 11, e1005609. [Google Scholar] [CrossRef]
- Vértessy, B.G.; Tóth, J. Keeping uracil out of DNA: Physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 2009, 42, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.N.; Leveles, I.; Vértessy, B.G. Preventive DNA repair by sanitizing the cellular (deoxy)nucleoside triphosphate pool. FEBS J. 2014, 281, 4207–4223. [Google Scholar] [CrossRef] [PubMed]
- Tormo-Más, M.Á.; Donderis, J.; García-Caballer, M.; Alt, A.; Mir-Sanchis, I.; Marina, A.; Penadés, J.R. Phage dUTPases control transfer of virulence genes by a proto-oncogenic G protein-like mechanism. Mol. Cell 2013, 49, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.L.L.; Dokland, T. The Type 2 dUTPase of bacteriophage φNM1 initiates mobilization of Staphylococcus aureus bovine pathogenicity island 1. J. Mol. Biol. 2016, 428, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.L.L.; Vlach, J.; Parker, L.K.; Christie, G.E.; Saad, J.S.; Dokland, T. Derepression of SaPIbov1 is independent of φNM1 type 2 dUTPase activity and is inhibited by dUTP and dUMP. J. Mol. Biol. 2017, 429, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Nyíri, K.; Vértessy, B.G. Perturbation of genome integrity to fight pathogenic microorganisms. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3593–3612. [Google Scholar] [CrossRef]
- Donderis, J.; Bowring, J.; Maiques, E.; Ciges-Tomas, J.R.; Alite, C.; Mehmedov, I.; Tormo-Mas, M.A.; Penadés, J.R.; Marina, A. Convergent evolution involving dimeric and trimeric dUTPases in pathogenicity island mobilization. PLoS Pathog. 2017, 13, e1006581. [Google Scholar] [CrossRef]
- Leveles, I.; Németh, V.; Szabó, J.E.; Harmat, V.; Nyíri, K.; Bendes, A.A.; Papp-Kádár, V.; Zagyva, I.; Róna, G.; Ozohanics, O.; et al. Structure and enzymatic mechanism of a moonlighting dUTPase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Bowring, J.; Neamah, M.M.; Donderis, J.; Mir-Sanchis, I.; Alite, C.; Ciges-Tomas, J.R.; Maiques, E.; Medmedov, I.; Marina, A.; Penadés, J.R. Pirating conserved phage mechanisms promotes promiscuous staphylococcal pathogenicity island transfer. Elife 2017, 6, e26487. [Google Scholar] [CrossRef] [Green Version]
- Nyíri, K.; Kőhegyi, B.; Micsonai, A.; Kardos, J.; Vértessy, B.G. Evidence-based structural model of the Staphylococcal repressor protein: Separation of functions into different domains. PLoS ONE 2015, 10, e0139086. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, Z.-Y. Application of hydrogen/deuterium exchange mass spectrometry to study protein tyrosine phosphatase dynamics, ligand binding, and substrate specificity. Methods 2007, 42, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.M.L.; Silva, L.; Schriemer, D.C.; Schryvers, A.B. Hydrogen–deuterium exchange coupled to mass spectrometry to investigate ligand–receptor interactions. In Neisseria Meningitidis—Advanced Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 237–252. [Google Scholar]
- Mistarz, U.H.; Brown, J.M.; Haselmann, K.F.; Rand, K.D. Probing the binding interfaces of protein complexes using gas-phase H/D exchange mass spectrometry. Structure 2016, 24, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Wales, T.E.; Engen, J.R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006, 25, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Engen, J.R. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem. 2009, 81, 7870–7875. [Google Scholar] [CrossRef] [PubMed]
- Nyíri, K.; Mertens, H.D.T.; Tihanyi, B.; Nagy, G.N.; Kőhegyi, B.; Matejka, J.; Harris, M.J.; Szabó, J.E.; Papp-Kádár, V.; Németh-Pongrácz, V.; et al. Structural model of human dUTPase in complex with a novel proteinaceous inhibitor. Sci. Rep. 2018, 8, 4326. [Google Scholar] [CrossRef] [PubMed]
- Houde, D.; Berkowitz, S.A.; Engen, J.R.; Fadgen, K.E.; Brown, J.; Engen, J.R.; Lee, C.T.; Steen, J.A.; Steen, H.; Mayer, M.P.; et al. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 2011, 100, 2071–2086. [Google Scholar] [CrossRef]
- Roberts, V.A.; Pique, M.E.; Hsu, S.; Li, S.; Slupphaug, G.; Rambo, R.P.; Jamison, J.W.; Liu, T.; Lee, J.H.; Tainer, J.A.; et al. Combining H/D exchange mass spectroscopy and computational docking reveals extended DNA-binding surface on uracil-DNA glycosylase. Nucleic Acids Res. 2012, 40, 6070–6081. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.A.; Mezulis, S.; Yates, C.; Wass, M.; Sternberg, M. The Phyre2 web portal for protein modelling, prediction, and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Benedek, A.; Temesváry-Kis, F.; Khatanbaatar, T.; Leveles, I.; Surányi, É.V.; Szabó, J.E.; Wunderlich, L.; Vértessy, B.G. The role of a key amino acid position in species-specific proteinaceous dUTPase inhibition. Biomolecules 2019, 9, 221. [Google Scholar] [CrossRef]
- Moroz, O.V.; Harkiolaki, M.; Galperin, M.Y.; Vagin, A.A.; González-Pacanowska, D.; Wilson, K.S. The crystal structure of a complex of Campylobacter jejuni dUTPase with substrate analogue sheds light on the mechanism and suggests the “basic module” for dimeric d(C/U)TPases. J. Mol. Biol. 2004, 342, 1583–1597. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Benedek, A.; Pölöskei, I.; Ozohanics, O.; Vékey, K.; Vértessy, B.G. The Stl repressor from Staphylococcus aureus is an efficient inhibitor of the eukaryotic fruitfly dUTPase. FEBS Open Biol. 2018, 8, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Moroz, O.V.; Wilson, K.S.; Murzin, A.G. House cleaning, a part of good housekeeping. Mol. Microbiol. 2006, 59, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kerepesi, C.; Szabó, J.E.; Papp-Kádár, V.; Dobay, O.; Szabó, D.; Grolmusz, V.; Vértessy, B.G. Life without dUTPase. Front. Microbiol. 2016, 7, 1768. [Google Scholar] [CrossRef] [PubMed]
- Hirmondo, R.; Lopata, A.; Suranyi, E.V.; Vertessy, B.G.; Toth, J. Differential control of dNTP biosynthesis and genome integrity maintenance by dUTPases. Sci. Rep. 2017, 7, 6043. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Hsu, K.C.; Yang, J.M.; Wu, M.L.; Ko, T.P.; Lin, S.R.; Wang, A.H.J. Staphylococcus aureus protein SAUGI acts as a uracil-DNA glycosylase inhibitor. Nucleic Acids Res. 2014, 42, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Mir-Sanchis, I.; Roman, C.A.; Misiura, A.; Pigli, Y.Z.; Boyle-Vavra, S.; Rice, P.A. Staphylococcal SCCmec elements encode an active MCM-like helicase and thus may be replicative. Nat. Struct. Mol. Biol. 2016, 23, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Yan, N.; O’Day, E.; Wheeler, L.A.; Engelman, A.; Lieberman, J. HIV DNA is heavily uracilated, which protects it from autointegration. Proc. Natl. Acad. Sci. USA 2011, 108, 9244–9249. [Google Scholar] [CrossRef] [Green Version]
- Weil, A.F.; Ghosh, D.; Zhou, Y.; Seiple, L.; McMahon, M.A.; Spivak, A.M.; Siliciano, R.F.; Stivers, J.T. Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proc. Natl. Acad. Sci. USA 2013, 110, E448–E457. [Google Scholar] [CrossRef] [Green Version]
- Papp-Kádár, V.; Szabó, J.E.; Nyíri, K.; Vertessy, B.G. In vitro analysis of predicted DNA-binding sites for the Stl repressor of the Staphylococcus aureus SaPIBov1 pathogenicity island. PLoS ONE 2016, 11, e0158793. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyíri, K.; Harris, M.J.; Matejka, J.; Ozohanics, O.; Vékey, K.; Borysik, A.J.; Vértessy, B.G. HDX and Native Mass Spectrometry Reveals the Different Structural Basis for Interaction of the Staphylococcal Pathogenicity Island Repressor Stl with Dimeric and Trimeric Phage dUTPases. Biomolecules 2019, 9, 488. https://doi.org/10.3390/biom9090488
Nyíri K, Harris MJ, Matejka J, Ozohanics O, Vékey K, Borysik AJ, Vértessy BG. HDX and Native Mass Spectrometry Reveals the Different Structural Basis for Interaction of the Staphylococcal Pathogenicity Island Repressor Stl with Dimeric and Trimeric Phage dUTPases. Biomolecules. 2019; 9(9):488. https://doi.org/10.3390/biom9090488
Chicago/Turabian StyleNyíri, Kinga, Matthew J. Harris, Judit Matejka, Olivér Ozohanics, Károly Vékey, Antoni J. Borysik, and Beáta G. Vértessy. 2019. "HDX and Native Mass Spectrometry Reveals the Different Structural Basis for Interaction of the Staphylococcal Pathogenicity Island Repressor Stl with Dimeric and Trimeric Phage dUTPases" Biomolecules 9, no. 9: 488. https://doi.org/10.3390/biom9090488