



Non-Coding RNA 2026, 12(3), 20; https://doi.org/10.3390/ncrna12030020 - 11 Jun 2026
Abstract
►
Show Figures
Background/Objectives: MicroRNAs are key post-transcriptional regulators involved in various diseases. Despite its status as the gold standard, real-time RT-PCR faces challenges arising from high sequence homology among closely related microRNAs and the substantial biomaterial required to enrich small RNA fractions. This study aimed
[...] Read more.
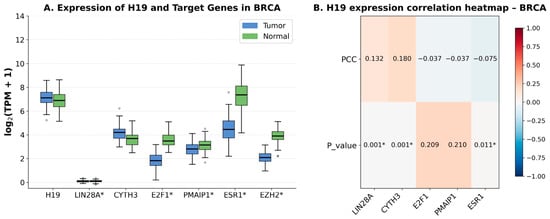
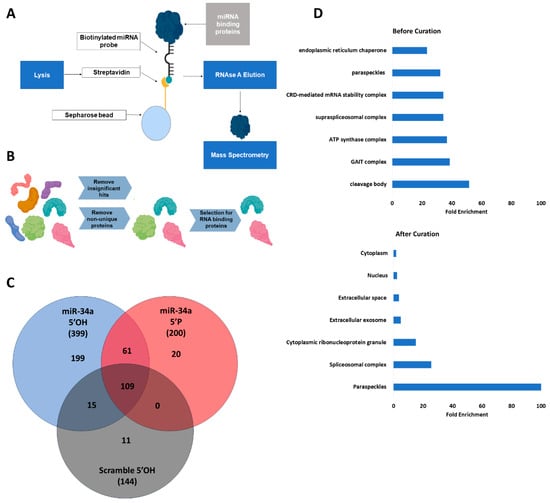
Background/Objectives: MicroRNAs are key post-transcriptional regulators involved in various diseases. Despite its status as the gold standard, real-time RT-PCR faces challenges arising from high sequence homology among closely related microRNAs and the substantial biomaterial required to enrich small RNA fractions. This study aimed to develop an optimized protocol for simultaneous analysis of microRNA and mRNA expression from a single total RNA sample using mouse (Mus musculus) brain tissue, avoiding dependence on pre-designed commercial assay panels. Methods: We optimized a real-time RT-PCR workflow enabling simultaneous analysis of mature microRNAs and mRNAs from a single total RNA sample. Modifications include a redesigned universal reverse primer, LNA-modified TaqMan probes, and omission of the 65 °C denaturation step during reverse transcription. The method was validated for five microRNAs in mouse brain tissue. Results: The assay showed high specificity, discriminating closely related miR-125a-5p and miR-125b-5p with a ΔCt difference of 6.7 ± 1.2 cycles. Co-analysis with Oligo(dT)18 and Random hexamer primers did not interfere with microRNA detection. Conclusions: The developed approach enables reliable detection of closely related microRNAs and parallel analysis of different RNA types, which is particularly important for studying regulatory networks when working with limited amounts of biomaterial. This protocol provides a complementary, accessible option for targeted studies in resource-limited settings or for non-cataloged miRNA targets.
Full article

Figure 1






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}