Solid-State NMR Characterization of the Structure of Self-Assembled Ile–Phe–OH

 ,
,  and
and
Abstract
1. Introduction
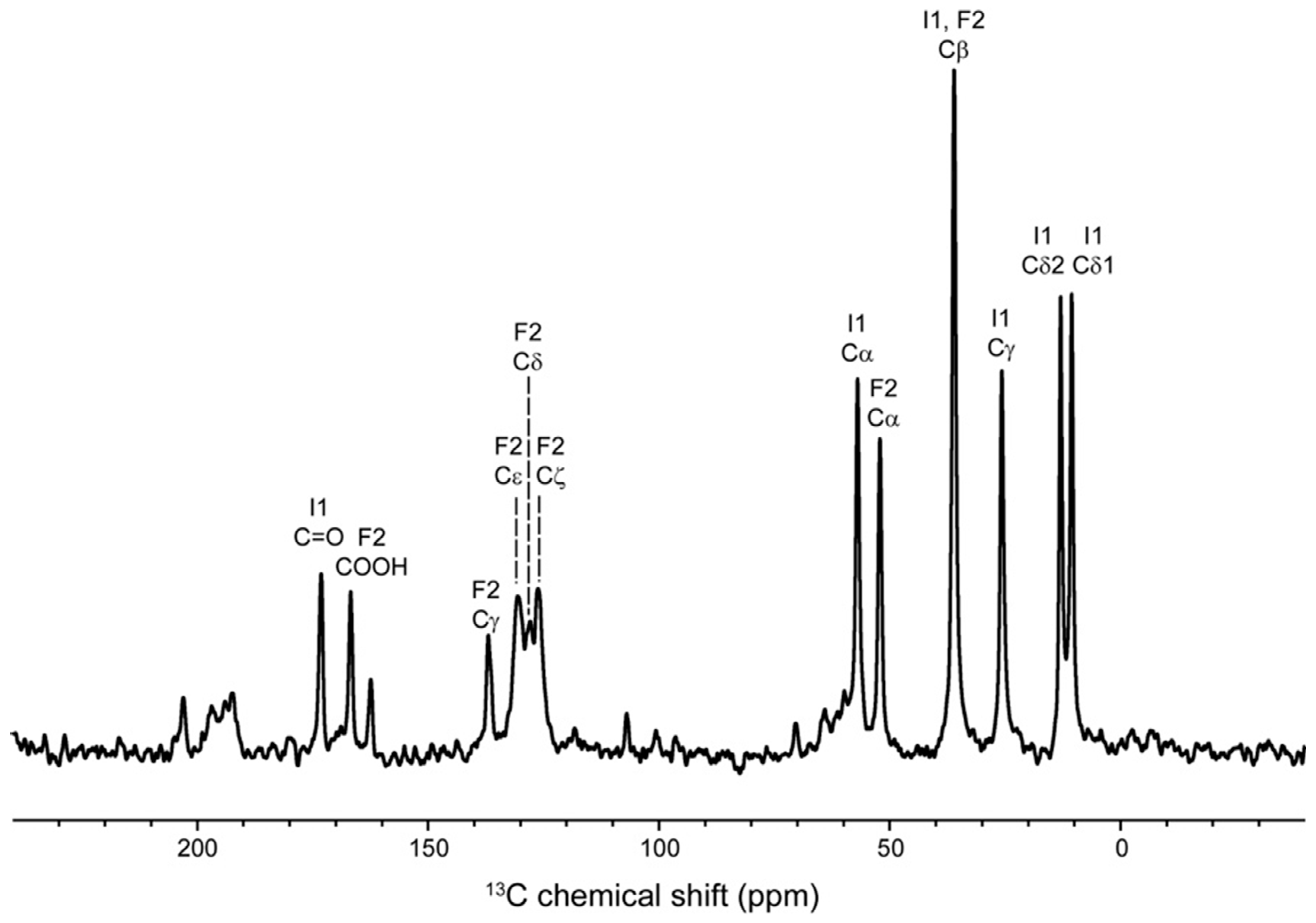
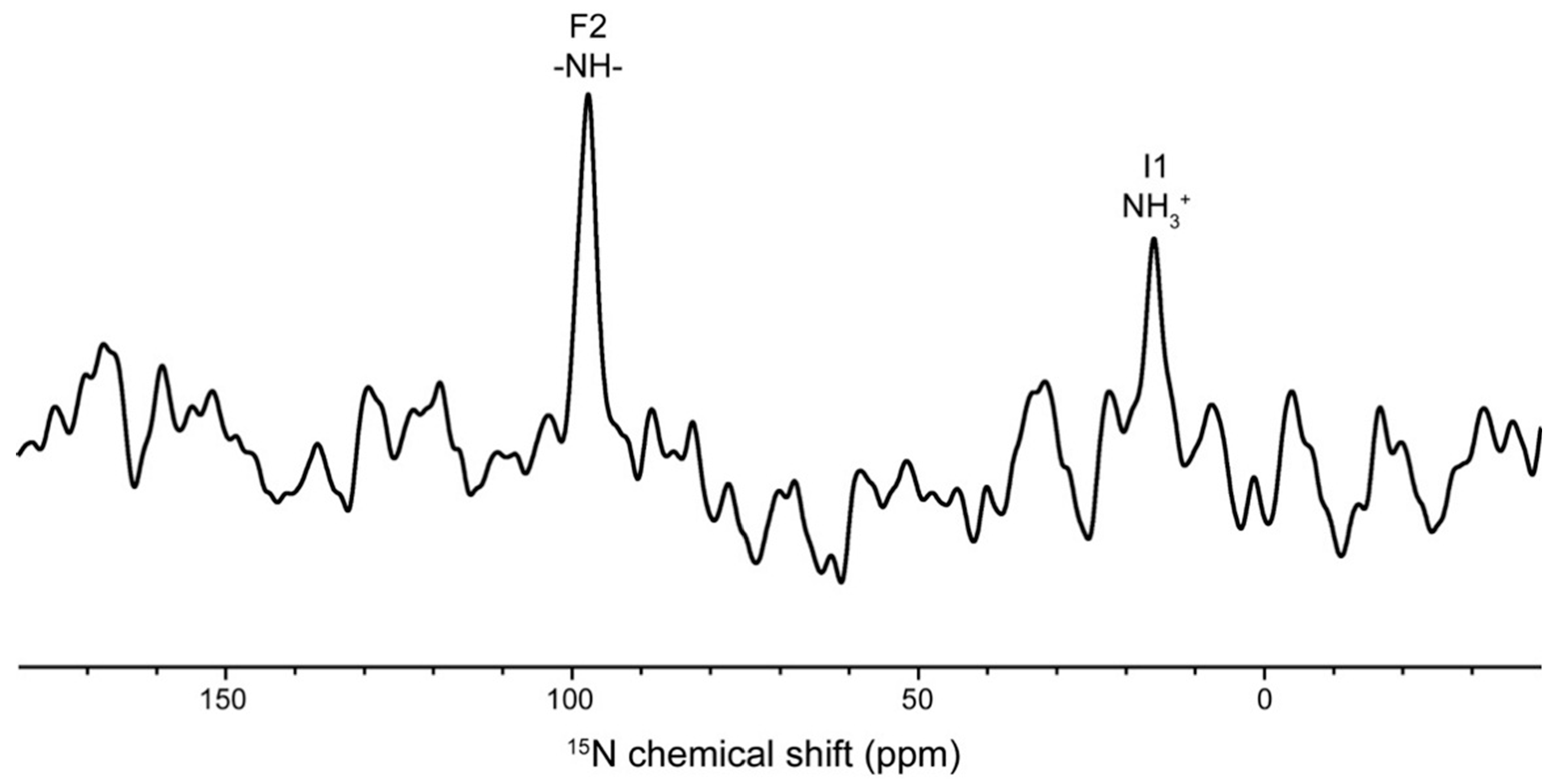
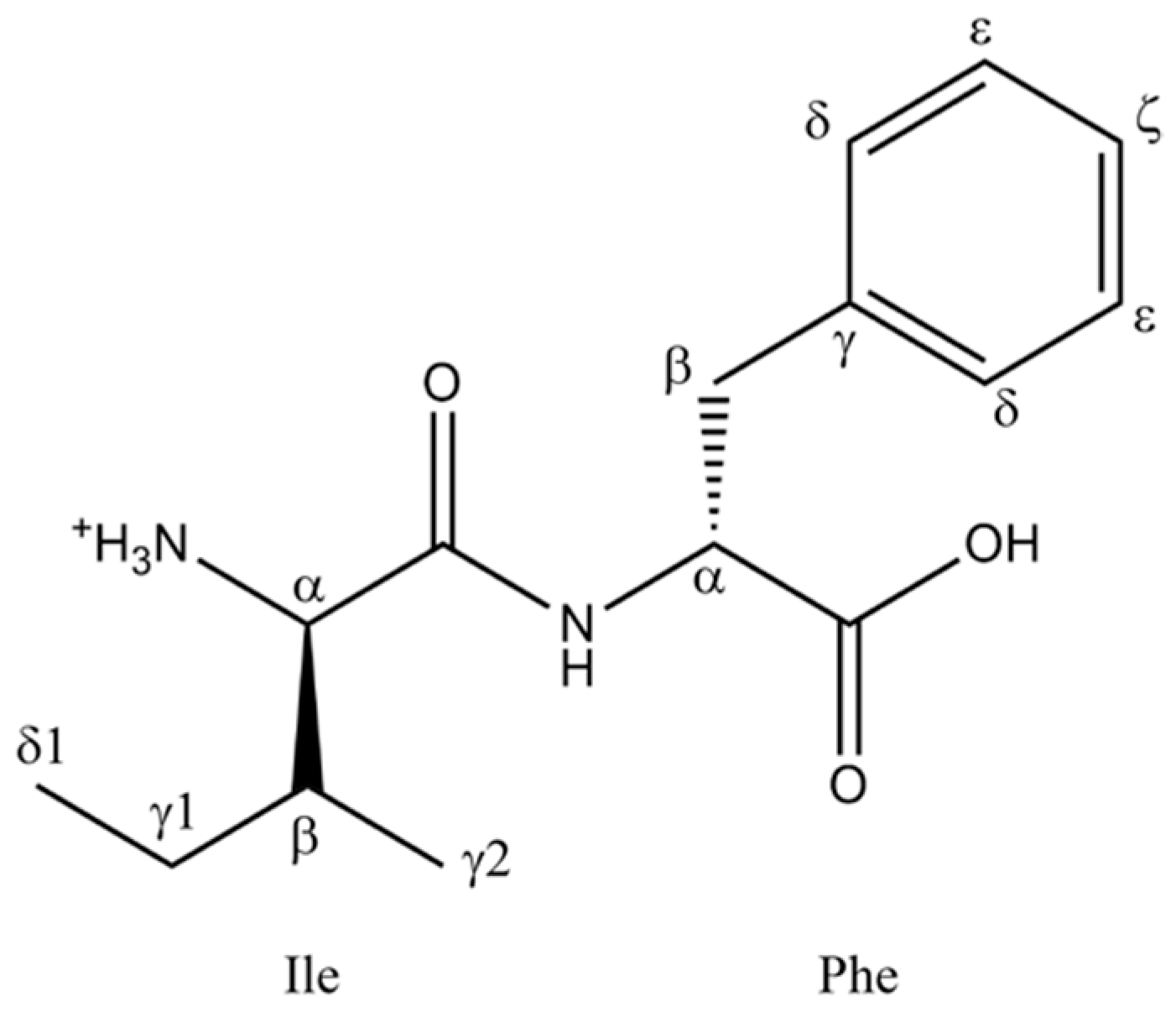
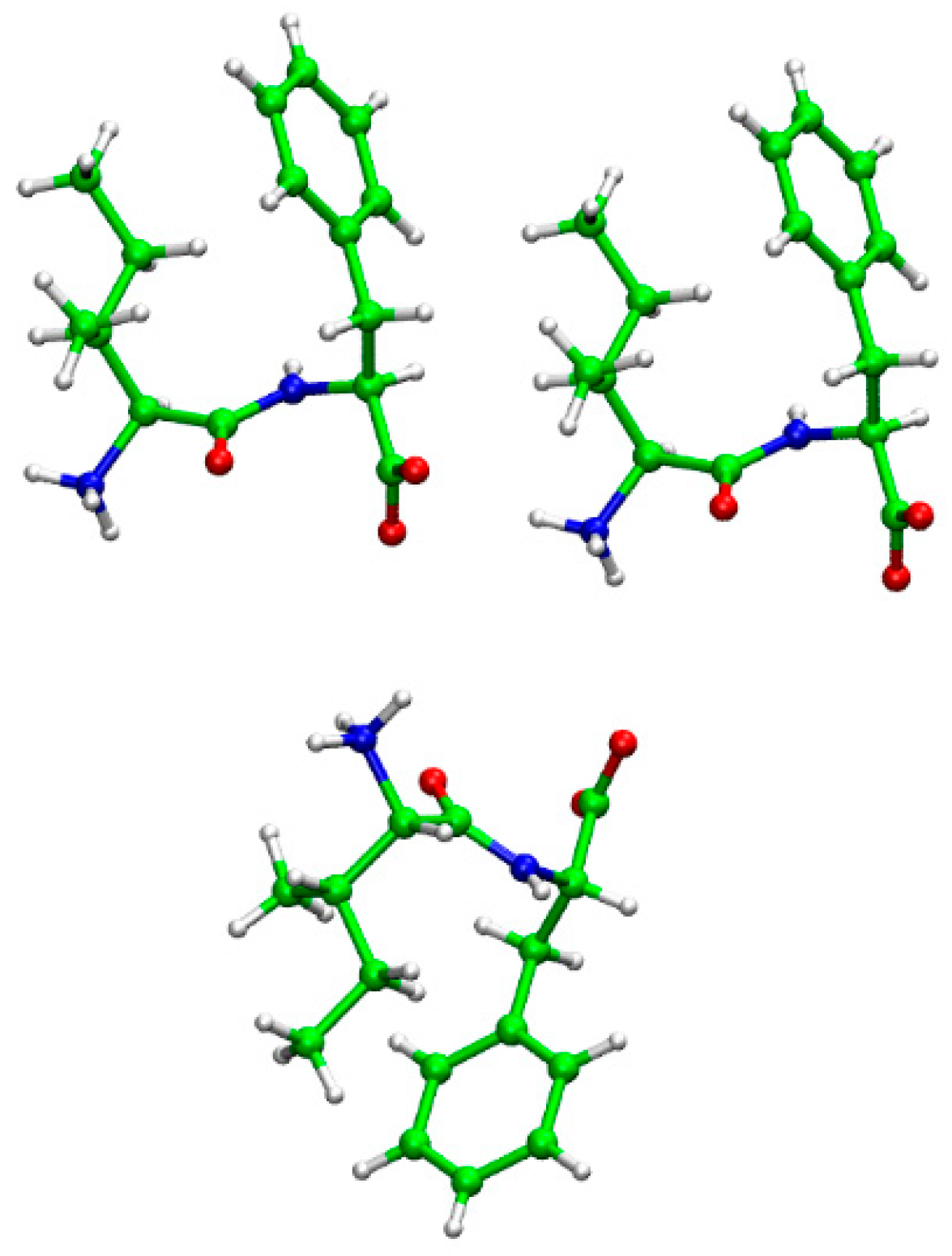
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Angle Names | Angles (degrees) |
|---|---|
| ϕ (C’1-N2- Cα2-C’2) | 49.49 |
| φ (N1- Cα1-C1’-N2) | 149.92 |
| ω (Cα1-C’1-N2- Cα2) | 170.64 |
| θ (Cβ1-Cα1···Cα2- Cβ2) | 16.02 |
References
- Shimizu, T.; Minamikawa, H.; Kogiso, M.; Aoyagi, M.; Kameta, N.; Ding, W.; Masuda, M. Self-organized nanotube materials and their application in bioengineering. Polym. J. 2014, 46, 831–858. [Google Scholar] [CrossRef]
- Reches, M.; Gazit, E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Görbitz, C.H. Microporus organic materials from hydrophobic dipeptides. Chem. Eur. J. 2007, 13, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Mason, T.O.; Chirgadze, D.Y.; Levin, A.; Adler-Abramovich, L.; Gazit, E.; Knowles, T.P.J.; Buell, A.K. Expanding the solvent chemical space for self-assembly of dipeptide nanostructures. ACS Nano 2014, 8, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Görbitz, C.H. The structure of nanotubes formed by diphenylalanine, the core recognition motif of Alzheimer’s β-amyloid polypeptide. Chem. Commun. 2006, 2332–2334. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, Y.; Qi, W.; Su, R.; He, Z. Temperature-induced reversible self-assembly of diphenylalanine peptide and the structural transition from organogel to crystalline nanowire. Nanoscale Res. Lett. 2014, 9, 653. [Google Scholar] [CrossRef] [PubMed]
- Adler-Abramovich, L.; Gazit, E. The physical properties of supramolecular peptide assembles: From building block association to technological applications. Chem. Soc. Rev. 2014, 43, 6881–6893. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhu, P.; Li, J. Self-assembly and application of diphenylalanine-based nanostructures. Chem. Soc. Rev. 2010, 39, 1877–1890. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Zhu, R.; Jenkins, K.; Yang, R. Self-assembly of diphenylalanine peptide with controlled polarization for power generation. Nat. Commun. 2016, 7, 13566. [Google Scholar] [CrossRef] [PubMed]
- Jeena, M.T.; Palanikumar, L.; Go, E.M.; Kim, I.; Kang, M.G.; Lee, S.; Park, S.; Choi, H.; Kim, C.; Jin, S.-M.; et al. Mitochondria localization induced self-assembly of peptide amphiphiles for cellular dysfunction. Nat. Commun. 2017, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Schnaider, L.; Brahmachari, S.; Schmidt, N.W.; Mensa, B.; Shaham-Niv, S.; Bychenko, D.; Adler-Abramovich, L.; Shimon, L.J.W.; Kolusheva, S.; DeGrado, W.F.; et al. Self-assembling dipeptide antibacterial nanostructures with membrane disrupting activity. Nat. Commun. 2017, 8, 1365. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.; Kawamura, I.; Javkhlantugs, N. Recent solid-state NMR studies of membrane bound peptides and proteins. In Annual Reports of NMR Spectroscopy; Webb, G.A., Ed.; Elsevier: New York, NY, USA, 2015; Volume 86, pp. 333–411. [Google Scholar] [CrossRef]
- Naito, A.; Kawamura, I. Solid-state NMR as a method to reveal structure and membrane-interaction of amyloidogenic proteins and peptides. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1900–1912. [Google Scholar] [CrossRef] [PubMed]
- Altheimer, B.D.; Mehta, A.M. Effects of structural differences on the NMR chemical shifts in isostructural dipeptides. J. Phys. Chem. A 2014, 118, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Bhate, M.P.; Woodard, J.C.; Mehta, M.A. Solvation and hydrogen bonding in alanine- and glycine-containing dipeptides probed using solution- and solid-state NMR spectroscopy. J. Am. Chem. Soc. 2009, 131, 9579–9589. [Google Scholar] [CrossRef] [PubMed]
- Jeziorna, A.; Stopczyk, K.; Shorupska, E.; Luberda-Durnas, K.; Oszajca, M.; Lasocha, W.; Górecki, M.; Frelek, J.; Potrzebowski, M.J. Cyclic dipeptides as building units of nano- and microdevices: Synthesis, properties, and structural studies. Cryst. Growth Des. 2015, 15, 5138–5148. [Google Scholar] [CrossRef]
- Jaworska, M.; Jeziorna, A.; Drabik, E.; Potrzebowski, M.J. Solid state NMR study of thermal processes in nanoassemblies formed by dipeptides. J. Phys. Chem. C 2012, 116, 12330–12338. [Google Scholar] [CrossRef]
- Saitô, H. Conformation-dependent 13C chemical shift: A new means of conformational characterization as obtained by high-resolution solid-state 13C NMR. Magn. Reson. Chem. 1986, 24, 835–852. [Google Scholar] [CrossRef]
- Saitô, H.; Ando, I.; Ramamoorthy, A. Chemical shift tensor–the heart of NMR: Insight into biological aspects of proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2010, 57, 181–228. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, I.; Ohmine, M.; Tanabe, J.; Tuzi, S.; Saitô, H.; Naito, A. Dynamic aspects of extracellular loop region as a proton release pathway of bacteriorhodopsin studied by relaxation time measurements by solid state NMR. Biochim. Biophys. Acta biomembr. 2007, 1768, 3090–3097. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, S.; Asakawa, N.; Ando, S.; Ando, I.; Shoji, A.; Ozaki, T. Hydrogen bond length and 15N NMR chemical shift of the glycine residue of some oligopeptide in the solid state. J. Mol. Struct. 1991, 245, 69–80. [Google Scholar] [CrossRef]
- Saitô, H.; Naito, A. NMR studies on fully hydrated membrane proteins, with emphasis on bacteriorhodopsin as a typical and prototype membrane protein. Biochim. Biophys. Acta 2007, 1768, 3145–3161. [Google Scholar] [CrossRef] [PubMed]
- Metz, G.; Siebert, F.; Engelhard, M. High-resolution solid state 13C NMR of bacteriorhodopsin: Characterization of [4-13C]asp resonances. Biochemistry 1992, 31, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.F.; Neudecker, P.; Kay, L.E. Determination of isoleucine side-chain conformations in ground and exited states of proteins from chemical shifts. J. Am. Chem. Soc. 2010, 132, 7589–7591. [Google Scholar] [CrossRef] [PubMed]
- de Groot, N.S.; Parella, T.; Aviles, F.X.; Vendrell, J.; Ventura, S. Ile-phe dipeptide self-assembly: Clues to amyloid formation. Biophys. J. 2007, 92, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Oldfield, E. Correlation between 15N NMR chemical shifts in proteins and secondary structure. J. Biomol. NMR 1994, 4, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Görbitz, C.H. L-isoleucyl-l-phenylalanine dihydrate. Acta Crystallogr. Sect. C 2004, 60, o371–o373. [Google Scholar] [CrossRef] [PubMed]
- Görbitz, C.H. Nanotube formation by hydrophobic dipeptides. Chem. Eur. J. 2001, 7, 5153–5159. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]




| Atom | Observed Chemical Shift (ppm) | Calculated Chemical Shift (ppm) | |
|---|---|---|---|
| Ile1 | 15NH3+ | 16.0 | 3.2 ± 1.3 |
| 13Cα | 57.1 | 60.1 ± 0.6 | |
| 13Cβ | 36.2 | 37.2 ± 0.2 | |
| 13Cγ1 | 25.8 | 23.3 ± 0.1 | |
| 13Cγ2 | 13.0 | −0.3 ± 0.3 | |
| 13Cδ1 | 10.7 | −0.6 ± 0.3 | |
| 13C=O | 173.2 | 150.1 ± 0.4 | |
| Phe2 | 15NH | 97.6 | 97.9 ± 2.5 |
| 13Cα | 52.3 | 53.4 ± 0.5 | |
| 13Cβ | 36.2 | 29.5 ± 0.3 | |
| 13Cγ | 137.1 | 141.7 ± 1.8 | |
| 13Cδ1 | 125.0 | 115.9 ± 0.6 | |
| 13Cδ2 | 121.9 ± 0.6 | ||
| 13Cε1 | 129.4 | 121.2 ± 0.4 | |
| 13Cε2 | 116.6 ± 0.4 | ||
| 13Cζ | 127.5 | 109.7 ± 1.2 | |
| 13COOH | 167.2 (COOH) | 159.4 ± 0.9 (COO−) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawamura, I.; Shirakata, H.; Ozawa, Y.; Mijiddorj, B.; Ueda, K. Solid-State NMR Characterization of the Structure of Self-Assembled Ile–Phe–OH. Magnetochemistry 2018, 4, 30. https://doi.org/10.3390/magnetochemistry4030030
Kawamura I, Shirakata H, Ozawa Y, Mijiddorj B, Ueda K. Solid-State NMR Characterization of the Structure of Self-Assembled Ile–Phe–OH. Magnetochemistry. 2018; 4(3):30. https://doi.org/10.3390/magnetochemistry4030030
Chicago/Turabian StyleKawamura, Izuru, Hiroki Shirakata, Yumi Ozawa, Batsaikhan Mijiddorj, and Kazuyoshi Ueda. 2018. "Solid-State NMR Characterization of the Structure of Self-Assembled Ile–Phe–OH" Magnetochemistry 4, no. 3: 30. https://doi.org/10.3390/magnetochemistry4030030
APA StyleKawamura, I., Shirakata, H., Ozawa, Y., Mijiddorj, B., & Ueda, K. (2018). Solid-State NMR Characterization of the Structure of Self-Assembled Ile–Phe–OH. Magnetochemistry, 4(3), 30. https://doi.org/10.3390/magnetochemistry4030030