The current study begins with MgNi-based alloy composition optimization, which is performed with thin film deposition using RF sputtering technique. After identifying the compositions that demonstrated the highest electrochemical capacities in the thin film form, bulk powder process consisting of MS + MA was adopted to reproduce the optimized compositions. Once the electrochemical performances of alloys in the bulk form were confirmed, the most suitable alloy composition in the bulk form was then selected for the investigation on modified electrolytes composed of various hydroxides. The abovementioned developments are discussed in detail in the following sections.
3.1. MgNi-Based Thin Film Prepared by Radio Frequency Sputtering
Thin film deposition was performed in a multi-target RF sputtering unit with a modified target and sample holder (
Figure 1). The deposition rate was approximately 0.5 μm·h
−1, and the resulting average film thickness was approximately 1 μm. In each run, 10–25 nickel substrates (1 cm × 1 cm) were deposited by thin films with different compositions, which were determined by energy-dispersive X-ray spectroscopy from the silicon witness samples placed among the nickel substrates. Results from the binary composition optimization are shown in
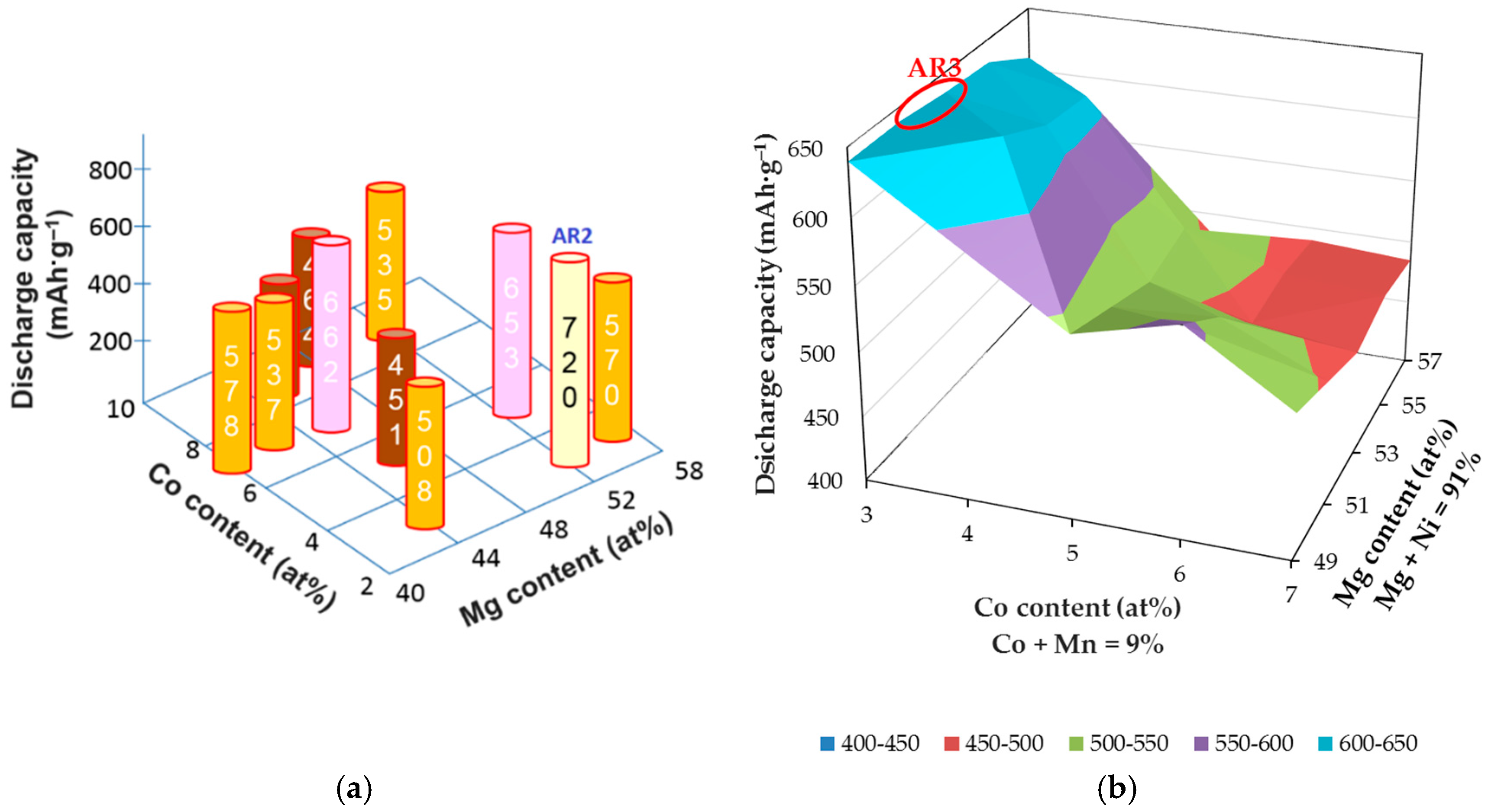
Table 2. Discharge capacity maximizes at the composition Mg
52Ni
48 (AR1), which is used as the basis for ternary Mg-Ni-Co and then quaternary Mg-Ni-Co-Mn composition optimizations (
Figure 2). Among all the compositions in the ternary and quaternary matrices, AR2 (Mg
52.1Ni
45.1Co
2.8) and AR3 (Mg
52Ni
39Co
3Mn
6) demonstrate the highest discharge capacities, respectively (
Table 3). Furthermore, the cycle stabilities of several RF-sputtered thin films are shown in
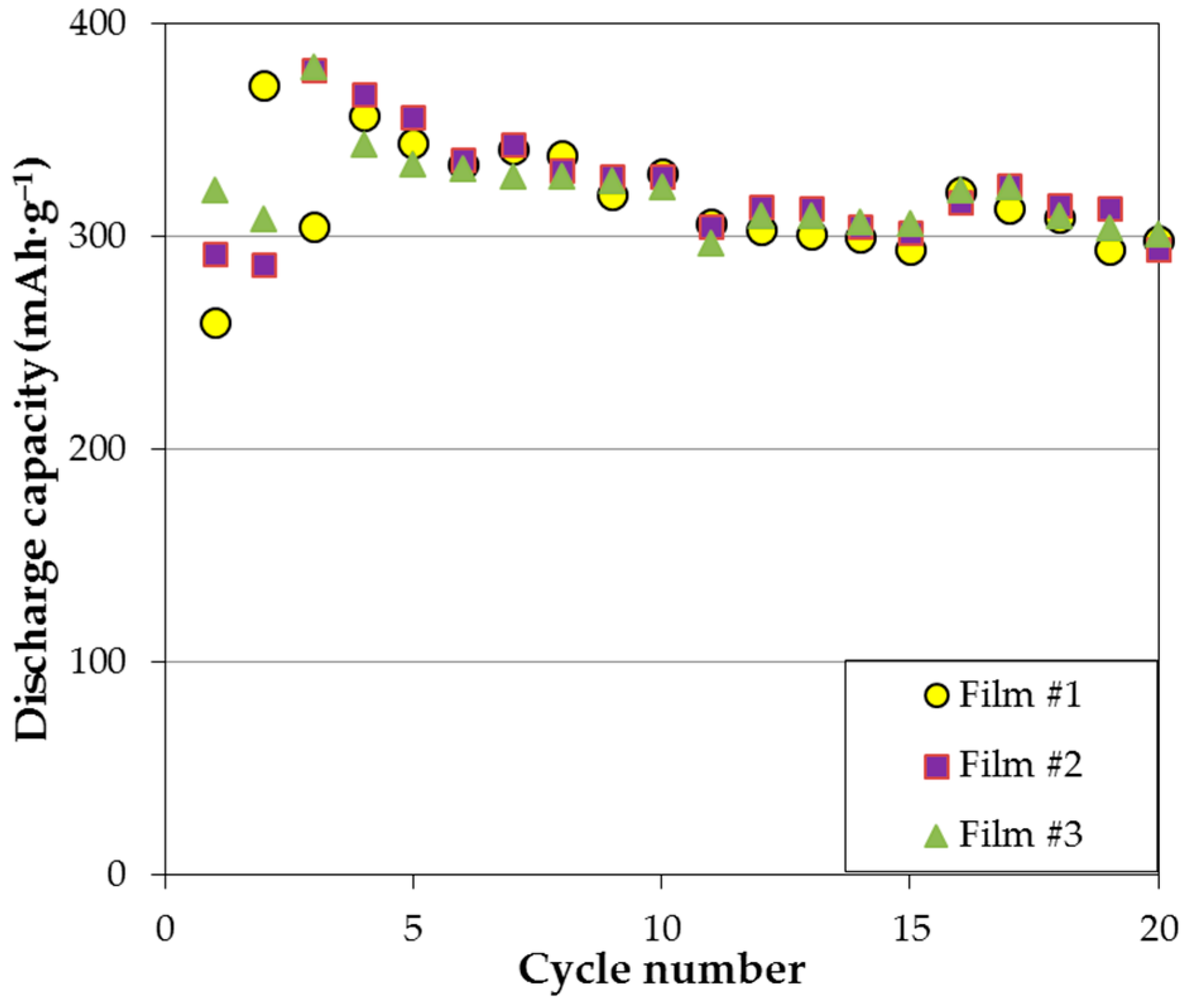
Figure 3, where three films with different chemical positions show similar capacity degradation in the first 20 cycles. More specifically, two or three activation cycles are needed for the thin film electrodes; once the electrodes are fully activated, steady capacity degradation is observed until around cycle 10; capacity remains stable thereafter. According to the XRD analysis, formation of Mg(OH)
2 on the surface impedes further electrochemical reaction and is the major source for capacity degradation (approximately 1.2% per cycle).
3.2. MgNi-Based Alloy Powder Prepared by Melt Spinning + Mechanical Alloying
According to the Mg-Ni binary phase diagram, AR2 and AR3 compositions cannot be reproduced by the conventional melt-and-cast method [
51]. Therefore, we used a powder fabrication method combining the MS and MA techniques. The former (see
Figure 4a for the MS system) produces ribbons with closely packed polycrystalline Mg
2Ni and MgNi
2 phases, and the latter (see
Figure 4b for the MA attritor system) produces powder with an amorphous MgNi phase and therefore reproduces the microstructure and electrochemical capacity of the thin film work.
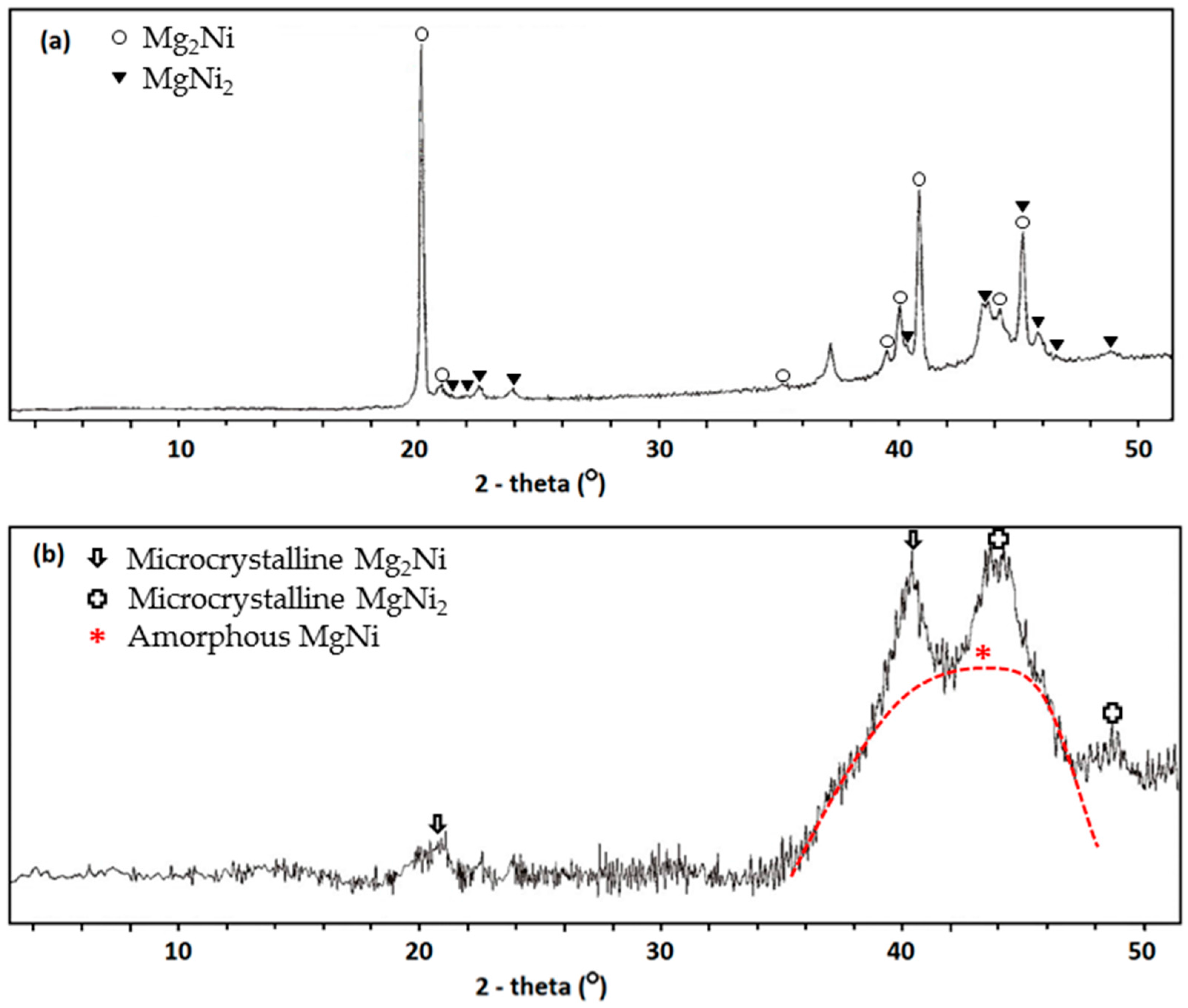
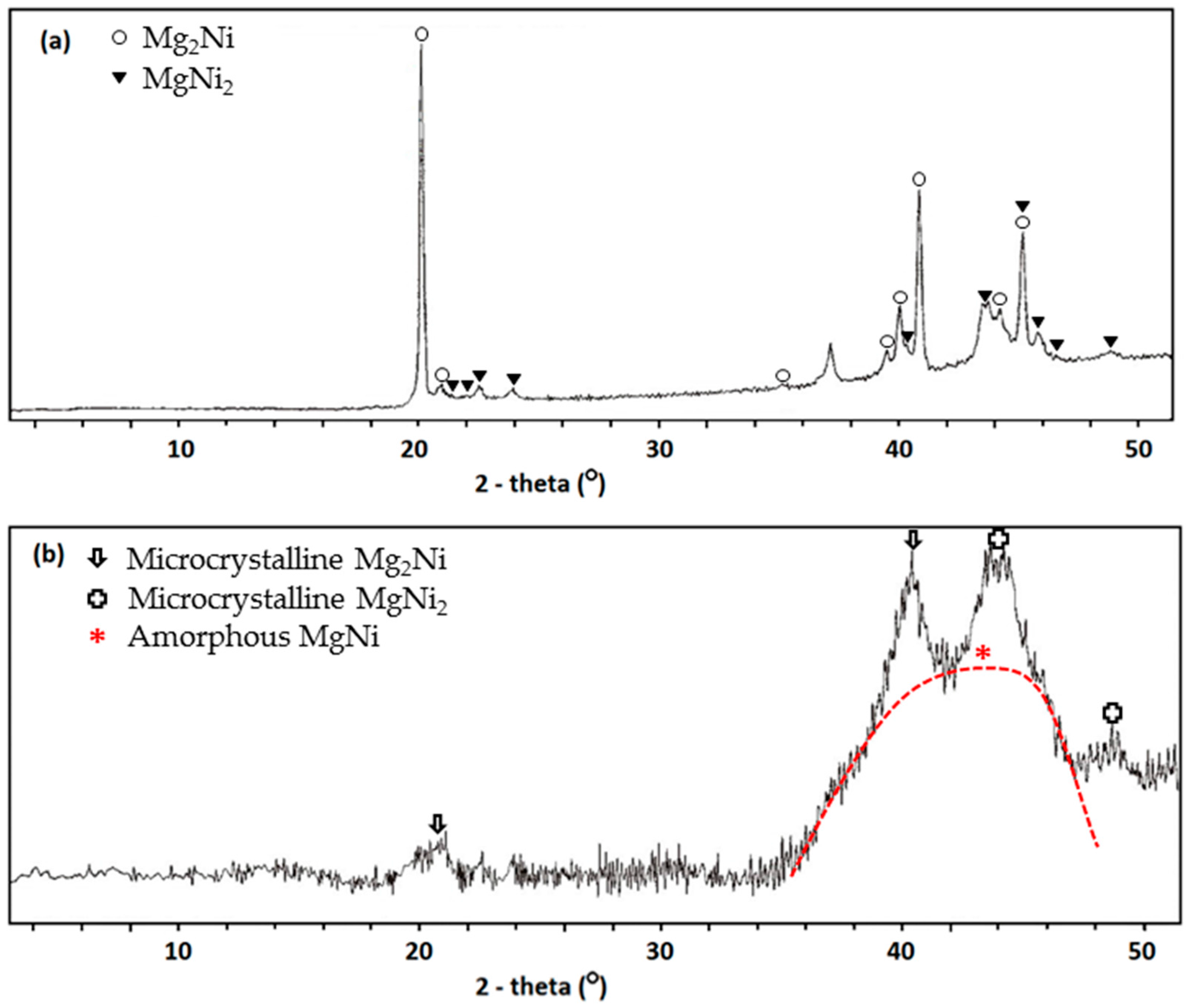
Compared to the method of using MA with raw material in elemental powder form alone, incorporation of MS in the alloy fabrication process can reduce the MA time from 72 h to 12 h and lower the material cost. The polycrystalline and amorphous natures of MS ribbon before and after the MA process, respectively, are revealed by XRD, as shown in
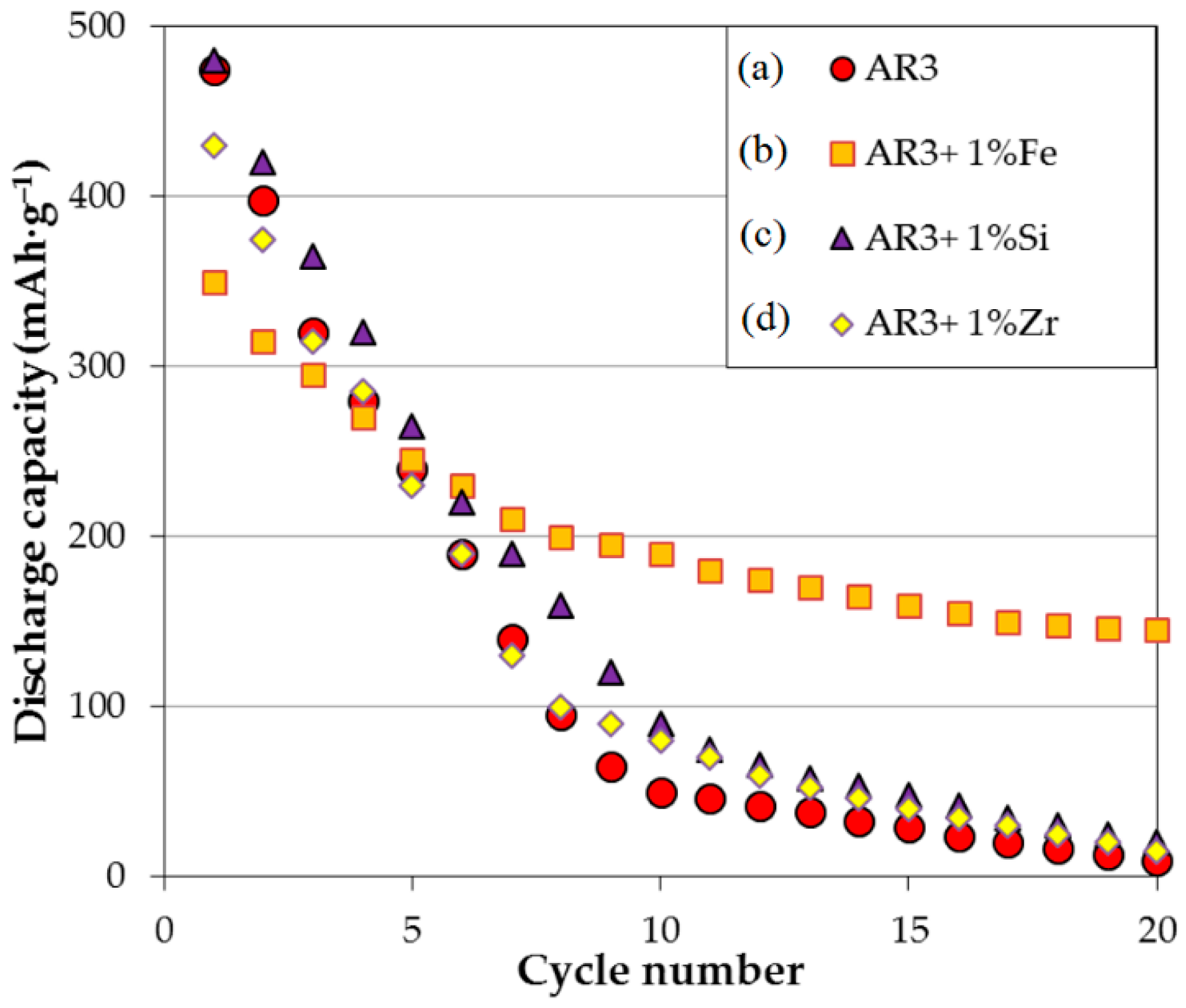
Figure 5. The cycle stability of the AR3 powder made by MS + MA, shown in
Figure 6, is much more severe than that of its thin film counterpart. More specifically, degradation of the bulk AR3 powder occurs at approximately 10% per cycle in the first 10 cycles. While the addition of 1 wt% Si or Zr in the MA process does not affect the cycling performance, the addition of 1 wt% Fe reduces the capacity degradation to around 4% per cycle in the first 10 cycles. Furthermore, although the initial capacity is decreased with the addition of Fe to AR3 powder, the maintained capacity at higher cycle number is much higher than the original or modified AR3 powder with other additions. Addition of Fe also shows significant improvement in cycle stability in our thin film results.
Figure 7 shows TEM micrographs of the AR3 powder sample made by MS + MA, and it reveals that the material density is not uniform. Large areas with lower density (brighter contrast) are found alongside the scroll-type denser regions (darker contrast). This type of microstructure cannot form the protective oxide layer that prevents further oxidation found in other MH alloys [
52] and consequently causes the more severe degradation observed in the bulk form compared to that the thin film form. Therefore, in order to magnify the effect of electrolyte composition modification, the AR3 powder made by MS + MA is the perfect candidate for the current study on corrosion of MH alloy in electrolytes composed of various hydroxides, despite the fact that we have developed a series of new compositions based on the quaternary AR3 composition, new surface treatments, coatings, binder/surfactant additions, and other methods to improve the cycle stability of MgNi-based MH alloys.
3.3. Conductivities of Various Hydroxide Electrolytes
Room temperature conductivities of different concentrations of KOH and other hydroxide aqueous solutions were measured and then normalized to that of 6 M KOH aqueous solution, and the results are summarized in
Table A1 in
Appendix. Due to its low solubility, no effect on conductivity is observed with the addition of Mg(OH)
2, Ca(OH)
2, Sr(OH)
2, or Ba(OH)
2 in 6 M KOH aqueous solution. Moreover, as a representative example, the electrolyte conductivities are plotted against the total OH
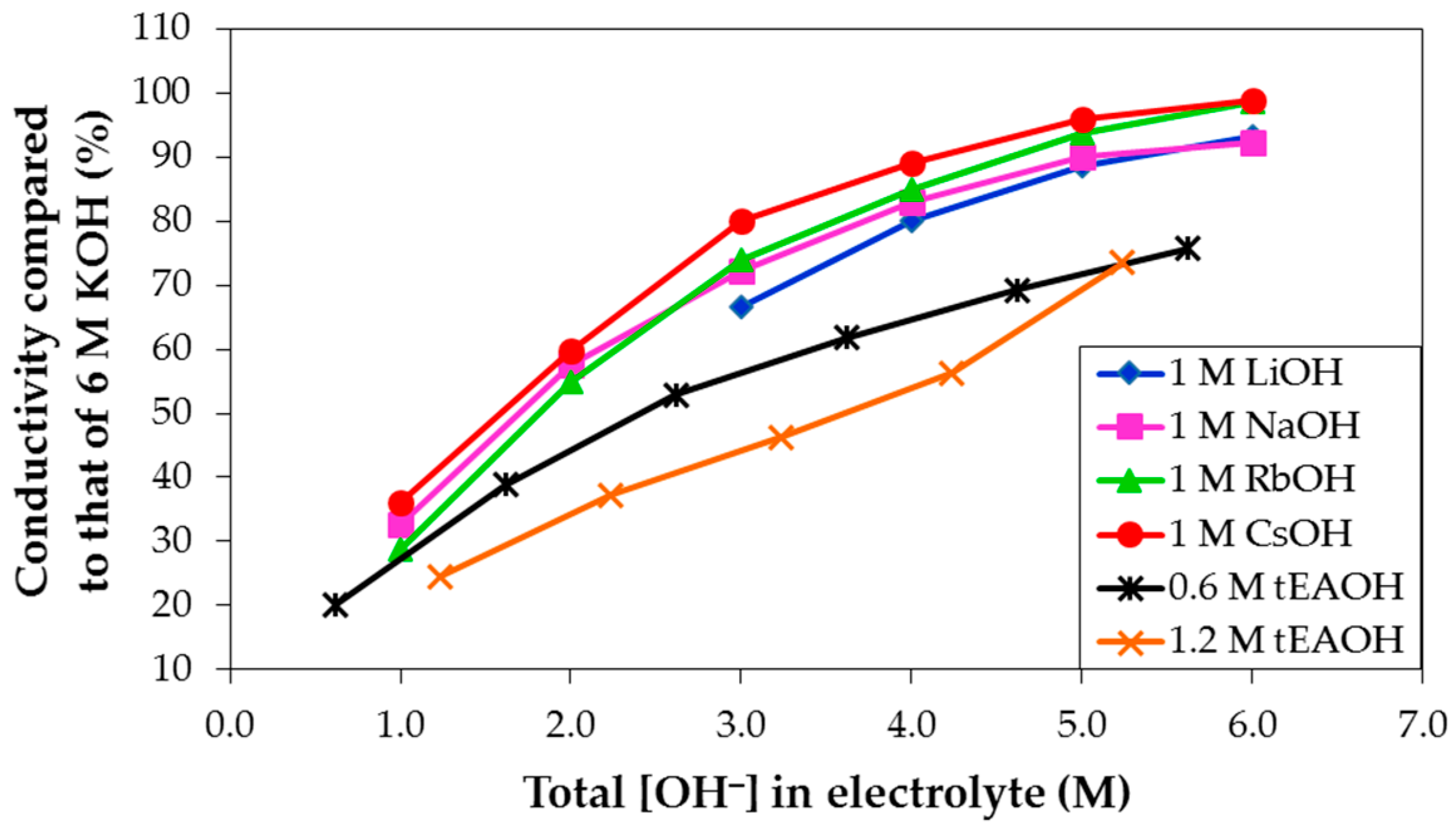
− concentration in solution for 1 M LiOH, NaOH, RbOH, and CsOH, and 0.6 M and 1.2 M tEAOH (tetraethylammonium hydroxide) in various concentrations of KOH aqueous solutions in
Figure 8. As the total OH
− concentration increases, the electrolyte conductivity increases. Also, at any fixed KOH concentration, the electrolyte conductivity increases in agreement with the following trend: tEAOH < LiOH < NaOH < RbOH < CsOH addition. However, none of the conductivities of mixed hydroxide electrolytes exceeds that of pure KOH aqueous solution. According to Stokes’ Law, as the object size increases, it experiences more friction or drag when moving through fluid, as indicated in the equation for a spherical particle object:
where ζ is the drag coefficient of object, η is the fluid viscosity, and
r is the object radius. The drag coefficient also presents in the Einstein relation:
where
D is the diffusion coefficient, k
B is the Boltzmann’s constant, and
T is the absolute temperature. Thus, the diffusion coefficient can be directly related to the size of object moving in fluid by combining Equations (1) and (2), which gives the Stokes-Einstein-Sutherland equation:
Furthermore, molar conductivity of each ionic species in solution can be linked to the diffusion coefficient by the Nernst-Einstein equation:
where
is the limiting molar conductivity (molar conductivity at the limit of infinite dilution) of ionic species, F is the Faraday constant, R is the gas constant,
zi is the charge number of ionic species, and
Di is the diffusion coefficient of ionic species. In the case of hydroxide solution, electrolyte conductivity is determined by the combination of contributions from both the cation and anion species, which can be expressed by the Kohlrausch’s law of independent migration of ions:
where
,
, and
are the limiting molar conductivities of electrolyte, cation, and anion, respectively, and
ν+ and
ν– are the stoichiometric coefficients of cation and anion, respectively. Since the anion species in the current study is unchanged, the observed differences in overall electrolyte conductivity are caused by the various cation species. As the cation size increases, the diffusion coefficient decreases, and the limiting molar conductivity consequently decreases. Among all the cation species in the current study, tEA
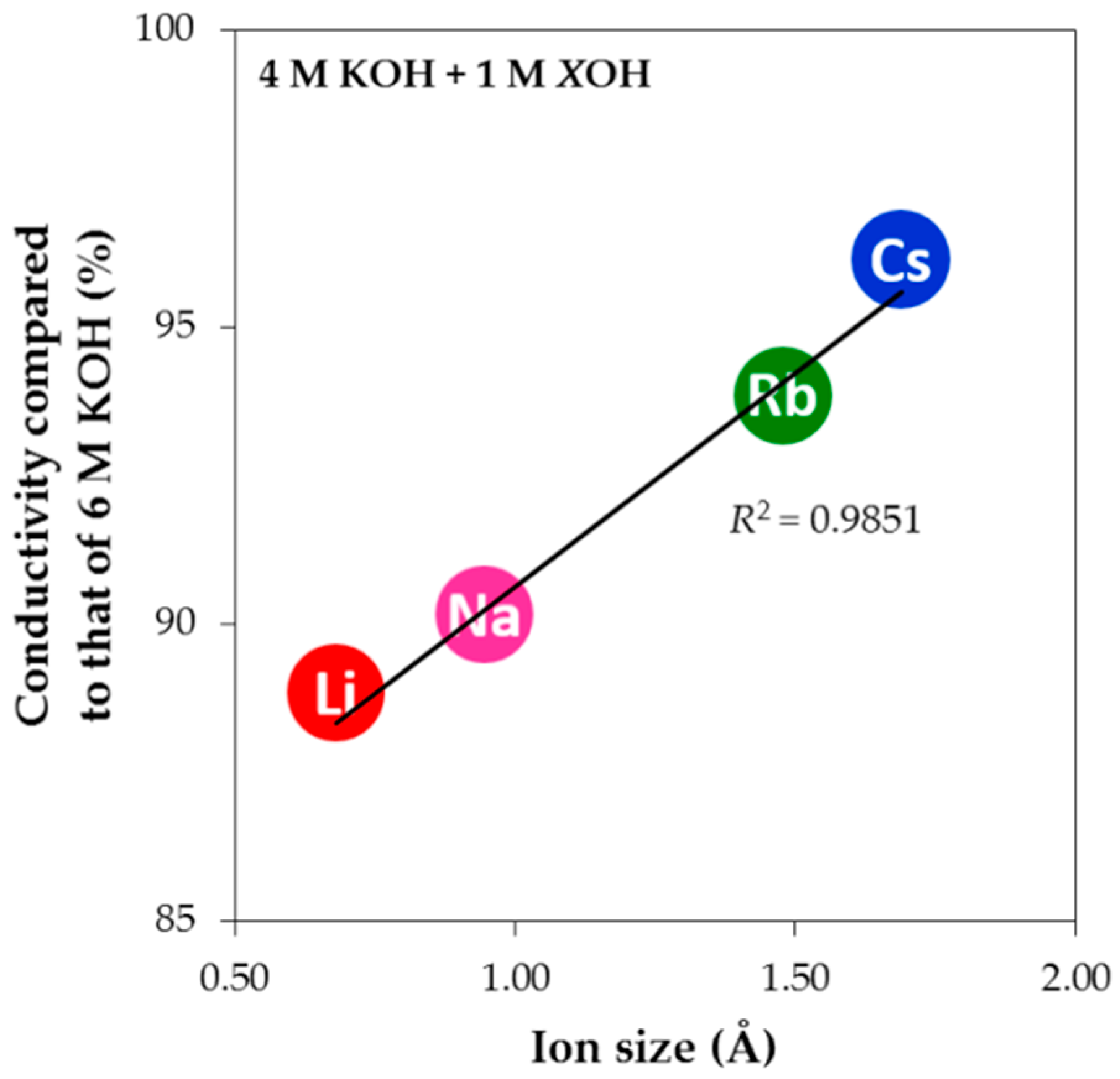
+ is the largest and results in the lowest conductivity upon tEAOH addition. However, this trend is not observed with the smaller alkali cations. When the cation size is very small, the cation has higher charge density and therefore shows a higher tendency to attract water molecules. In other words, a smaller cation has a larger amount of surrounding water, which makes its transport in solution more difficult (higher drag coefficient and lower diffusion constant) and contributes to the observed lower electrolyte conductivity. The normalized electrolyte conductivities of alkali hydroxides at the same level of addition in 4 M KOH aqueous solution are plotted against the cation radii in
Figure 9, and a linear dependency with a very high correlation factor (
R2) is clearly demonstrated. In addition, adding hydroxides with other alkali cations in KOH aqueous solution reduces the electrolyte conductivity slightly compared to pure KOH aqueous solution (5 M KOH aqueous solution has approximately the same conductivity as 6 M KOH aqueous solution at room temperature, which has the normalized conductivity value of 100%), indicating that a pure electrolyte system may have higher electrolyte conductivity than a heterogeneous electrolyte system at the same OH
− solution concentration. Such a phenomenon is interesting to observe and deserves further investigation of, for example, the effect of interaction between cations in a heterogeneous hydroxide electrolyte system on conductivity.
3.4. Corrosion Performances in Various Hydroxide Electrolytes
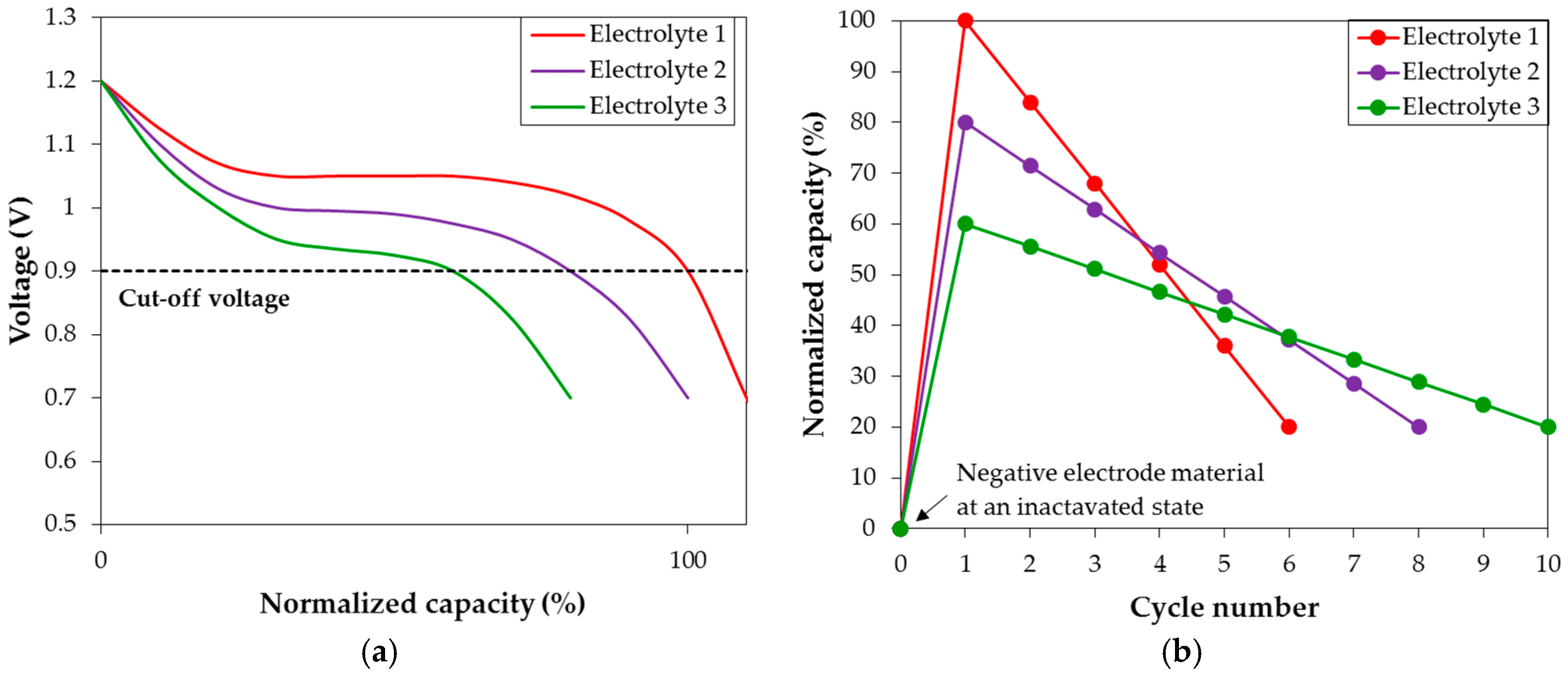
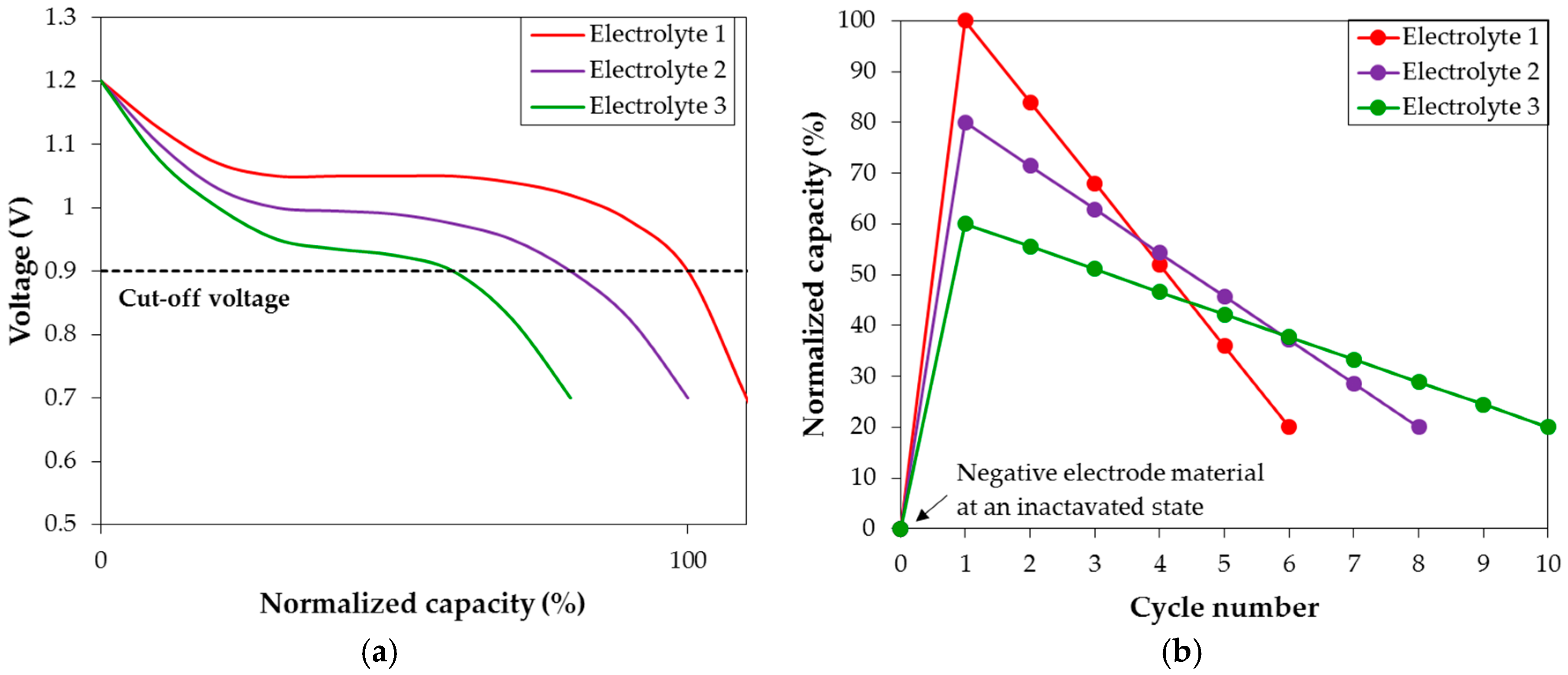
The AR3 powder made by MS + MA is used as the negative electrode material for the corrosion study in various hydroxide aqueous solutions. Before studying the relative corrosion strengths of different electrolytes, an evaluation of capacity degradation must be established. We use three hypothetic electrolytes (Electrolytes 1, 2, and 3) in
Figure 10 as the representatives to explain and describe the differences in discharge characteristics in various hydroxide aqueous solutions observed in the current study. Looking closely at the first cycle discharge voltage profiles from half-cells with the three different electrolytes (
Figure 10a), the largest initial discharge capacity is observed with Electrolyte 1. The difference in discharge voltage curve length indicates that Electrolyte 1 has the strongest activation power compared to Electrolytes 2 and 3. In other words, Electrolyte 1 activates the largest portion of MH alloy within the same initial charging period as in Electrolytes 2 and 3, which is the main reason for its superior initial capacity performance. However, higher activation power also corresponds to stronger corrosion strength, increasing in the order of Electrolyte 3 < Electrolyte 2 < Electrolyte 1. The discharge capacities are plotted against the cycle number in
Figure 10b, and the degradation in Electrolyte 1 is worse (higher capacity loss per cycle) compared to that in Electrolyte 3. Therefore, in order to separate the contribution of activation to degradation, degradation calculation in this study is defined as:
where
Caphigh and
Caplow are the highest and lowest capacities throughout cycling (
Caphigh is usually the initial capacity in the current study), respectively, and
nhigh and
nlow are the corresponding cycle numbers. It is worth noting that a positive correlation is found between the activation power and electrolyte conductivity in the current study, i.e., electrolyte conductivity increases in the order of Electrolyte 3 < Electrolyte 2 < Electrolyte 1. The highest conductivity of Electrolyte 1 (or the lowest internal resistance, as observed from its highest voltage plateau in
Figure 10a) is also a contributing factor for its highest initial capacity, albeit minor compared to the influence of activation power. Therefore, the contribution of conductivity on capacity is neglected in the degradation calculation.
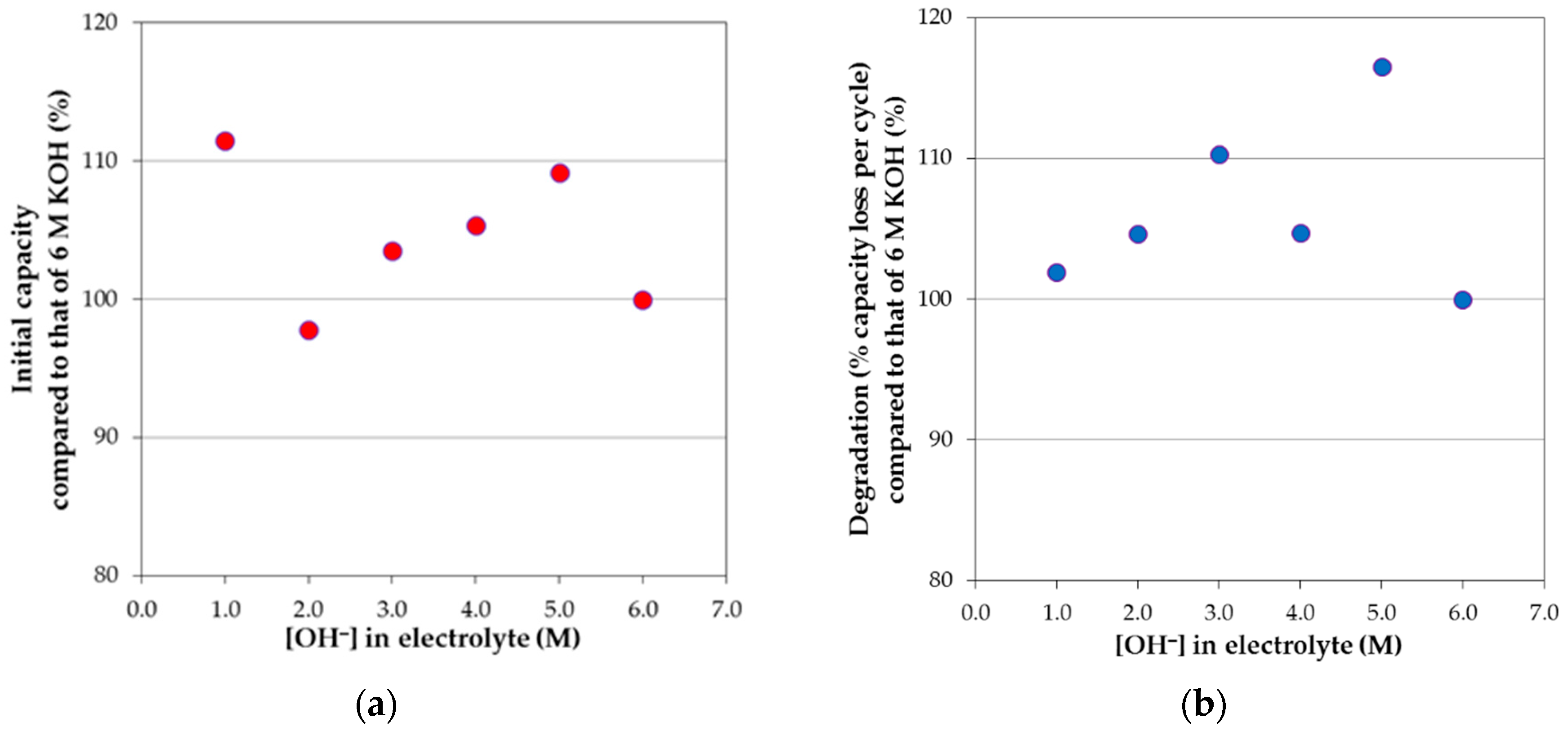
Room temperature degradation of different concentrations of KOH and/or other hydroxide aqueous solutions were measured, and the results are summarized in
Table A2 in
Appendix. Due to its low solubility, no effect on degradation is observed with the addition of Mg(OH)
2, Ca(OH)
2, Sr(OH)
2, or Ba(OH)
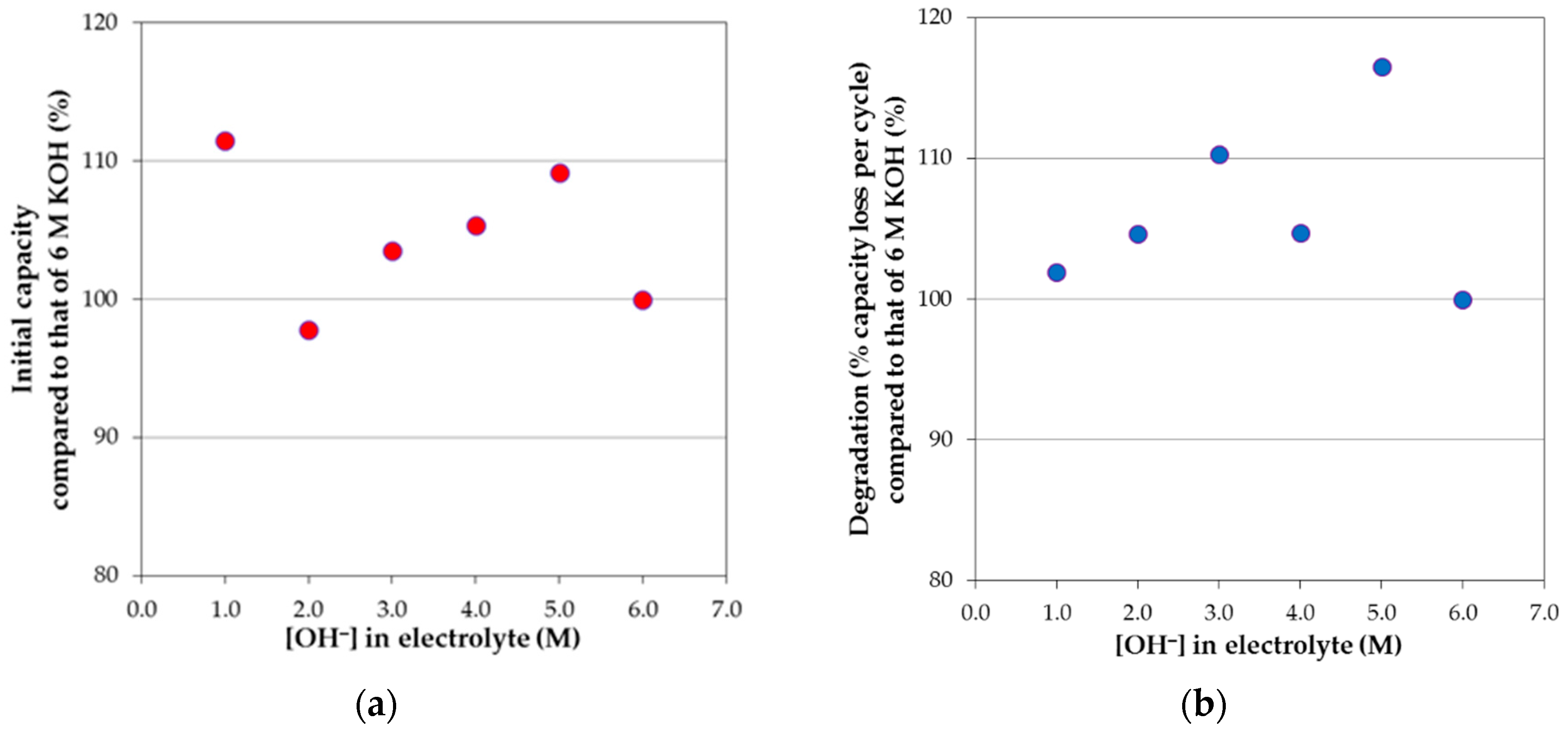
2 in 6 M KOH aqueous solution. For the pure KOH aqueous solution systems at various concentrations, the initial capacities and degradations normalized to those of 6 M KOH aqueous solution are plotted against the total OH
− concentration in solution in
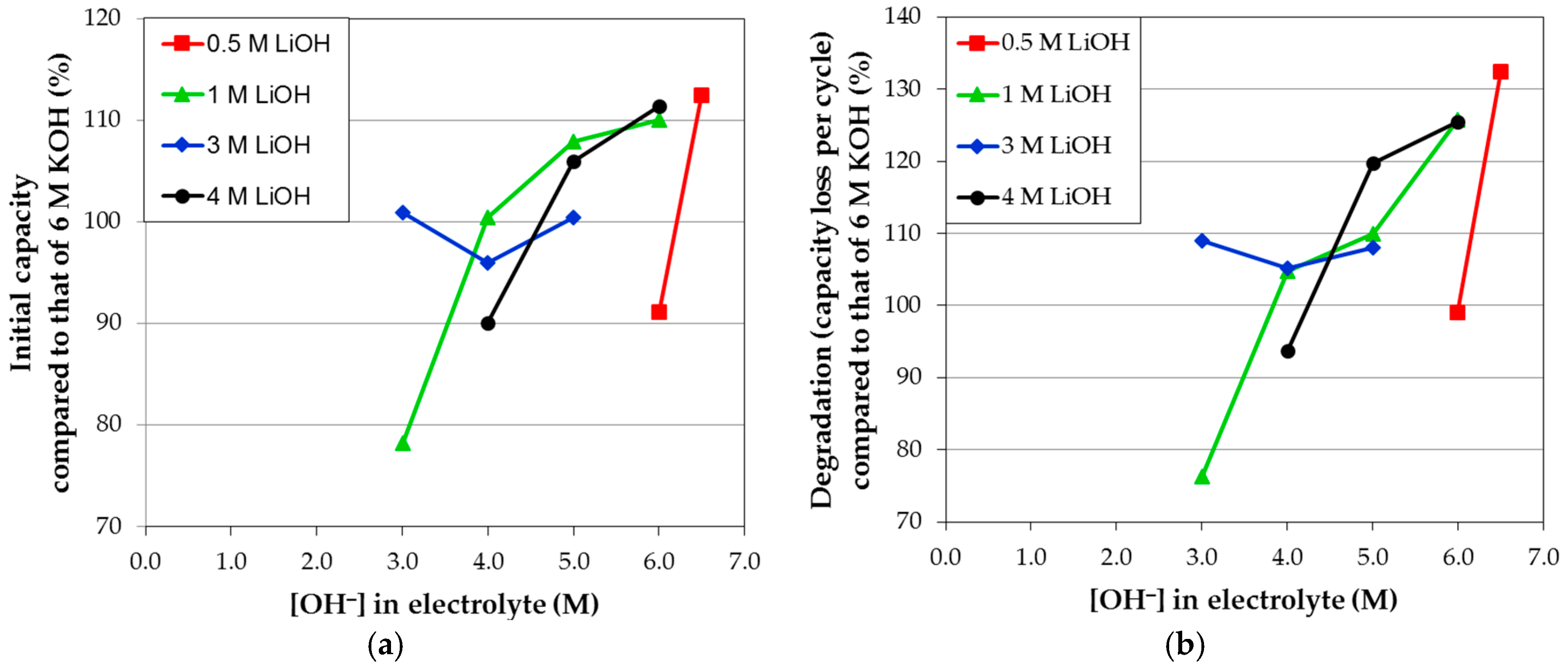
Figure 11a,b, respectively. Even after the attempt to eliminate the contribution of activation on degradation, a strong similarity between the initial capacity and degradation performances is still present, meaning that activation and corrosion are very closely related and cannot be completely separated by our calculation for KOH-based electrolytes. Such correlation can also be seen in mixed electrolytes. For example, a resemblance between the initial capacity (
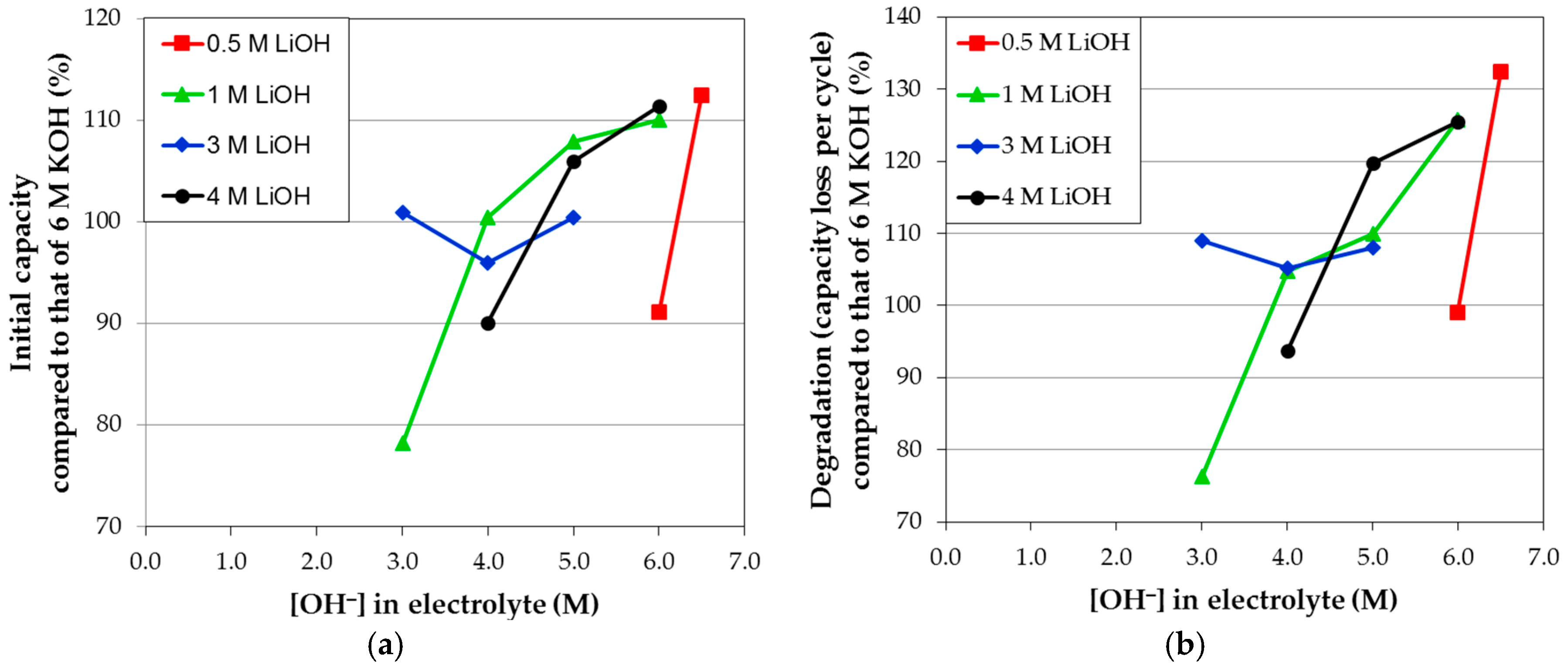
Figure 12a) and degradation (
Figure 12b) performances in various electrolyte mixtures of KOH and LiOH can be observed. It is also interesting to find that higher OH
− concentrations in electrolytes do not necessarily correspond to higher corrosion.
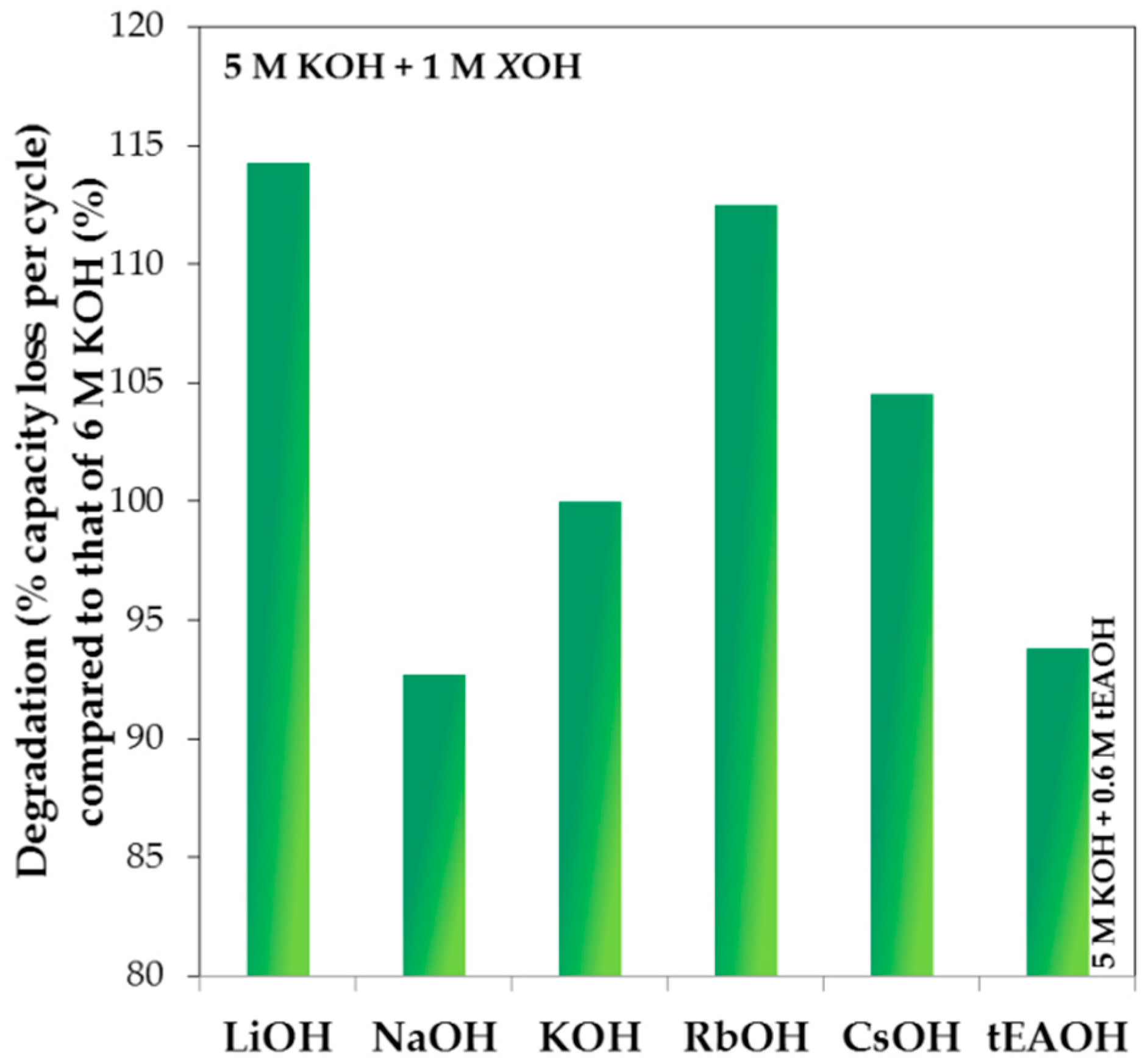
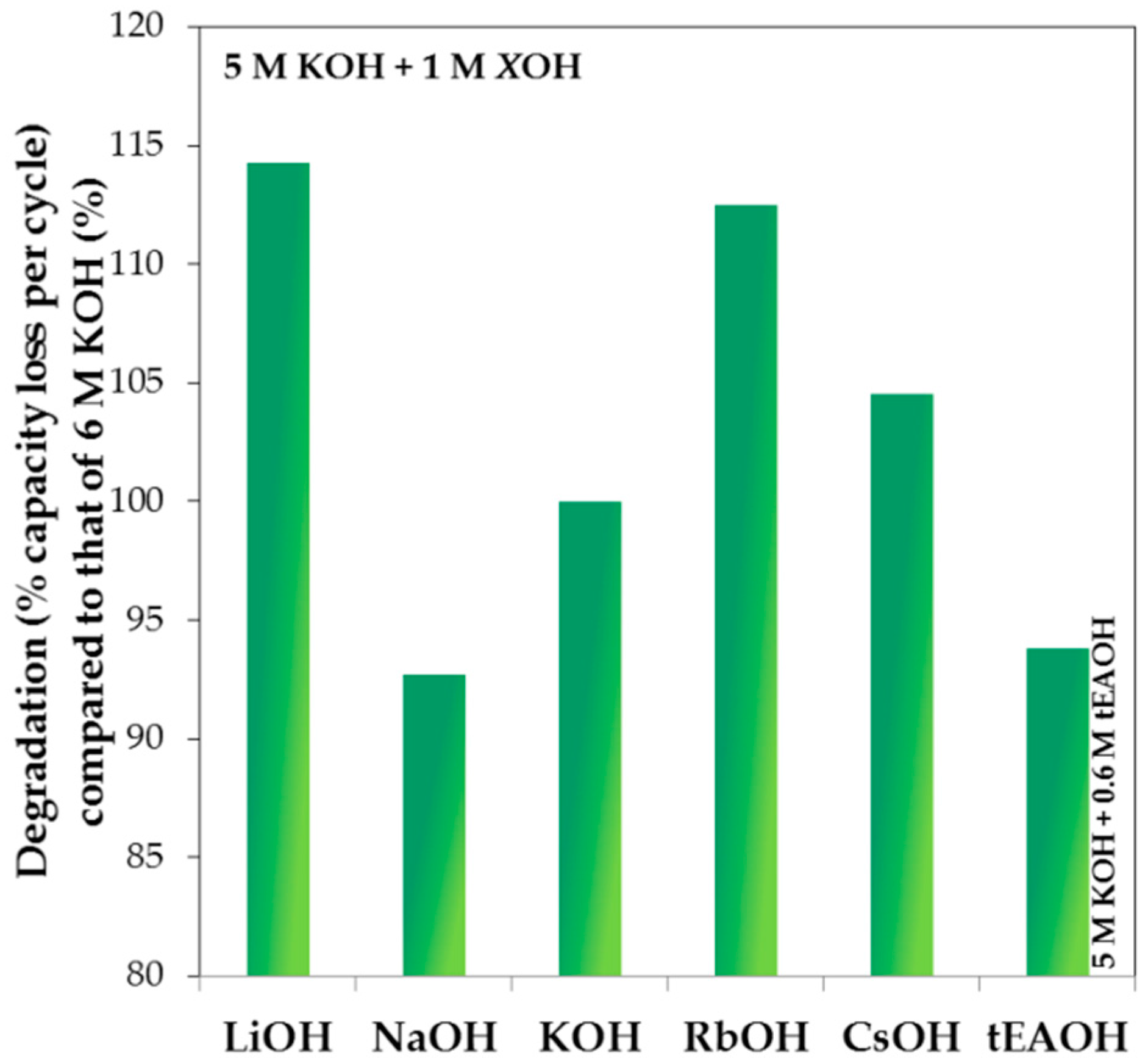
As a representative example, the degradation performances compared to that of 6 M KOH aqueous solution are plotted against the total OH
− concentration in solution for 1 M LiOH, NaOH, RbOH, and CsOH, and 0.6 M tEAOH in 5 M KOH aqueous solution in
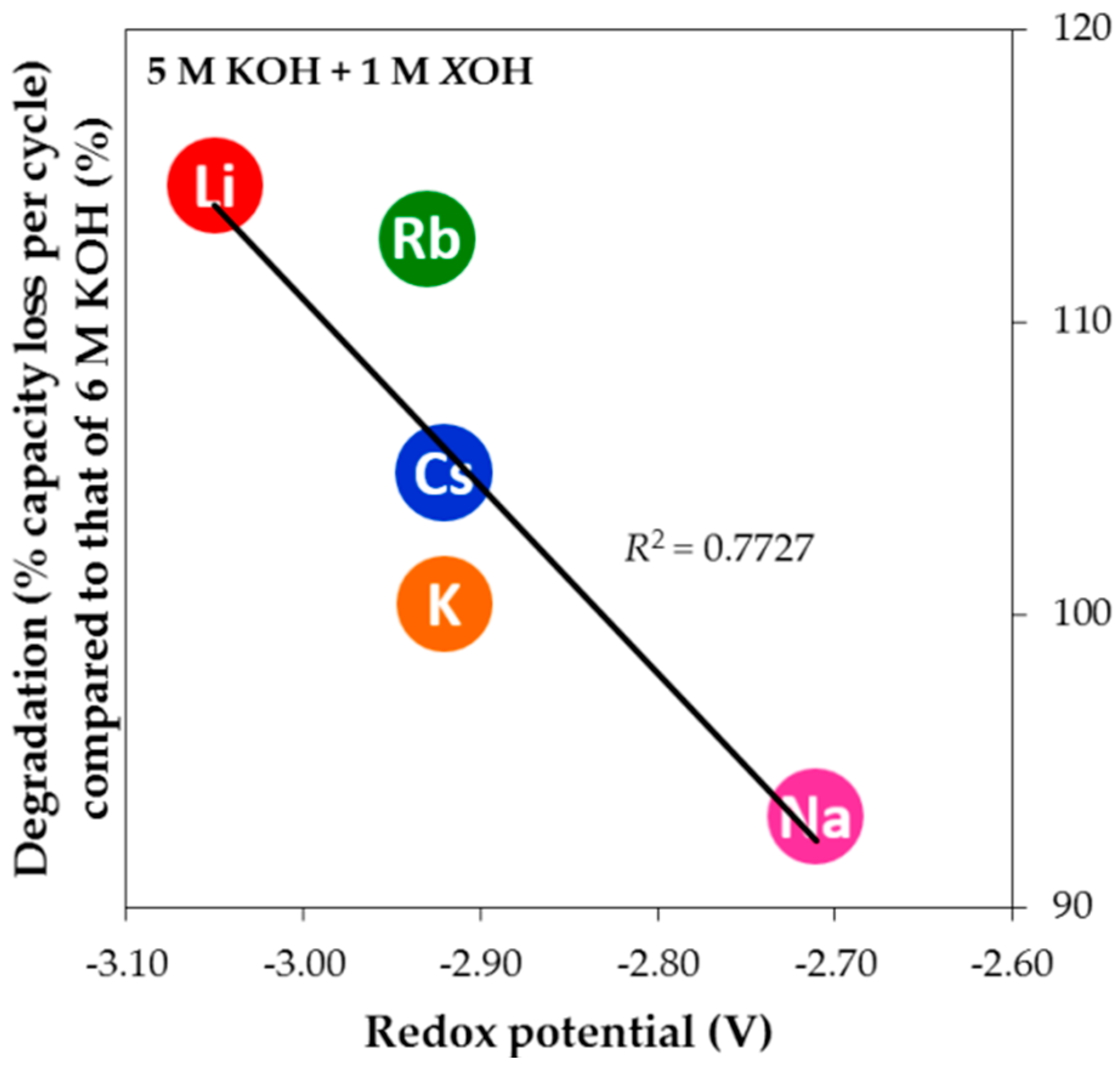
Figure 13. With regard to lowering the corrosion strength of pure KOH aqueous solution, both NaOH and tEAOH additions are beneficial while CsOH, RbOH, and LiOH additions worsen the degradation behavior. More specifically, the degradation performance improves in the trend of LiOH < RbOH < CsOH < KOH < tEAOH < NaOH addition. Since the anion species in the current study is unchanged, the differences in degradation are caused by the various cation species. The trend in reactivity series of alkali elements was considered to be responsible for the observed degradation trend, which increases in the order of Cs < Rb < K < Na < Li. Interestingly, it is clear that the two trends do not correlate very well. Determination of reactivity relies on the element characteristics alone, so the trend in reactivity series is similar to another qualitative measure: ionization energy. However, the element’s reaction partner also affects the reactivity of reaction. For example, for the reaction of an alkali element in water, the element’s characteristics and interaction with water, such as enthalpy of sublimation (
), ionization energy (
), and enthalpy of dissolution (
), must be taken into consideration in predicting how reactive the reaction is. Therefore, the qualitative electrochemical series is used to correlate with the degradation trend. The degradation performances of alkali hydroxides at the same level of addition in 5 M KOH aqueous solution are plotted against the standard redox potential in
Figure 14, resulting in a linear dependency with a high
R2. Due to the lowest and highest redox potentials of Li and Na, respectively, the addition of LiOH appears to be the most corrosive while the addition of NaOH retards degradation. Consequently, NaOH is used as an electrolyte supplement for HT applications, where corrosion is much more aggressive, and LiOH is added in electrolyte to increase electrolyte activity for the LT discharge performance in Ni/MH battery [
53].
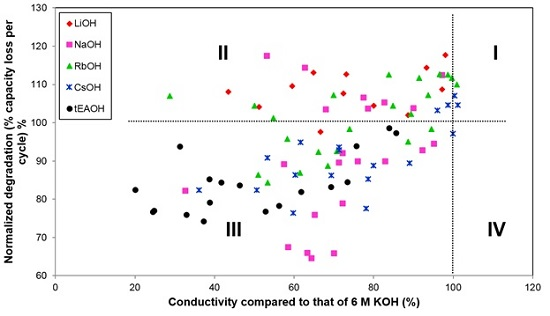
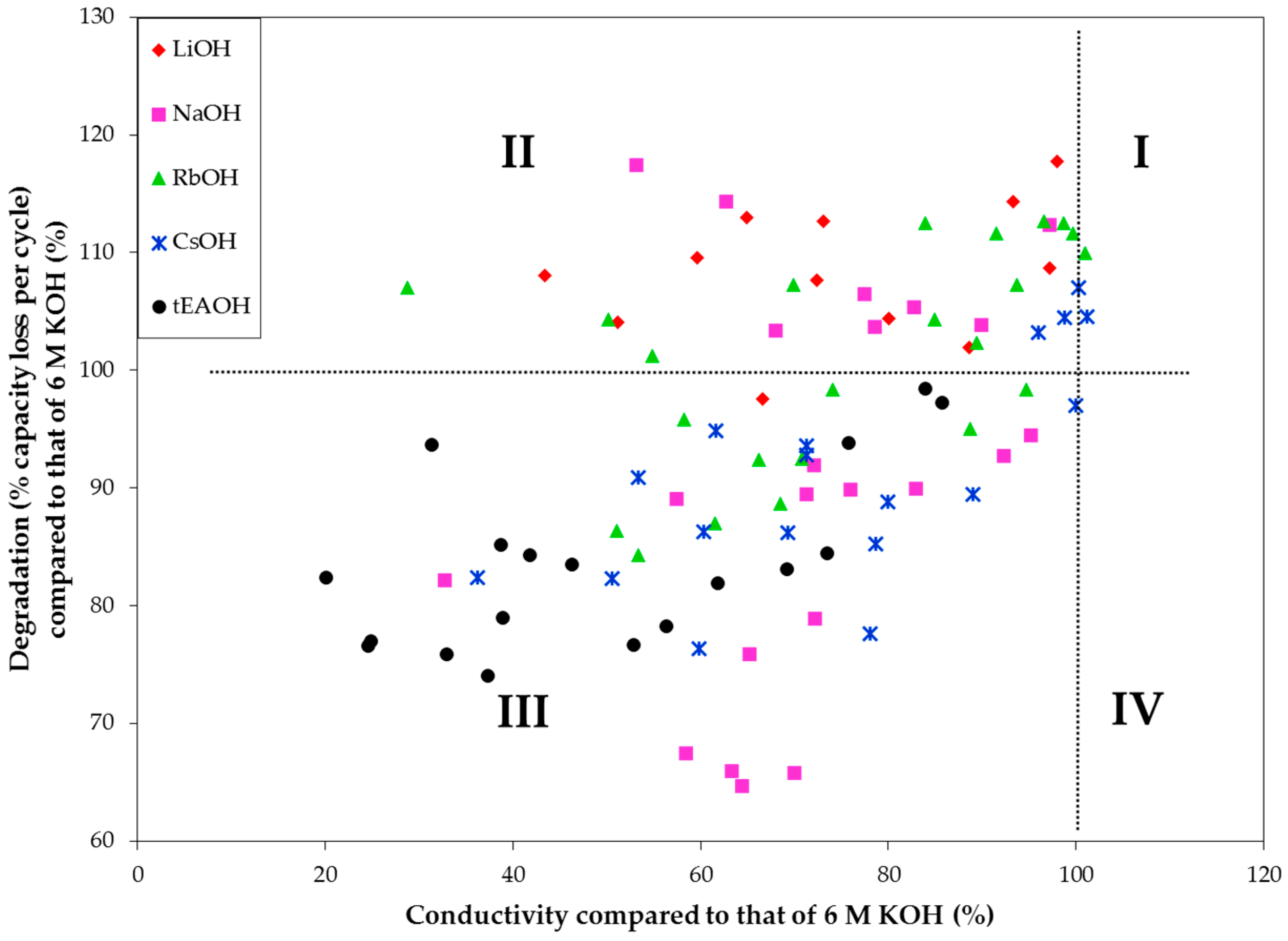
The entire data sets from
Appendix are plotted in
Figure 15. Most of the data plot to Quadrant III (with lower degradation but also lower conductivity). Those points in Quadrant II show lower conductivity and higher degradation and represent the most unsuitable electrolytes. From this chart, we also find that while NaOH and CsOH additions can reduce the corrosive nature of an electrolyte without sacrificing excessive amounts of conductivity, tEAOH is also a good candidate if electrolyte conductivity is not a major concern for the application (e.g., high energy Ni/MH battery without strict power requirements). We plan to continue this electrolyte study with additions of other organic hydroxides and amphoteric hydroxides/oxides, such as quaternary ammonium hydroxides, bis(ethylenediamine) copper(II) hydroxide, choline base solution, tetrabutylphosphonium hydroxide, hexamethonium hydroxide, and zinc oxide/hydroxide, in order to find possible candidates that fall in Quadrant IV.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}