Increasing Uniformity of Biosurfactant Production in Starmerella bombicola via the Expression of Chimeric Cytochrome P450s

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Culture Conditions
2.2. Molecular Techniques
2.2.1. General Techniques
2.2.2. Vector Construction
2.2.3. Heterologous Expression of Wild-Type Ustilago Maydis CYP1
2.2.4. CYP1 Chimeric Construct
2.2.5. Validation of CYP1BMR Chimeric Construct
2.3. Sampling and Analysis
2.3.1. Follow-Up of Growth and Glucose Consumption
2.3.2. Analysis of Sophorolipid Production
2.3.3. Purification of C16:0 Sophorolipids from Culture Broth
2.3.4. NMR Analysis
3. Results
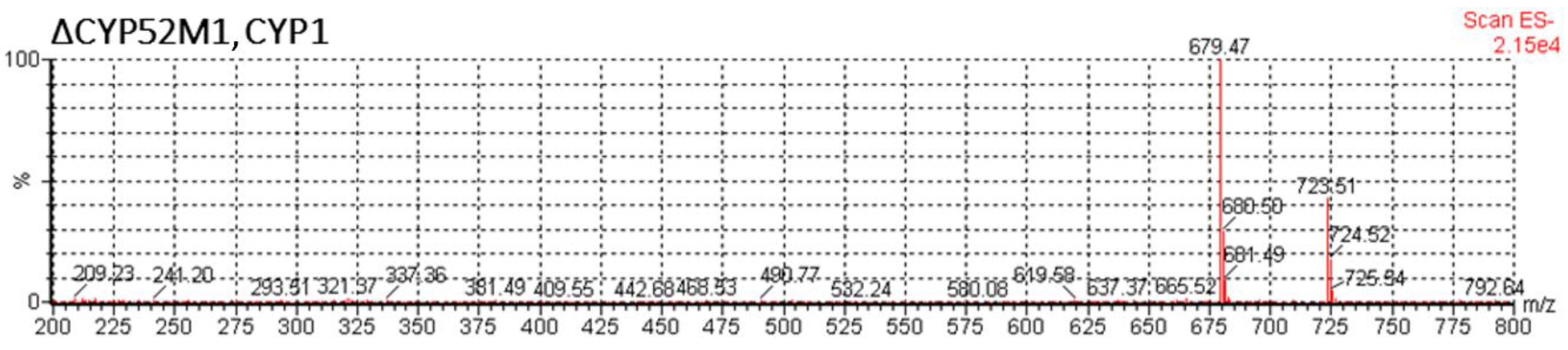
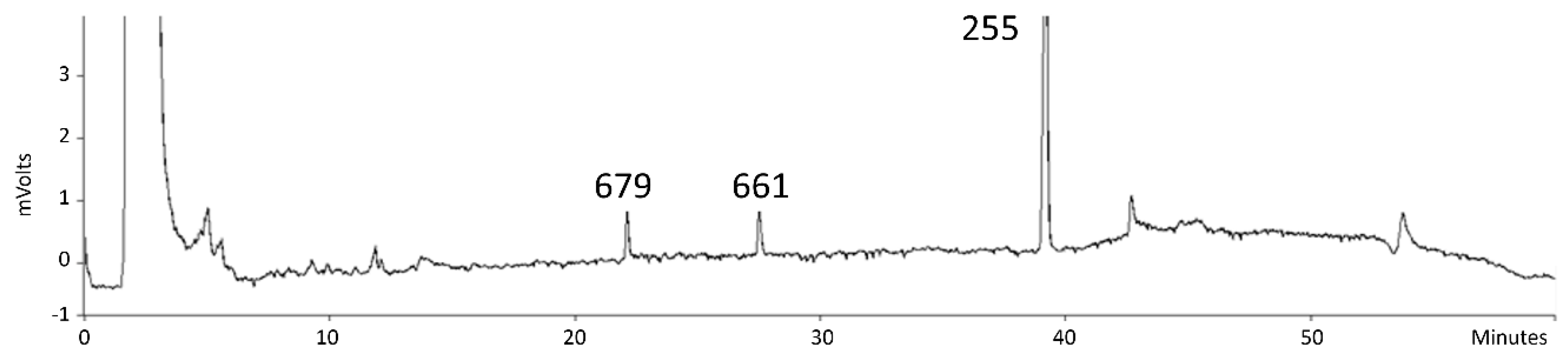
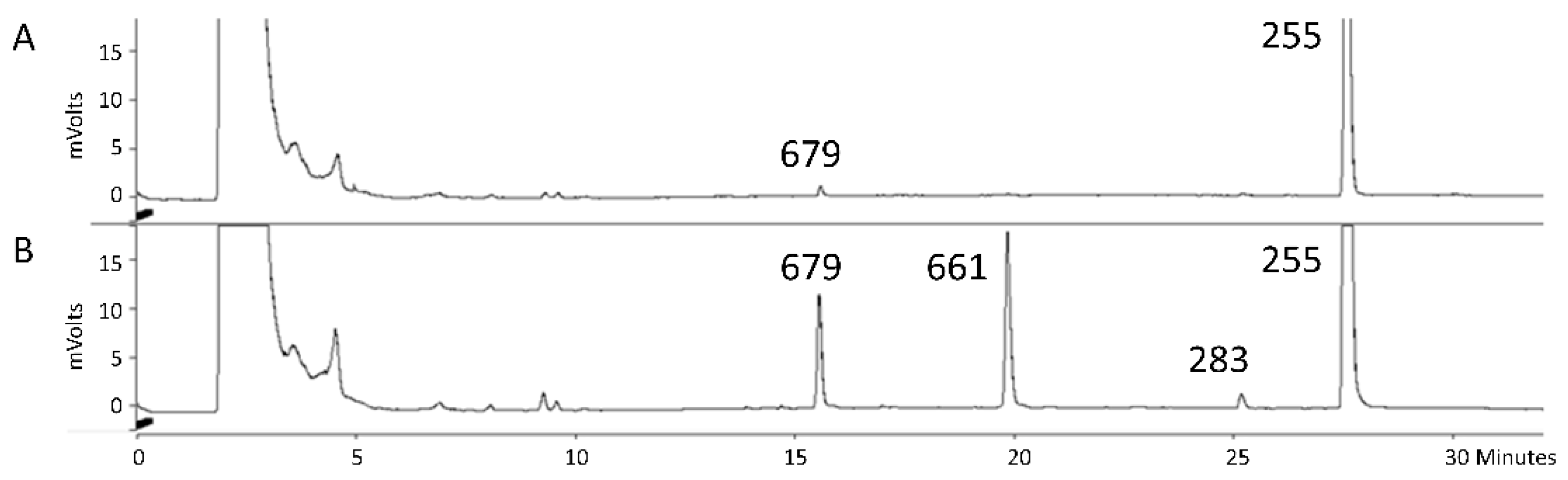
3.1. Expression of Wild-Type CYP1 from U. maydis in S. bombicola
3.2. Production Analysis of the CYP1 Chimeric P450
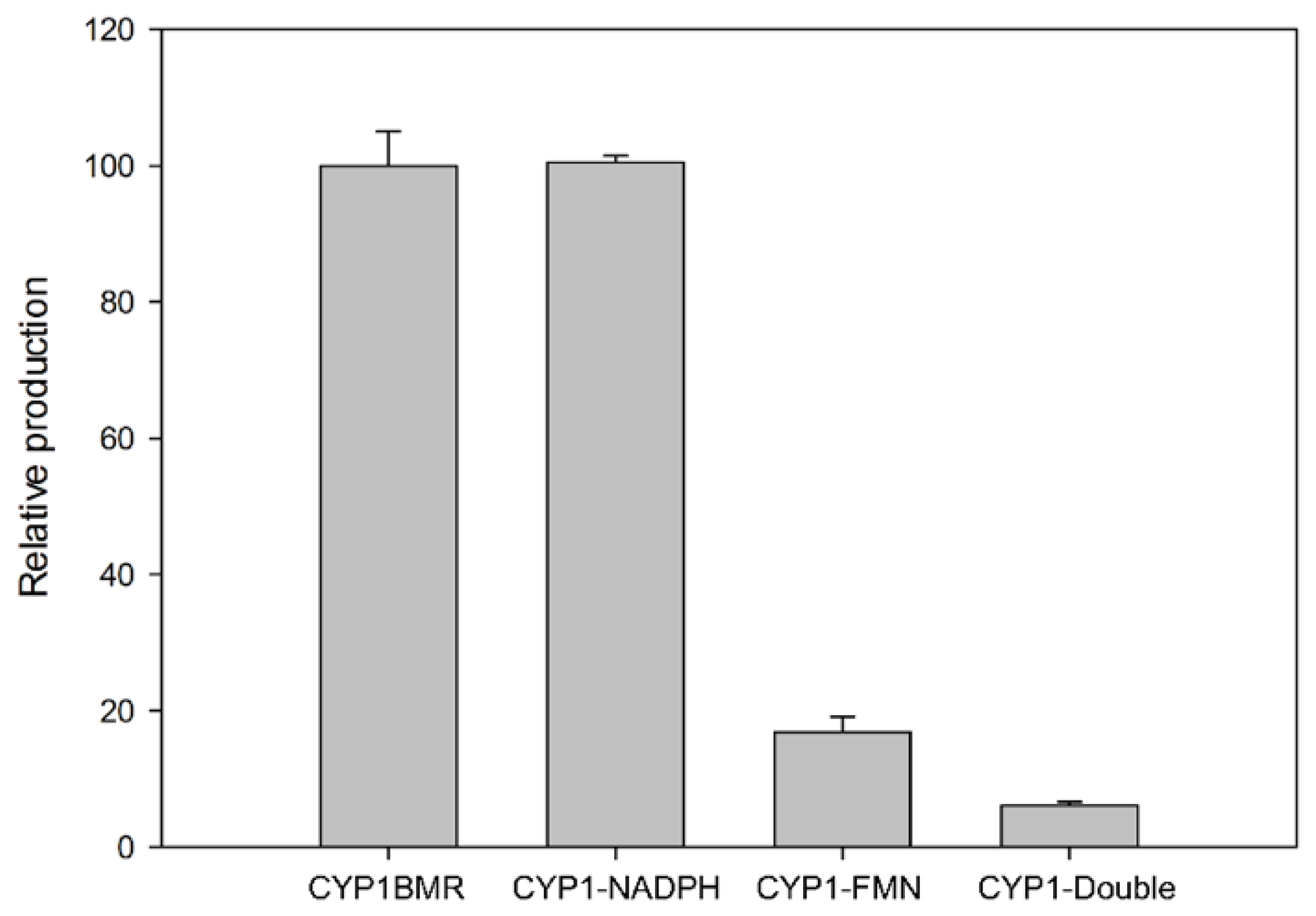
3.3. Validation of the CPR_BMR Functionality
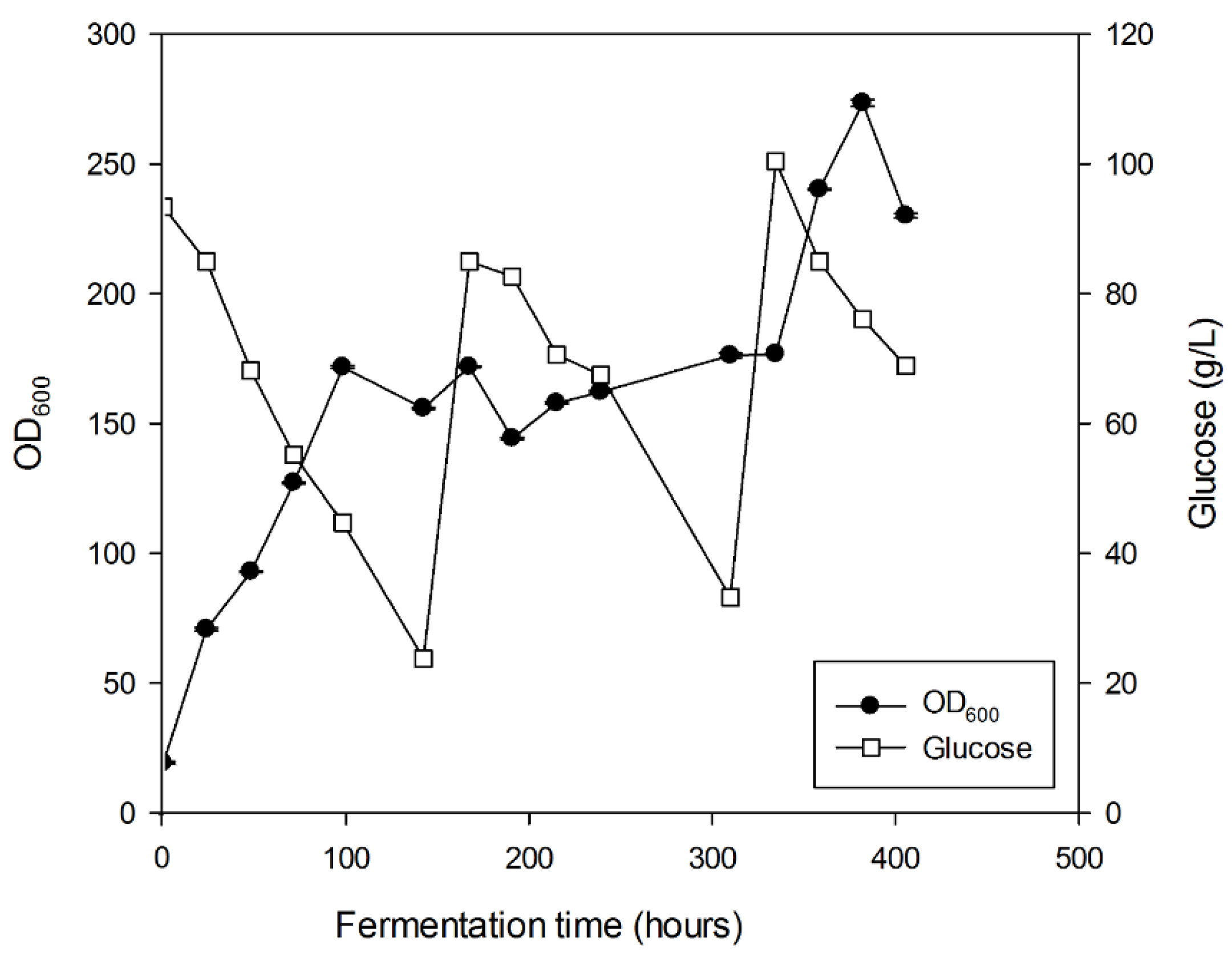
3.4. Substrate and Fermentation Optimization of CYP1BMR
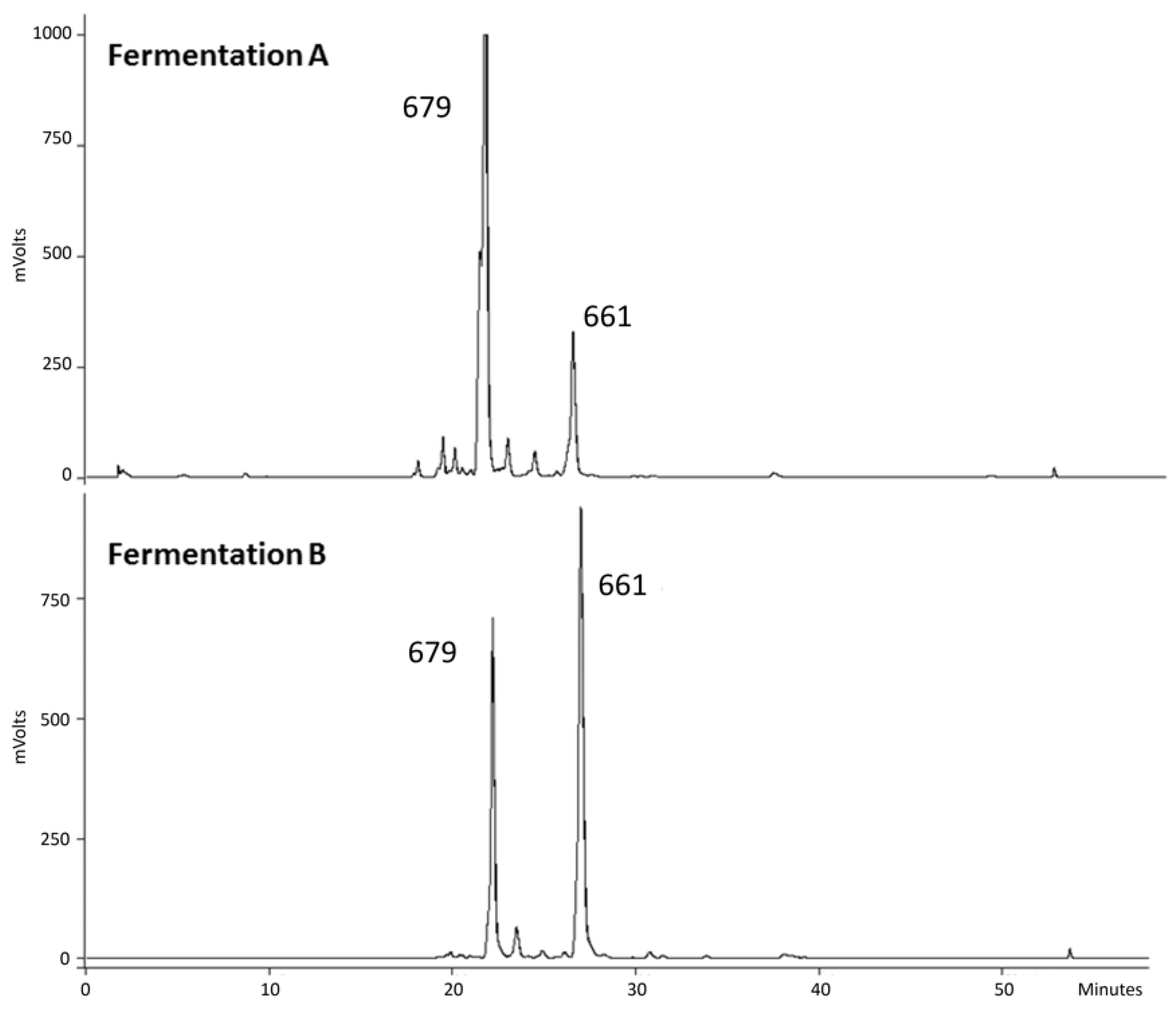
3.5. Scale-Up of the CYP1BMR Chimeric Strain
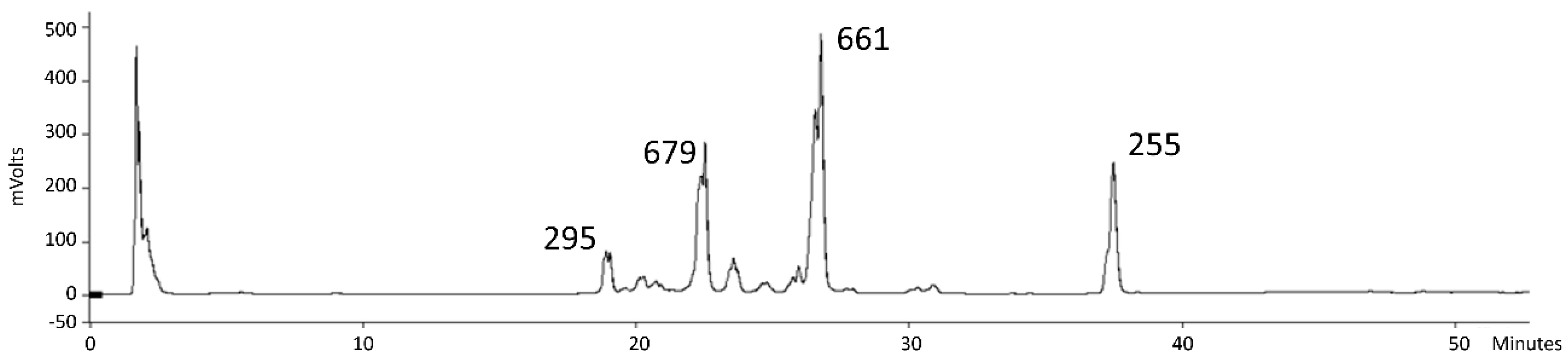
3.6. NMR Structure Elucidation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davila, A.-M.M.; Marchal, R.; Vandecasteele, J.-P.P. Sophorose lipid fermentation with differentiated substrate supply for growth and production phases. Appl. Microbiol. Biotechnol. 1997, 47, 496–501. [Google Scholar] [CrossRef]
- Van Bogaert, I.N.A.; Holvoet, K.; Roelants, S.L.K.W.; Li, B.; Lin, Y.-C.C.; Van de Peer, Y.; Soetaert, W. The biosynthetic gene cluster for sophorolipids: A biotechnological interesting biosurfactant produced by Starmerella bombicola. Mol. Microbiol. 2013, 88, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.M.; Kers, J.A.; Johnson, E.G.; Song, L.; Aston, P.R.; Patel, B.; Krasnoff, S.B.; Crane, B.R.; Gibson, D.M.; Loria, R.; et al. Cytochrome P450–catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat. Chem. Biol. 2012, 8, 814–816. [Google Scholar] [CrossRef] [PubMed]
- Saerens, K.M.J.; Van Bogaert, I.N.A.; Soetaert, W. Characterization of sophorolipid biosynthetic enzymes from Starmerella bombicola. FEMS Yeast Res. 2015, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Delbeke, E.I.P.; Movsisyan, M.; Van Geem, K.M.; Stevens, C.V. Chemical and enzymatic modification of sophorolipids. Green Chem. 2016, 18, 76–104. [Google Scholar] [CrossRef]
- Van Bogaert, I.; Fleurackers, S.; Van Kerrebroeck, S.; Develter, D.; Soetaert, W. Production of new-to-nature sophorolipids by cultivating the yeast Candida bombicola on unconventional hydrophobic substrates. Biotechnol. Bioeng. 2011, 108, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Tehlivets, O.; Scheuringer, K.; Kohlwein, S.D. Fatty acid synthesis and elongation in yeast. Biochim. Biophys. Acta–Mol. Cell Biol. Lipids 2007, 1771, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Brakemeier, A.; Wullbrandt, D.; Lang, S. Microbial alkyl-sophorosides based on 1-dodecanol or 2-, 3- or 4-dodecanones. Biotechnol. Lett. 1998, 20, 215–218. [Google Scholar] [CrossRef]
- Van Bogaert, I.N.A.; Sabirova, J.; Develter, D.; Soetaert, W.; Vandamme, E.J. Knocking out the MFE-2 gene of Candida bombicola leads to improved medium-chain sophorolipid production. FEMS Yeast Res. 2009, 9, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, F.; Igarashi, K.; Hagihara, H. Identification of the fatty alcohol oxidase FAO1 from Starmerella bombicola and improved novel glycolipids production in an FAO1 knockout mutant. Appl. Microbiol. Biotechnol. 2016, 100, 9519–9528. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, B.; Linne, U.; Hewald, S.; Marahiel, M.A.; Bölker, M. A biosynthetic gene cluster for a secreted cellobiose lipid with antifungal activity from Ustilago maydis. Mol. Microbiol. 2007, 66, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Lodens, S.; De Graeve, M.; Roelants, S.L.K.W.K.W.; De Maeseneire, S.L.; Soetaert, W. Transformation of an exotic yeast species into a platform organism: A case study for engineering glycolipid production in the yeast Starmerella bombicola. In Synthetic Biology: Methods and Protocols; Braham, J.C., Ed.; Springer Humana Press: New York, NY, USA, 2018; pp. 95–123. ISBN 9781493977949. [Google Scholar]
- Lang, S.; Brakemeier, A.; Heckmann, R.; Spöckner, S.; Rau, U. Production of native and modified sophorose lipids. Chim Oggi 2000, 18, 76–79. [Google Scholar]
- Saerens, K.M.J.; Saey, L.; Soetaert, W. One-Step Production of Unacetylated Sophorolipids by an Acetyltransferase Negative Candida bombicola. Biotechnol. Bioeng. 2011, 108, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbour Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O.; Hutchison, C.A., III; America, N. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Asmer, H.-J.; Lang, S.; Wagner, F.; Wray, V. Microbial production, structure elucidation and bioconversion of sophorose lipids. J. Am. Oil Chem. Soc. 1988, 65, 1460–1466. [Google Scholar] [CrossRef]
- Petersen, B.O.; Vinogradov, E.; Kay, W.; Würtz, P.; Nyberg, N.T.; Duus, J.; Sørensen, O.W. H2BC: A new technique for NMR analysis of complex carbohydrates. Carbohydr. Res. 2006, 341, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Gheysen, K.; Mihai, C.; Conrath, K.; Martins, J.C. Rapid identification of common hexapyranose monosaccharide units by a simple TOCSY matching approach. Chem. A Eur. J. 2008, 14, 8869–8878. [Google Scholar] [CrossRef] [PubMed]
- Kleine, M.L.; Fulco, A.J. Critical residues involved in FMN binding and catalytic activity in cytochrome P450BM-3. J. Biol. Chem. 1993, 268, 7553–7561. [Google Scholar]
- Cirino, P.C.C.; Arnold, F.H. Regioselectivity and Activity of Cytochrome P450 BM-3 and Mutant F87A in Reactions Driven by Hydrogen Peroxide. Adv. Synth. Catal. 2002, 344, 932–937. [Google Scholar] [CrossRef]
- Dietrich, M.; Do, T.A.; Schmid, R.D.; Pleiss, J.; Urlacher, V.B. Altering the regioselectivity of the subterminal fatty acid hydroxylase P450 BM-3 towards gamma- and delta-positions. J. Biotechnol. 2009, 139, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Davila, A.-M.; Marchal, R.; Vandecasteele, J.-P. Kinetics and balance of a fermentation free from product inhibition: Sophorose lipid production by Candida bombicola. Appl. Microbiol. Biotechnol. 1992, 38, 6–11. [Google Scholar] [CrossRef]
- Van Bogaert, I.N.A.; Saerens, K.; De Muynck, C.; Develter, D.; Soetaert, W.; Vandamme, E.J.; Van Bogaert, I.N.A.; Saerens, K.; De Muynck, C.; Develter, D.; et al. Microbial production and application of sophorolipids. Appl. Microbiol. Biotechnol. 2007, 76, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.R.; Camilios-neto, D.; Baldo, C.; Magri, A.; Celligoi, M.A.P.C. Biosynthesis and Production of Sophorolipids. Int. J. Sci. Technol. Res. 2014, 3, 133–143. [Google Scholar]
- Ratsep, P.; Shah, V. Identification and quantification of sophorolipid analogs using ultra-fast liquid chromatography–mass spectrometry. J. Microbiol. Methods 2009, 78, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Guilmanov, V.; Ballistreri, A.; Impallomeni, G.; Gross, R.A.; Catania, Á.; Doria, V.A.; Chimiche, S. Oxygen transfer rate and sophorose lipid production by Candida bombicola. Biotechnol. Bioeng. 2002, 77, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Van Renterghem, L.; Roelants, S.L.K.W.; Baccile, N.; Uytersprot, K.; Taelman, M.C.; Everaert, B.; Mincke, S.; Ledegen, S.; Debrouwer, S.; Scholtens, K.; et al. From lab to market: An integrated bioprocess design approach for new-to-nature biosurfactants produced by Starmerella bombicola. Biotechnol. Bioeng. 2018, 115, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Roelants, S.L.K.W.; Ciesielska, K.; De Maeseneire, S.L.; Moens, H.; Everaert, B.; Verweire, S.; Denon, Q.; Vanlerberghe, B.; Van Bogaert, I.N.A.; Van der Meeren, P.; et al. Towards the industrialization of new biosurfactants: Biotechnological opportunities for the lactone esterase gene from Starmerella bombicola. Biotechnol. Bioeng. 2016, 113, 550–559. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| P52 | ATATGTACTTTTCAATATGATAAACGGAGAAATAACG |
| P69 | GTTTCTTAGCCTCCCATGGAAG |
| P395 | TGAAAAGTACATATGGTACCATGTCGCTCAAAGTGCAG |
| P396 | AACGCTAGCTTGGCGTTATCTAGTGCCTTCCTTGCAAC |
| P722 | AAAATTCCGCTTGGCGGTATTCCTTCACC |
| P723 | GGGAGGCTAAGAAACTTACCCAGCCCACACGTCT |
| P788 | GCCAAGCGGAATTTTTCTAGTGCCTTCCTTGCAACATCGAAGGG |
| P847 | CGTTGTCAAGTCCTAAGGTAT |
| P887 | AAGCGTGAAGCTCCTCTGACAATC |
| P1439 | GTTTTCTGCCTTTTTGCGTAC |
| P1641 | CGGAGACGGAAGCCAAATGGCACCTGCCGTTGAAGCAACG |
| P1642 | CAAATACGGAGTAGCGAACGCCTTTTACTTCATCAGCA |
| P1643 | CGTTGCTTCAACGGCAGGTGCCATTTGGCTTCCGTCTCCG |
| P1644 | TGCTGATGAAGTAAAAGGCGTTCGCTACTCCGTATTTG |
| P1645 | GCAAAACGTTTAACAATGCTTGAACTGCTTGAAAAATACC |
| P1646 | CCGGGTATTTTTCAAGCAGTTCAAGCATTGTTAAACGTTTTGCC |
| Strain | Characteristics |
|---|---|
| S. bombicola Δcyp52m1 | Knock-out strain of cyp52m1 [2] |
| S. bombicola cyp1 | Knock-in of cyp1 at the cyp52m1 locus |
| S. bombicola cyp1-bmr | Knock-in of cyp1bmr at the cyp52m1 locus |
| S. bombicola cyp1-nadph | Knock-in of cyp1bmr with a mutation in the NADPH binding domain at the cyp52m1 locus |
| S. bombicola cyp1-fmn | Knock-in of cyp1bmr with a mutation in the FMN domain at the cyp52m1 locus |
| S. bombicola cyp1-double | Knock-in of cyp1bmr with a mutation in both the NADPH binding domain and the FMN domain at the cyp52m1 locus |
| Fermentation A (Manual Feed) | Fermentation B (Automatic Feed) | |
|---|---|---|
| Aeration | ±1.2 L/min (approx. 0.1 vvm) | ±5 L/min (approx. 0.3 vvm) |
| Stirring rate | 600 rpm | 600 rpm |
| Total glucose consumption | 3492 g | 2577 g |
| Total ethyl palmitate | 54.85 g | 162 g |
| Duration | 448 h | 359 h |
| Average optical density | 131.9 ± 16.7 | 120.9 ± 13.6 |
| Sophorolipid titers | 9.0 ± 0.2 g | 12.1 ± 0.2 g |
| Productivity | 20 ± 0.4 mg/L·h | 33 ± 0.5 mg/L·h |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geys, R.; De Graeve, M.; Lodens, S.; Van Malderen, J.; Lemmens, C.; De Smet, M.; Mincke, S.; Van Bogaert, I.N.A.; Stevens, C.; De Maeseneire, S.L.; et al. Increasing Uniformity of Biosurfactant Production in Starmerella bombicola via the Expression of Chimeric Cytochrome P450s. Colloids Interfaces 2018, 2, 42. https://doi.org/10.3390/colloids2040042
Geys R, De Graeve M, Lodens S, Van Malderen J, Lemmens C, De Smet M, Mincke S, Van Bogaert INA, Stevens C, De Maeseneire SL, et al. Increasing Uniformity of Biosurfactant Production in Starmerella bombicola via the Expression of Chimeric Cytochrome P450s. Colloids and Interfaces. 2018; 2(4):42. https://doi.org/10.3390/colloids2040042
Chicago/Turabian StyleGeys, Robin, Marilyn De Graeve, Sofie Lodens, Jeroen Van Malderen, Christophe Lemmens, Margaux De Smet, Stein Mincke, Inge N. A. Van Bogaert, Christian Stevens, Sofie L. De Maeseneire, and et al. 2018. "Increasing Uniformity of Biosurfactant Production in Starmerella bombicola via the Expression of Chimeric Cytochrome P450s" Colloids and Interfaces 2, no. 4: 42. https://doi.org/10.3390/colloids2040042
APA StyleGeys, R., De Graeve, M., Lodens, S., Van Malderen, J., Lemmens, C., De Smet, M., Mincke, S., Van Bogaert, I. N. A., Stevens, C., De Maeseneire, S. L., Roelants, S. L. K. W., & Soetaert, W. K. G. (2018). Increasing Uniformity of Biosurfactant Production in Starmerella bombicola via the Expression of Chimeric Cytochrome P450s. Colloids and Interfaces, 2(4), 42. https://doi.org/10.3390/colloids2040042