Molecular Dynamics Simulation of Protein Biosurfactants


Abstract
:1. Introduction
2. Molecular Simulation
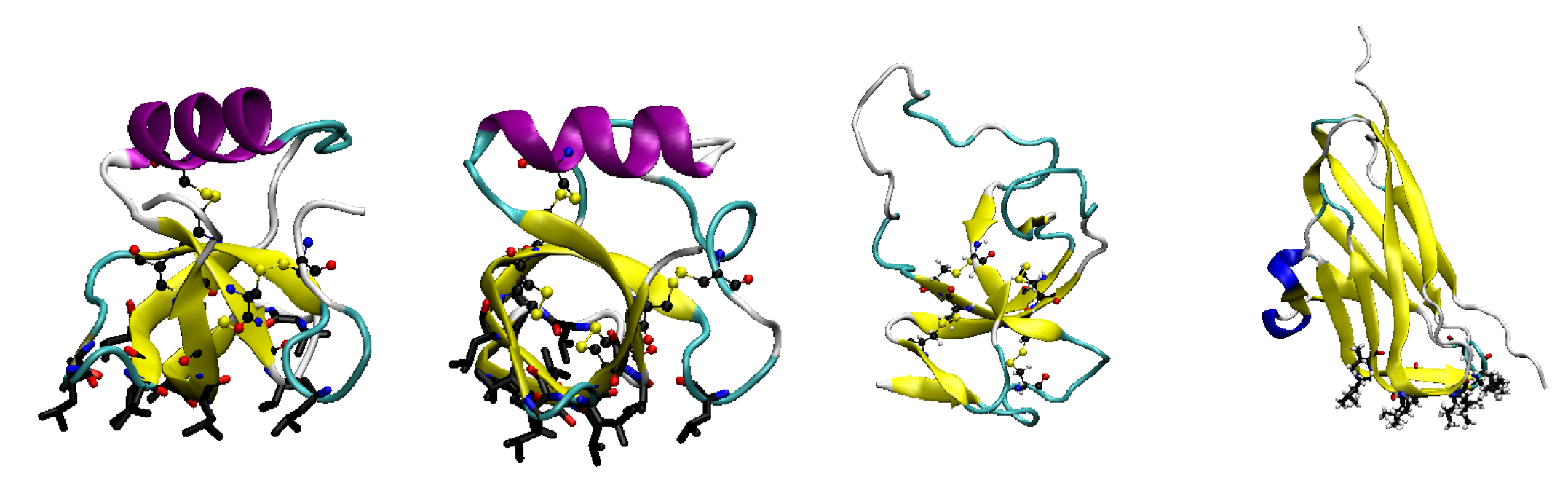
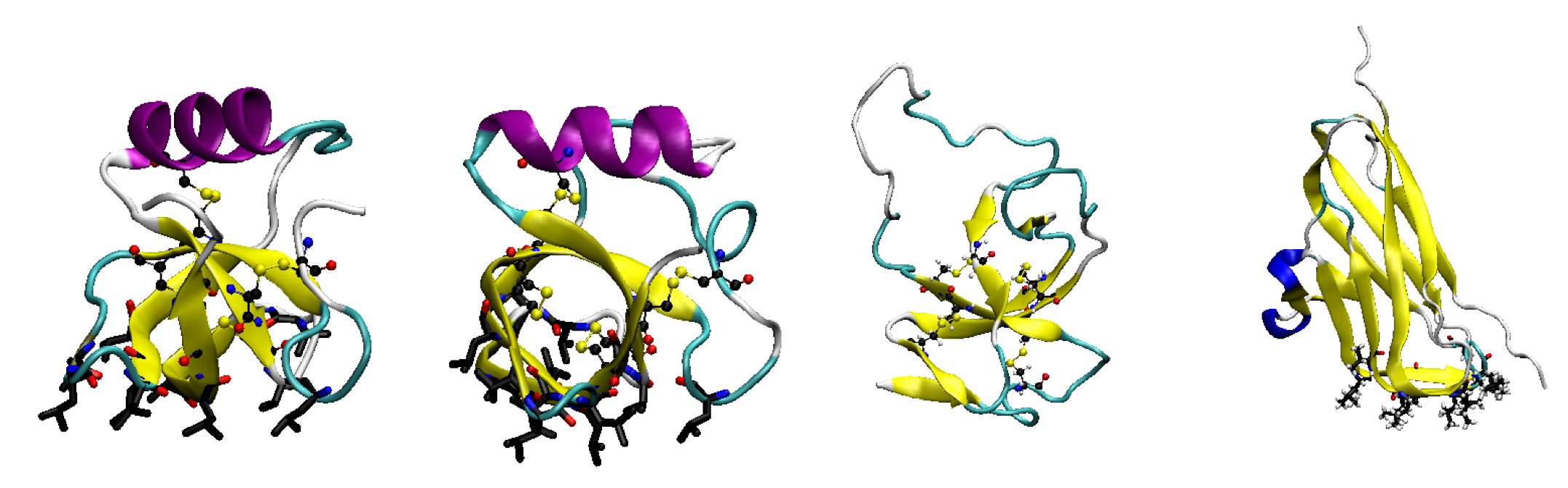
3. Hydrophobins
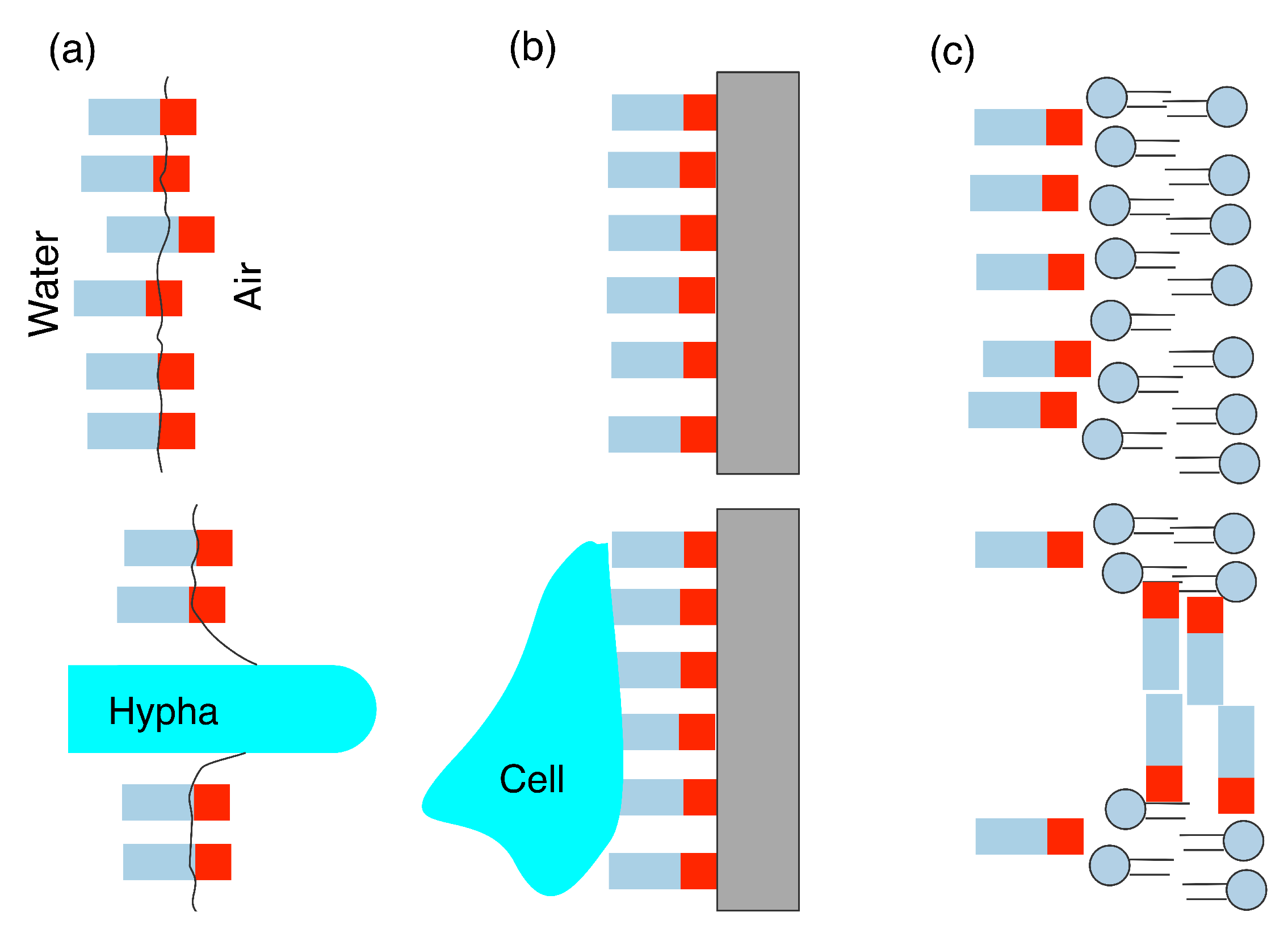
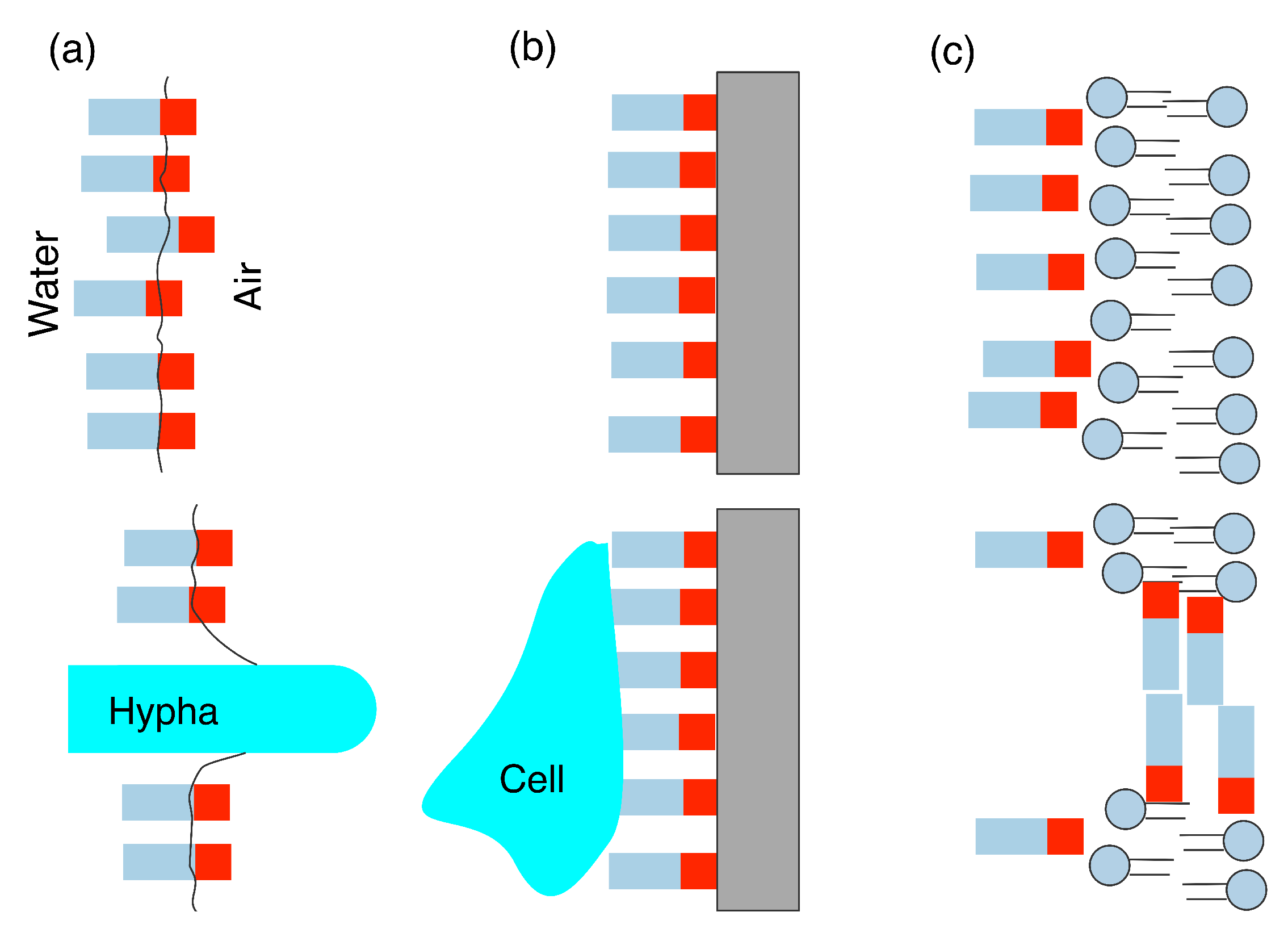
3.1. Hydrophobin Behaviour at Fluid (Air–Water or Oil–Water) Interfaces
3.2. Adsorption onto Surfaces
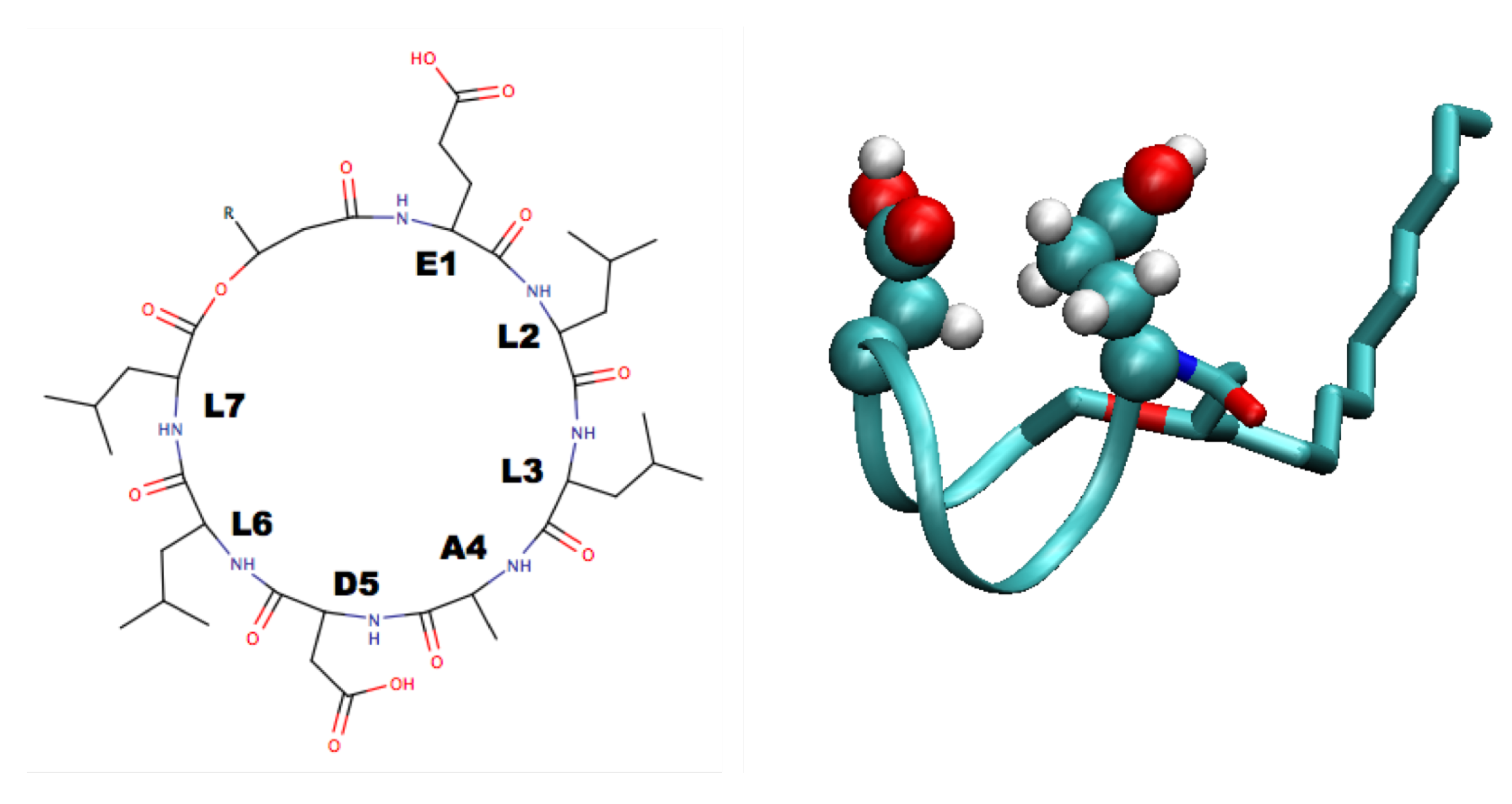
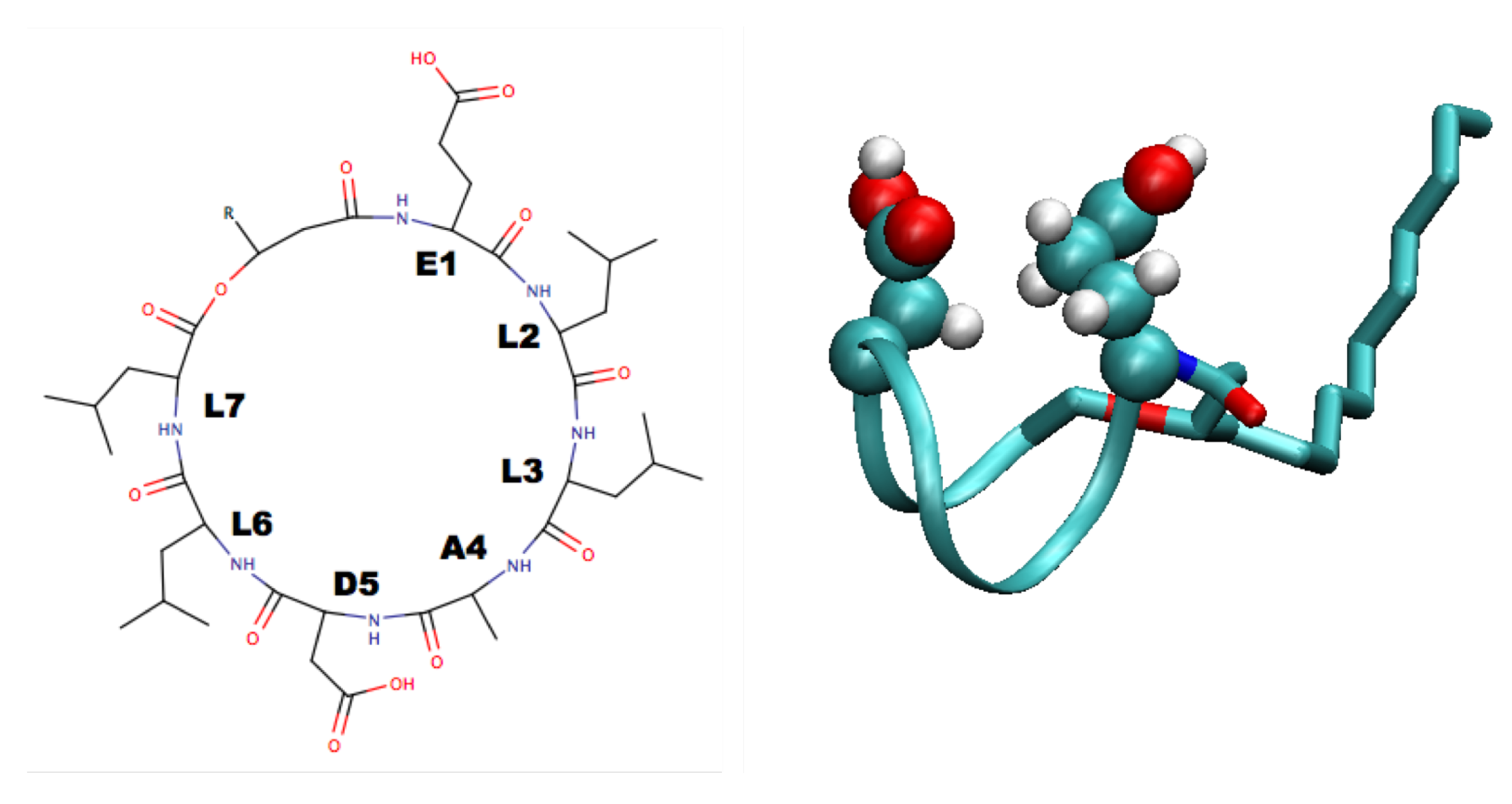
4. Surfactin and Other Lipopeptides
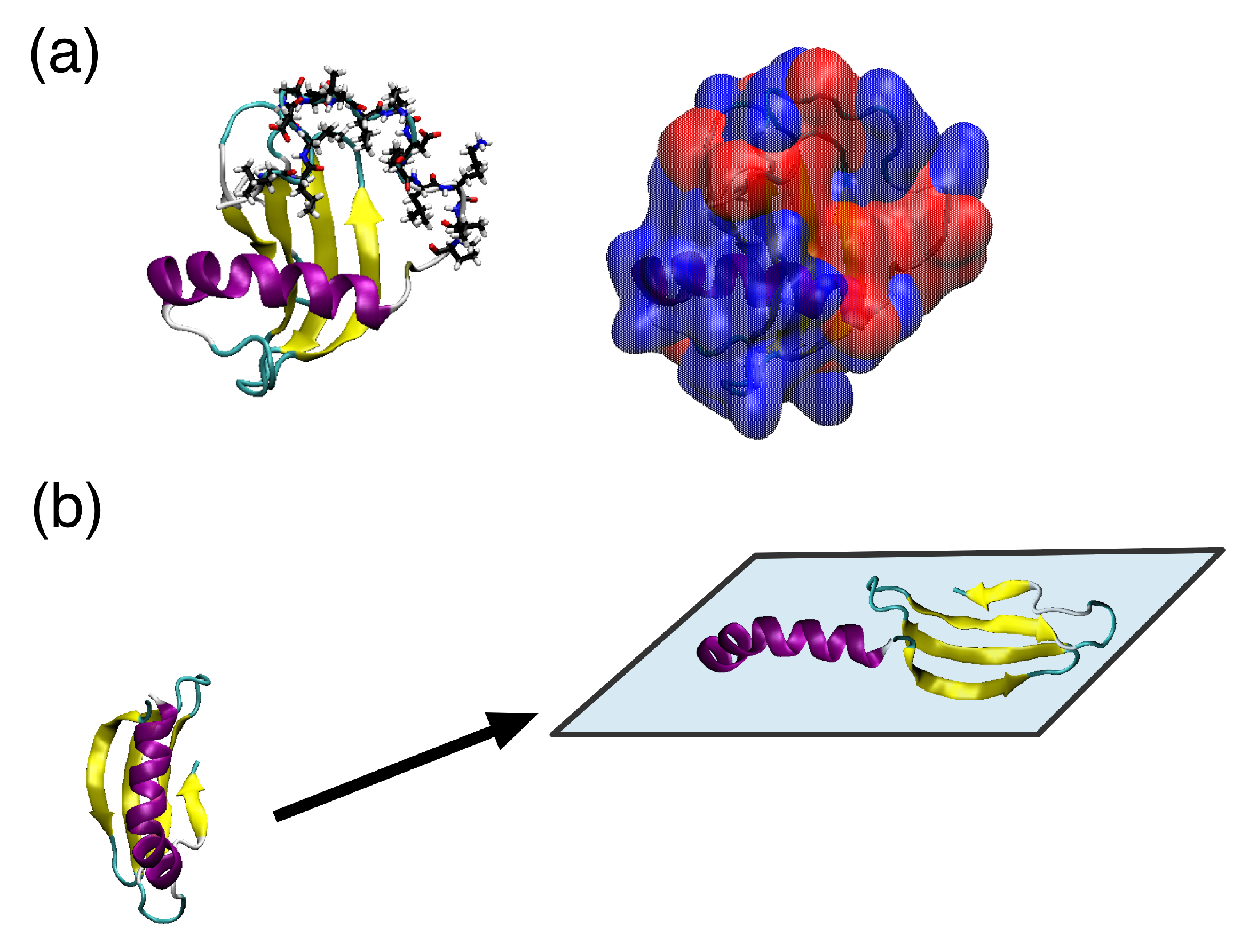
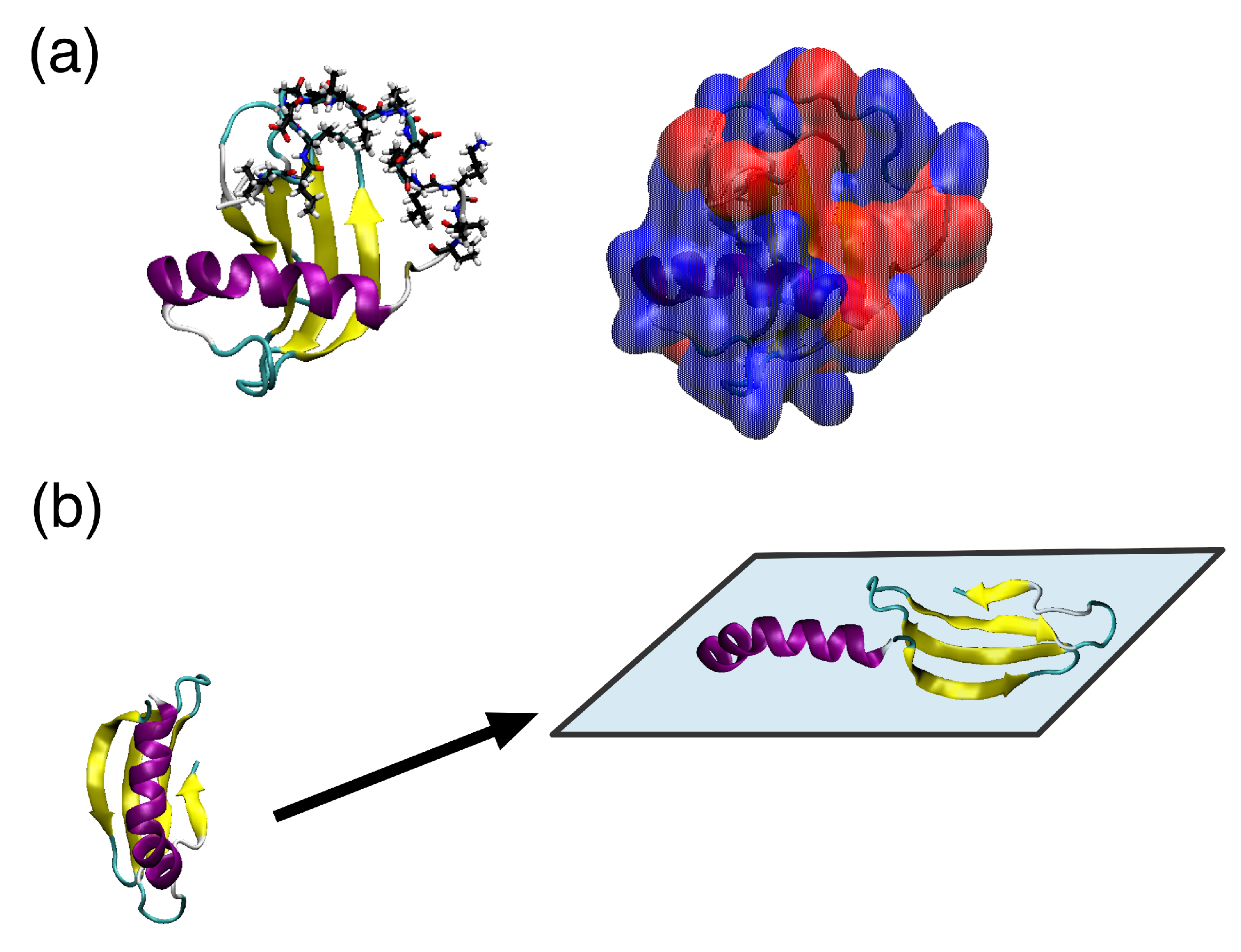
5. Ranaspumin
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CG | Coarse-Grain |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| EM | Electron Microscopy |
| GPU | Graphics Processing Unit |
| IRRAS | Infra-Red Reflection Adsorption Spectropscopy |
| MD | Molecular Dynamics |
| MM-PBSA | Molecular Mechanics Poisson–Boltzmann Solvation Approximation |
| NMR | Nuclear Magnetic Resonance |
| OPLS | Optimised Potentials for Liquid Simulation |
| PDB | Protein Data Bank |
| PLUNC | Palate Lung Nasal Epithelial Clone |
| SAM | Self-Assembled Monolayer |
| SANS | Small Angle Neutron Scattering |
| VMD | Visual Molecular Dynamics |
References
- Ron, E.Z.; Rosenberg, E. Natural roles of biosurfactants. Environ. Microbiol. 2001, 3, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.M.; De Bruijn, I.; Nybroe, O.; Ongena, M. Natural functions of lipopeptides from Bacillus and Pseudomonas: More than surfactants and antibiotics. FEMS Microbiol. Rev. 2010, 34, 1037–1062. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.; Van Wetter, M.A.; Lugones, L.G.; Van der Mei, H.C.; Busscher, H.J.; Wessels, J.G. How a fungus escapes the water to grow into the air. Curr. Biol. 1999, 9, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Elliot, M.A.; Talbot, N.J. Building filaments in the air: Aerial morphogenesis in bacteria and fungi. Curr. Opin. Microbiol. 2004, 7, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Arnaouteli, S.; MacPhee, C.E.; Stanley-Wall, N.R. Just in case it rains: Building a hydrophobic biofilm the Bacillus subtilis way. Curr. Opin. Microbiol. 2016, 34, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.; Schuren, F.H.; Wessels, J.G. Interfacial self-assembly of a hydrophobin into an amphipathic protein membrane mediates fungal attachment to hydrophobic surfaces. EMBO J. 1994, 13, 5848–5854. [Google Scholar] [CrossRef] [PubMed]
- Cameotra, S.S.; Makkar, R.S. Recent applications of biosurfactants as biological and immunological molecules. Curr. Opin. Microbiol. 2004, 7, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V.; et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, K.; Chi, L.; Fuchs, H.; Pötter, M.; Steinbüchel, A. Studies on the influence of phasins on accumulation and degradation of PHB and nanostructure of PHB granules in Raistonia eutropha H16. Biomacromolecules 2007, 8, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Schor, M.; Reid, J.L.; MacPhee, C.E.; Stanley-Wall, N.R. The Diverse Structures and Functions of Surfactant Proteins. Trends Biochem. Sci. 2016, 41, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, M.; Pham, C.L.; Kwan, A.H. Molecular Characteristics and Biological Functions of Surface-Active and Surfactant Proteins. Annu. Rev. Biochem. 2017, 86, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.F. Kinetics of protein unfolding at interfaces. J. Phys. Condens. Matter 2012, 24, 503101. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.L.; Day, L.; Aguilar, M.I.; Wooster, T.J. Protein folding at emulsion oil/water interfaces. Curr. Opin. Colloid Interface Sci. 2013, 18, 257–271. [Google Scholar] [CrossRef]
- Cooper, A.; Kennedy, M.W. Biofoams and natural protein surfactants. Biophys. Chem. 2010, 151, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wösten, H.A.; Scholtmeijer, K. Applications of hydrophobins: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Vance, S.J.; Smith, B.O.; Kennedy, M.W. Frog foams and natural protein surfactants. Colloids Surf. A Physicochem. Eng. Asp. 2017, 534, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Tchuenbou-Magaia, F.; Norton, I.; Cox, P. Hydrophobins stabilised air-filled emulsions for the food industry. Food Hydrocoll. 2009, 23, 1877–1885. [Google Scholar] [CrossRef]
- Cox, A.R.; Aldred, D.L.; Russell, A.B. Exceptional stability of food foams using class II hydrophobin HFBII. Food Hydrocoll. 2009, 23, 366–376. [Google Scholar] [CrossRef]
- Nishinari, K.; Fang, Y.; Guo, S.; Phillips, G.O. Soy proteins: A review on composition, aggregation and emulsification. Food Hydrocoll. 2014, 39, 301–318. [Google Scholar] [CrossRef]
- Dickinson, E. Structure formation in casein-based gels, foams, and emulsions. Colloids Surf. A Physicochem. Eng. Asp. 2006, 288, 3–11. [Google Scholar] [CrossRef]
- Valo, H.K.; Laaksonen, P.H.; Peltonen, L.J.; Linder, M.B.; Hirvonen, J.T.; Laaksonen, T.J. Multifunctional hydrophobin: Toward functional coatings for drug nanoparticles. ACS Nano 2010, 4, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Kimpel, F.; Schmitt, J.J. Review: Milk Proteins as Nanocarrier Systems for Hydrophobic Nutraceuticals. J. Food Sci. 2015, 80, R2361–R2366. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Ebersbacher, C.F.; Myung, N.V.; Montemagno, C.D. Synthesis of nanoparticles with frog foam nest proteins. J. Nanoparticle Res. 2012, 14, 1–13. [Google Scholar] [CrossRef]
- Santhiya, D.; Burghard, Z.; Greiner, C.; Jeurgens, L.P.H.; Subkowski, T.; Bill, J. Bioinspired deposition of TiO2 thin films induced by hydrophobins. Langmuir 2010, 26, 6494–6502. [Google Scholar] [CrossRef] [PubMed]
- Maity, J.P.; Lin, T.J.; Cheng, H.P.H.; Chen, C.Y.; Reddy, A.S.; Atla, S.B.; Chang, Y.F.; Chen, H.R.; Chen, C.C. Synthesis of brushite particles in reverse microemulsions of the biosurfactant surfactin. Int. J. Mol. Sci. 2011, 12, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Whang, L.M.; Liu, P.W.G.; Ma, C.C.; Cheng, S.S. Application of biosurfactants, rhamnolipid, and surfactin, for enhanced biodegradation of diesel-contaminated water and soil. J. Hazard. Mater. 2008, 151, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Laaksonen, P.; Walther, A.; Malho, J.M.; Kainlauri, M.; Ikkala, O.; Linder, M.B. Genetic engineering of biomimetic nanocomposites: Diblock proteins, graphene, and nanofibrillated cellulose. Angew. Chem. Int. Ed. 2011, 50, 8688–8691. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Gravagnuolo, A.M.; Maddalena, P.; Altucci, C.; Giardina, P.; Gesuele, F. Green synthesis of luminescent and defect-free bio-nanosheets of MoS2: Interfacing two-dimensional crystals with hydrophobins. RSC Adv. 2017, 7, 22400–22408. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife 2016, 5, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and simulation of protein-surface interactions: Achievements and challenges. Q. Rev. Biophys. 2016, 49, 1–45. [Google Scholar] [CrossRef] [PubMed]
- James, J.J.; Lakshmi, B.S.; Seshasayee, A.S.N.; Gautam, P. Activation of Candida rugosa lipase at alkane-aqueous interfaces: A molecular dynamics study. FEBS Lett. 2007, 581, 4377–4383. [Google Scholar] [CrossRef] [PubMed]
- Kubiak, K.; Mulheran, P.A. Molecular dynamics simulations of hen egg white lysozyme adsorption at a charged solid surface. J. Phys. Chem. B 2009, 113, 12189–12200. [Google Scholar] [CrossRef] [PubMed]
- Arooj, M.; Gandhi, N.S.; Kreck, C.A.; Arrigan, D.W.; Mancera, R.L. Adsorption and Unfolding of Lysozyme at a Polarized Aqueous-Organic Liquid Interface. J. Phys. Chem. B 2016, 120, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L. Adsorption and conformations of lysozyme and α-lactalbumin at a water–octane interface. J. Chem. Phys. 2017, 147, 195101. [Google Scholar] [CrossRef] [PubMed]
- Zare, D.; McGrath, K.M.; Allison, J.R. Deciphering β-Lactoglobulin Interactions at an Oil–Water Interface: A Molecular Dynamics Study. Biomacromolecules 2015, 16, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Zare, D.; Allison, J.R.; McGrath, K.M. Molecular Dynamics Simulation of Beta-Lactoglobulin at Different Oil/Water Interfaces. Biomacromolecules 2016, 17, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, L.; Bussi, G.; Di Felice, R.; Corni, S. Fibrillation-prone conformations of the amyloid-β-42 peptide at the gold/water interface. Nanoscale 2017, 9, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms To Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Mackerell, A.D. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; MacKerell, A.D.J. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Tieleman, D.P. Perspective on the martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periole, X. Interplay of G protein-coupled receptors with the membrane: Insights from supra-atomic coarse grain molecular dynamics simulations. Chem. Rev. 2017, 117, 156–185. [Google Scholar] [CrossRef] [PubMed]
- Euston, S.R. Molecular dynamics simulation of protein adsorption at fluid interfaces: A comparison of all-atom and coarse-grained models. Biomacromolecules 2010, 11, 2781–2787. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, W.; DeVane, R.; Klein, M.L. Multi-property fitting and parameterization of a coarse grained model for aqueous surfactants. Mol. Simul. 2007, 33, 27–36. [Google Scholar] [CrossRef]
- DeVane, R.; Shinoda, W.; Moore, P.B.; Klein, M.L. Transferable Coarse Grain Nonbonded Interaction Model for Amino Acids. J. Chem. Theory Comput. 2009, 5, 2115–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica exchange molecular dynamics method for protein folding simulation. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Earl, D.J.; Deem, M.W. Parallel tempering: Theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 2005, 7, 3910–3916. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L. Conformations of Myoglobin-Derived Peptides at the Air–Water Interface. Langmuir 2016, 32, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Xie, Y.; Zhou, J. Computer simulations of fibronectin adsorption on hydroxyapatite surfaces. RSC Adv. 2014, 4, 15759–15769. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Piana, S.; Laio, A. A Bias-Exchange Approach to Protein Folding. J. Phys. Chem. B 2007, 111, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603/1–4. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Colombi Ciacchi, L. Specific material recognition by small peptides mediated by the interfacial solvent structure. J. Am. Chem. Soc. 2012, 134, 2407–2413. [Google Scholar] [CrossRef] [PubMed]
- Deighan, M.; Pfaendtner, J. Exhaustively Sampling Peptide Adsorption with Metadynamics. Langmuir 2013, 29, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.A.; Fischer, S.A.; Shea, J.E.; Pfaendtner, J. Trp-Cage Folding on Organic Surfaces. J. Phys. Chem. B 2015, 119, 10417–10425. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.B.; Palafox-Hernandez, J.P.; Rodger, P.M.; Corni, S.; Walsh, T.R. Facet selectivity in gold binding peptides: Exploiting interfacial water structure. Chem. Sci. 2015, 6, 5204–5214. [Google Scholar] [CrossRef] [PubMed]
- Huai Han, M.; Cheng Chiu, C. Fast estimation of protein conformational preference at air/water interface via molecular dynamics simulations. J. Taiwan Inst. Chem. Eng. 2018. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Park, S.; Schulten, K. Calculating potentials of mean force from steered molecular dynamics simulations. J. Chem. Phys. 2004, 120, 5946–5961. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.B.; Wang, M.; Yan, C.; Oloyede, A. Molecular dynamics simulation of mechanical behavior of osteopontin-hydroxyapatite interfaces. J. Mech. Behav. Biomed. Mater. 2014, 36, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Abbott, N.L.; Pablo, J.J.D.; Miller, C.A.; Abbott, N.L.; Pablo, J.J.D. Surface Activity of Amphiphilic Helical beta-Peptides from Molecular Dynamics Simulation Surface Activity of Amphiphilic Helical-Peptides from Molecular Dynamics Simulation. Langmuir 2009, 25, 2811–2823. [Google Scholar] [CrossRef] [PubMed]
- Engin, O.; Villa, A.; Sayar, M.; Hess, B. Driving forces for adsorption of amphiphilic peptides to the air–water interface. J. Phys. Chem. B 2010, 114, 11093–11101. [Google Scholar] [CrossRef] [PubMed]
- Engin, O.; Sayar, M. Adsorption, folding, and packing of an amphiphilic peptide at the air/water interface. J. Phys. Chem. B 2012, 116, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.B. Hydrophobins: Proteins that self assemble at interfaces. Curr. Opin. Colloid Interface Sci. 2009, 14, 356–363. [Google Scholar] [CrossRef]
- Linder, M.B.; Szilvay, G.R.; Nakari-Setälä, T.; Penttilä, M.E. Hydrophobins: The protein-amphiphiles of filamentous fungi. FEMS Microbiol. Rev. 2005, 29, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Temple, B.; Horgen, P.A.; Bernier, L.; Hintz, W.E. Cerato-ulmin, a hydrophobin secreted by the causal agents of Dutch elm disease, is a parasitic fitness factor. Fungal Genet. Biol. 1997, 22, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.; Szilvay, G.R.; Nakari-Setälä, T.; Söderlund, H.; Penttilä, M. Surface adhesion of fusion proteins containing the hydrophobins HFBI and HFBII from Trichoderma reesei. Protein Sci. 2002, 11, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Hobley, L.; Ostrowski, A.; Rao, F.V.; Bromley, K.M.; Porter, M.; Prescott, A.R.; MacPhee, C.E.; van Aalten, D.M.F.; Stanley-Wall, N.R. BslA is a self-assembling bacterial hydrophobin that coats the Bacillus subtilis biofilm. Proc. Natl. Acad. Sci. USA 2013, 110, 13600–13605. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zangi, R.; de Vocht, M.L.; Robillard, G.T.; Mark, A.E. Molecular dynamics study of the folding of hydrophobin SC3 at a hydrophilic/hydrophobic interface. Biophys. J. 2002, 83, 112–124. [Google Scholar] [CrossRef]
- Euston, S.R. Molecular simulation of adsorption of hydrophobin HFBI to the air–water, DPPC-water and decane-water interfaces. Food Hydrocoll. 2013, 42, 66–74. [Google Scholar] [CrossRef]
- Zhao, Y.; Cieplak, M. Proteins at air-Water and oil-water interfaces in an all-atom model. Phys. Chem. Chem. Phys. 2017, 19, 25197–25206. [Google Scholar] [CrossRef] [PubMed]
- Raffaini, G.; Milani, R.; Ganazzoli, F.; Resnati, G.; Metrangolo, P. Atomistic simulation of hydrophobin HFBII conformation in aqueous and fluorous media and at the water/vacuum interface. J. Mol. Graph. Model. 2016, 63, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Wang, X.; Zhu, J.; Robillard, G.T.; Mark, A.E. Molecular dynamics simulations of the hydrophobin SC3 at a hydrophobic/hydrophilic interface. Proteins 2006, 64, 863–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askolin, S.; Linder, M.; Scholtmeijer, K.; Tenkanen, M.; Penttilä, M.; de Vocht, M.L.; Wösten, H.A.B. Interaction and comparison of a class I hydrophobin from Schizophyllum commune and class II hydrophobins from Trichoderma reesei. Biomacromolecules 2006, 7, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Penfold, J.; Thomas, R.K.; Tucker, I.M.; Petkov, J.T.; Bent, J.; Cox, A.; Campbell, R.A. Adsorption behavior of hydrophobin and hydrophobin/surfactant mixtures at the air–water interface. Langmuir 2011, 27, 11316–11323. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.H.Y.; Winefield, R.D.; Sunde, M.; Matthews, J.M.; Haverkamp, R.G.; Templeton, M.D.; Mackay, J.P. Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. USA 2006, 103, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Kitchen, C.; Kwan, A.H.; Sunde, M.; Dobson, C.M.; Frenkel, D. Intrinsic disorder modulates protein self-assembly and aggregation. Proc. Natl. Acad. Sci. USA 2012, 109, 6951–6956. [Google Scholar] [CrossRef] [PubMed]
- Jean, L.; Lee, C.F.; Vaux, D.J. Enrichment of amyloidogenesis at an air–water interface. Biophys. J. 2012, 102, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Campioni, S.; Carret, G.; Jordens, S.; Nicoud, L.; Mezzenga, R.; Riek, R. The Presence of an Air-Water Interface Affects Formation and Elongation of α-Synuclein Fibrils. J. Am. Chem. Soc. 2013, 136, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Fioroni, M.; Linder, M.B.; Nessel, A.; Bocola, M.; Subkowski, T.; Schwaneberg, U.; Böker, A.; Rodríguez-Ropero, F. Exploring the mineralization of hydrophobins at a liquid interface. Soft Matter 2012, 8, 11343–11352. [Google Scholar] [CrossRef]
- Schulz, A.; Liebeck, B.M.; John, D.; Heiss, A.; Subkowski, T.; Böker, A. Protein—Mineral hybrid capsules from emulsions stabilized with an amphiphilic protein. J. Mater. Chem. 2011, 21, 9731–9736. [Google Scholar] [CrossRef]
- Cheung, D.L. Molecular simulation of hydrophobin adsorption at an oil-water interface. Langmuir 2012, 28, 8730–8736. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Glogowski, E.; Emrick, T.; Russell, T.P.; Dinsmore, A.D. Adsorption Energy of Nano- and Microparticles at Liquid-Liquid Interfaces. Langmuir 2010, 26, 12518–12522. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L.; Carbone, P. How stable are amphiphilic dendrimers at the liquid–liquid interface? Soft Matter 2013, 9, 6841–6850. [Google Scholar] [CrossRef] [Green Version]
- Bromley, K.M.; Morris, R.J.; Hobley, L.; Brandani, G.; Gillespie, R.M.C.; McCluskey, M.; Zachariae, U.; Marenduzzo, D.; Stanley-Wall, N.R.; MacPhee, C.E. Interfacial self-assembly of a bacterial hydrophobin. Proc. Natl. Acad. Sci. USA 2015, 112, 5419–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, D.L.; Bon, S.A.F. Stability of Janus nanoparticles at fluid interfaces. Soft Matter 2009, 5, 3969–3976. [Google Scholar] [CrossRef] [Green Version]
- Brandani, G.B.; Schor, M.; Morris, R.; Stanley-Wall, N.; MacPhee, C.E.; Marenduzzo, D.; Zachariae, U. The Bacterial Hydrophobin BslA is a Switchable Ellipsoidal Janus Nanocolloid. Langmuir 2015, 31, 11558–11563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, C.; Thompson, D. Molecular dynamics of the “hydrophobic patch” that immobilizes hydrophobin protein HFBII on silicon. J. Mol. Model. 2011, 17, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Mereghetti, P.; Wade, R.C. Diffusion of hydrophobin proteins in solution and interactions with a graphite surface. BMC Biophys. 2011, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Szilvay, G.R.; Kisko, K.; Serimaa, R.; Linder, M.B. The relation between solution association and surface activity of the hydrophobin HFBI from Trichoderma reesei. FEBS Lett. 2007, 581, 2721–2726. [Google Scholar] [CrossRef] [PubMed]
- Mrksich, M.; Whitesides, G.M. Using Self-Assembled Monolayers to Understand the Interactions of Man-made Surfaces with Proteins and Cells. Annu. Rev. Biophys. Biomol. Struct. 2003, 25, 55–78. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.; O’Dwyer, C.; Nijhuis, C.A.; Greer, J.C.; Quinn, A.J.; Thompson, D. Nanoscale dynamics and protein adhesivity of alkylamine self-assembled monolayers on graphene. Langmuir 2013, 29, 7271–7282. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Liu, J.; Zhao, D.; Zhou, J. Adsorption of hydrophobin on different self-assembled monolayers: The role of the hydrophobic dipole and the electric dipole. Langmuir 2014, 30, 11401–11411. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, M.; Feng, X.; Shao, X.; Cai, W. Adsorption behavior of hydrophobin proteins on polydimethylsiloxane substrates. J. Phys. Chem. B 2012, 116, 12227–12234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Penfold, J.; Thomas, R.K.; Tucker, I.M.; Petkov, J.T.; Bent, J.; Cox, A. Adsorption behavior of hydrophobin and hydrophobin/surfactant mixtures at the solid-solution interface. Langmuir 2011, 27, 10464–10474. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Christofferson, A.; Penna, M.; Winkler, D.; Maclaughlin, S.; Yarovsky, I. Surface-water Interface Induces Conformational Changes Critical for Protein Adsorption: Implications for Monolayer Formation of EAS Hydrophobin. Front. Mol. Biosci. 2015, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, L.; Zhao, C.; Zheng, J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef]
- Sheikh, S.; Blaszykowski, C.; Nolan, R.; Thompson, D.; Thompson, M. On the hydration of subnanometric antifouling organosilane adlayers: A molecular dynamics simulation. J. Colloid Interface Sci. 2015, 437, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Penna, M.; Ley, K.; Maclaughlin, S.; Yarovsky, I. Surface heterogeneity: A friend or foe of protein adsorption—Insights from theoretical simulations. Faraday Discuss. 2016, 191, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Bonmatin, J.; Genest, M.; Labbé, H.; Ptak, M. Solution three-dimensional structure of surfactin: A cyclic lipopeptide studied by 1H-nmr, distance geometry, and molecular dynamics. Biopolymers 1994, 34, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Kinsinger, R.F.; Shirk, M.C.; Fall, R. Rapid surface motility in Bacillus subtilis is dependent on extracellular surfactin and potassium ion. J. Bacteriol. 2003, 185, 5627–5631. [Google Scholar] [CrossRef] [PubMed]
- Seydlová, G.; Svobodová, J. Review of surfactin chemical properties and the potential biomedical applications. Central Eur. J. Med. 2008, 3, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Gallet, X.; Deleu, M.; Razafindralambo, H.; Jacques, P.; Thonart, P.; Paquot, M.; Brasseur, R. Computer Simulation of Surfactin Conformation at a Hydrophobic/Hydrophilic Interface. Langmuir 1999, 15, 2409–2413. [Google Scholar] [CrossRef]
- Nicolas, J.P. Molecular dynamics simulation of surfactin molecules at the water-hexane interface. Biophys. J. 2003, 85, 1377–1391. [Google Scholar] [CrossRef]
- Binks, B.P. Particles as surfactants—Similarities and differences. Curr. Opin. Colloid Interface Sci. 2002, 7, 21–41. [Google Scholar] [CrossRef]
- Gang, H.Z.; Liu, J.F.; Mu, B.Z. Interfacial behavior of surfactin at the decane/water interface: A molecular dynamics simulation. J. Phys. Chem. B 2010, 114, 14947–14954. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Fernández, J.; Darré, L.; Kohlmeyer, A.; Thomas, R.K.; Shen, H.H.; Domene, C. Surfactin at the Water/Air Interface and in Solution. Langmuir 2015, 31, 11097–11104. [Google Scholar] [CrossRef] [PubMed]
- Gang, H.Z.; Liu, J.F.; Mu, B.Z. Molecular dynamics simulation of surfactin derivatives at the decane/water interface at low surface coverage. J. Phys. Chem. B 2010, 114, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, A.; Matsumoto, M.; Ishiguro, R.; Murata, K.; Fujiyoshi, Y.; Ishigami, Y.; Osman, M. Electron cryo-microscopic studies on micellar shape and size of surfactin, an anionic lipopeptide. Colloids Surf. B Biointerfaces 1995, 5, 43–48. [Google Scholar] [CrossRef]
- Han, Y.; Huang, X.; Cao, M.; Wang, Y. Micellization of surfactin and its effect on the aggregate conformation of amyloid β(1–40). J. Phys. Chem. B 2008, 112, 15195–15201. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.H.; Thomas, R.K.; Chen, C.Y.; Darton, R.C.; Baker, S.C.; Penfold, J. Aggregation of the naturally occurring lipopeptide, surfactin, at interfaces and in solution: An unusual type of surfactant? Langmuir 2009, 25, 4211–4218. [Google Scholar] [CrossRef] [PubMed]
- She, A.Q.; Gang, H.Z.; Mu, B.Z. Temperature Influence on the Structure and Interfacial Properties of Surfactin Micelle: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2012, 116, 12735–12743. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.A.; Swope, W.C.; Jordan, K.E.; Warren, P.B.; Noro, M.G.; Bray, D.J.; Anderson, R.L. Toward a Standard Protocol for Micelle Simulation. J. Phys. Chem. B 2016, 120, 6337–6351. [Google Scholar] [CrossRef] [PubMed]
- Loison, C.; Nasir, M.N.; Benichou, E.; Besson, F.; Brevet, P.F. Multi-scale modeling of mycosubtilin lipopeptides at the air/water interface: Structure and optical second harmonic generation. Phys. Chem. Chem. Phys. 2014, 16, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Cen, M.; Fan, J.F.; Liu, D.Y.; Song, X.Z.; Liu, J.; Zhou, W.Q.; Xiao, H.M. Cyclo-hexa-peptides at the water/cyclohexane interface: A molecular dynamics simulation. J. Mol. Model. 2013, 19, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; DeVane, R.H.; Kohlmeyer, A.; Klein, M.L. Probing Peptide Nanotube Self-Assembly at a Liquid-Liquid Interface with Coarse-Grained Molecular Dynamics. Nano Lett. 2008, 8, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Kennedy, M.W.; Fleming, R.I.; Wilson, E.H.; Videler, H.; Wokosin, D.L.; Su, T.J.; Green, R.J.; Lu, J.R. Adsorption of frog foam nest proteins at the air–water interface. Biophys. J. 2005, 88, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, C.D.; Smith, B.O.; Meister, A.; Blume, A.; Zhao, X.; Lu, J.R.; Kennedy, M.W.; Cooper, A. Ranaspumin-2: Structure and function of a surfactant protein from the foam nests of a tropical frog. Biophys. J. 2009, 96, 4984–4992. [Google Scholar] [CrossRef] [PubMed]
- Brandani, G.B.; Vance, S.J.; Schor, M.; Cooper, A.; Kennedy, M.W.; Smith, B.O.; MacPhee, C.E.; Cheung, D.L. Adsorption of the natural protein surfactant Rsn-2 onto liquid interfaces. Phys. Chem. Chem. Phys. 2017, 19, 8584–8594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, J.; Wooster, T.J.; Hoffmann, S.V.; Lee, T.H.; Augustin, M.A.; Aguilar, M.I. Structural rearrangement of β-lactoglobulin at different oil-water interfaces and its effect on emulsion stability. Langmuir 2011, 27, 9227–9236. [Google Scholar] [CrossRef] [PubMed]
- Go, N. Theoretical Studies of Protein Folding. Annu. Rev. Biophys. Bioeng. 1983, 12, 183–210. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.J.; Brandani, G.B.; Desai, V.; Smith, B.O.; Schor, M.; MacPhee, C.E. The Conformation of Interfacially Adsorbed Ranaspumin-2 is an Arrested State on the Unfolding Pathway. Biophys. J. 2016, 111, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.M.; Visser, A.J.W.G.; van Mierlo, C.P.M. Conformation and orientation of a protein folding intermediate trapped by adsorption. Proc. Natl. Acad. Sci. USA 2004, 101, 11316–11321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, G.; Vellore, N.A.; Yancey, J.A.; Stuart, S.J.; Latour, R.A. Comparison between empirical protein force fields for the simulation of the adsorption behavior of structured LK peptides on functionalized surfaces. Biointerphases 2012, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.K.; Vellore, N.A.; Yancey, J.A.; Kucukkal, T.G.; Collier, G.; Brooks, B.R.; Stuart, S.J.; Latour, R.A. Simulation of multiphase systems utilizing independent force fields to control intraphase and interphase behavior. J. Comput. Chem. 2012, 33, 1458–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating Molecular Dynamic Simulation on Graphics Processing Units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.E.; Fleming, R.I.; Beeley, J.G.; Bovell, D.L.; Lu, J.R.; Zhao, X.; Cooper, A.; Kennedy, M.W. Latherin: A surfactant protein of horse sweat and saliva. PLoS ONE 2009, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vance, S.J.; Mcdonald, R.E.; Cooper, A.; Smith, B.O.; Kennedy, M.W. The structure of latherin, a surfactant allergen protein from horse sweat and saliva. J. R. Soc. Interface 2013, 10, 20130453. [Google Scholar] [CrossRef] [PubMed]
- Gakhar, L.; Bartlett, J.A.; Penterman, J.; Mizrachi, D.; Singh, P.K.; Mallampalli, R.K.; Ramaswamy, S.; McCray, P.B. PLUNC is a novel airway surfactant protein with anti-biofilm activity. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Ning, F.; Wang, C.; Berry, K.Z.; Kandasamy, P.; Liu, H.; Murphy, R.C.; Voelker, D.R.; Nho, C.W.; Pan, C.H.; Dai, S.; et al. Structural characterization of the pulmonary innate immune protein SPLUNC1 and identification of lipid ligands. FASEB J. 2014, 28, 5349–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, W.G.; Ahmad, S.; Little, M.S.; Kim, C.S.; Tyrrell, J.; Lin, Q.; Di, Y.P.; Tarran, R.; Redinbo, M.R. Structural Features Essential to the Antimicrobial Functions of Human SPLUNC1. Biochemistry 2016, 55, 2979–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, K.; Fujisawa, T.; Hill, J.P.; Ariga, K. Conformational interchange of a carbohydrate by mechanical compression at the air–water interface. Phys. Chem. Chem. Phys. 2014, 16, 10286–10294. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, D.; Zhao, C.X.; Middelberg, A.P.J. Interfacial Biomimetic Synthesis of Silica Nanocapsules Using a Recombinant Catalytic Modular Protein. Langmuir 2015, 31, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, D.; Mori, T.; Yonamine, Y.; Nakanishi, W.; Cheung, D.L.; Hill, J.P.; Ariga, K. Mechanochemical Tuning of the Binaphthyl Conformation at the Air-Water Interface. Angew. Chem. Int. Ed. 2015, 54, 8988–8991. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; He, L.; Middelberg, A.P.J.; Mark, A.E.; Poger, D. Determining the Structure of Interfacial Peptide Films: Comparing Neutron Reflectometry and Molecular Dynamics Simulations. Langmuir 2014, 30, 10080–10089. [Google Scholar] [CrossRef] [PubMed]
- Knecht, V.; Reiter, G.; Schlaad, H.; Reiter, R. Structure Formation in Langmuir Peptide Films As Revealed from Coarse-Grained Molecular Dynamics Simulations. Langmuir 2017, 33, 6492–6502. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, B.; Mazzuca, C.; Conflitti, P.; Venanzi, M.; Palleschi, A. Behavior of a Peptide during a Langmuir-Blodgett Compression Isotherm: A Molecular Dynamics Simulation Study. J. Phys. Chem. C 2018, 122, 515–521. [Google Scholar] [CrossRef]
- Thøgersen, L.; Schiøtt, B.; Vosegaard, T.; Nielsen, N.C.; Tajkhorshid, E. Peptide aggregation and pore formation in a lipid bilayer: A combined coarse-grained and all atom molecular dynamics study. Biophys. J. 2008, 95, 4337–4347. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Bevan, D.R. Aggregation of alzheimer’s amyloid β-peptide in biological membranes: A molecular dynamics study. Biochemistry 2013, 52, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Solernou, A.; Hanson, B.S.; Richardson, R.A.; Welch, R.; Read, D.J.; Harlen, O.G.; Harris, S.A. Fluctuating Finite Element Analysis (FFEA): A continuum mechanics software tool for mesoscale simulation of biomolecules. PLoS Comput. Biol. 2018, 14, 1–29. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Protein(s) | References |
|---|---|---|
| Emulsion/foam stabilisers in food | HFBII (hydrophobin) | [17,18] |
| Globulin | [19] | |
| Casein | [20] | |
| Drug delivery | HFBI (hydrophobin) | [21] |
| Casein | [22] | |
| Nanoparticle synthesis | Rsn-2 | [23] |
| Biomineralization | H* Protein B (hydrophobin) | [24] |
| Surfactin | [25] | |
| Remediation | Surfactin | [26] |
| Exfoliation of 2D materials | HFBI (hydrophobin) | [27] |
| Vmh2 (hydrophobin) | [28] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheung, D.L.; Samantray, S. Molecular Dynamics Simulation of Protein Biosurfactants. Colloids Interfaces 2018, 2, 39. https://doi.org/10.3390/colloids2030039
Cheung DL, Samantray S. Molecular Dynamics Simulation of Protein Biosurfactants. Colloids and Interfaces. 2018; 2(3):39. https://doi.org/10.3390/colloids2030039
Chicago/Turabian StyleCheung, David L., and Suman Samantray. 2018. "Molecular Dynamics Simulation of Protein Biosurfactants" Colloids and Interfaces 2, no. 3: 39. https://doi.org/10.3390/colloids2030039
APA StyleCheung, D. L., & Samantray, S. (2018). Molecular Dynamics Simulation of Protein Biosurfactants. Colloids and Interfaces, 2(3), 39. https://doi.org/10.3390/colloids2030039