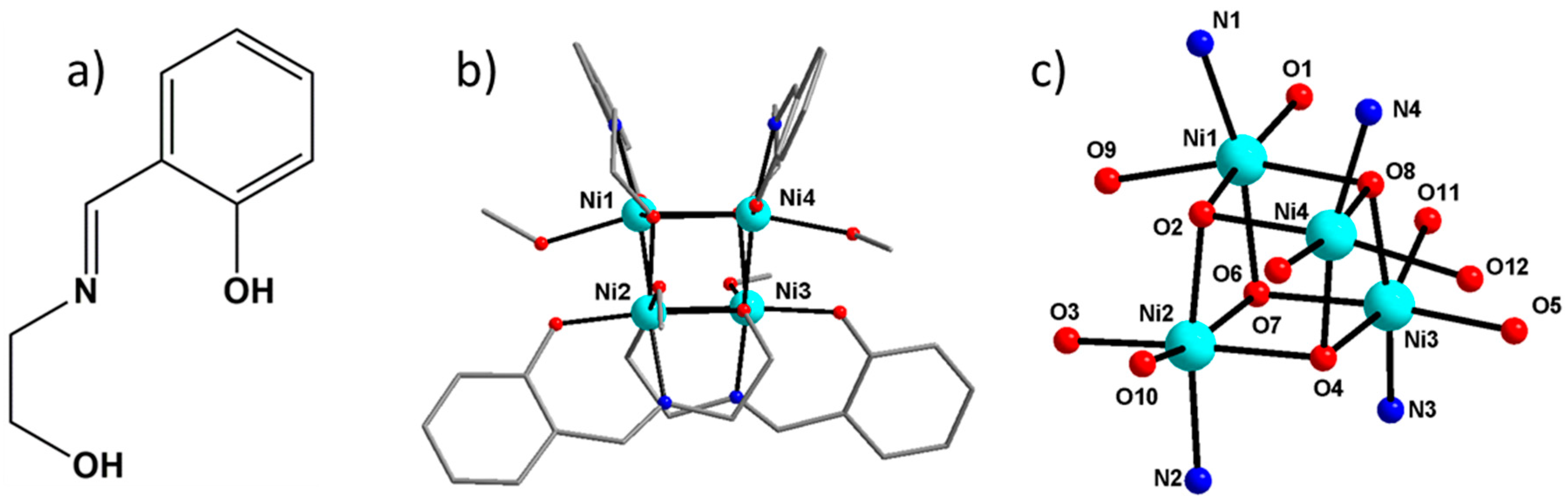
3.1. Bulk Magnetic Anisotropy from Magnetometry
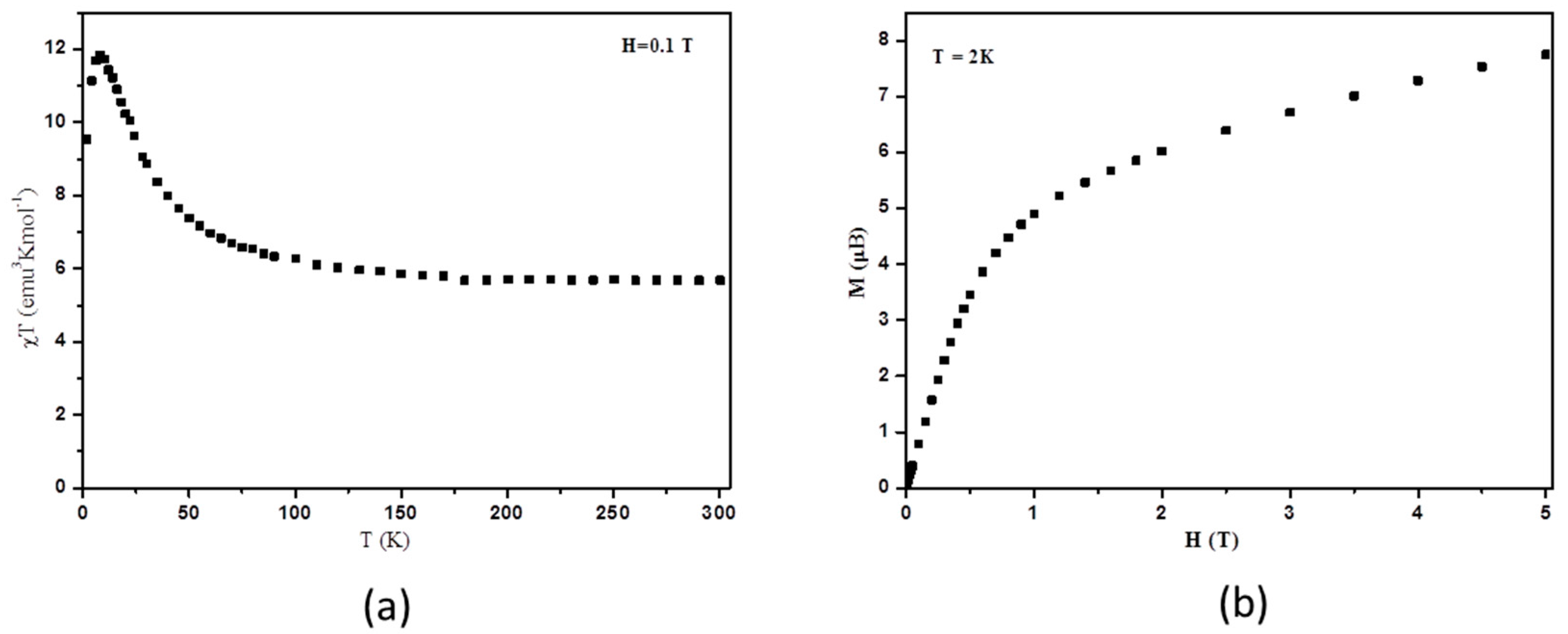
Magnetic measurements on a polycrystalline sample are in agreement with the previously reported study [
3], as shown on
Figure 2. In a magnetic field of 0.1 T the temperature dependence of the product of the molar magnetic susceptibility with temperature (χ
T) is 5.67 emu·K·moL
−1 (
Figure 2a), which is higher than the expected value for four uncorrelated Ni
2+ ions. The χ
T product continuously increases upon cooling to reach a maximum of 11.83 cm
3·K·moL
−1 at 8 K, indicating the dominant ferromagnetic interactions within the cluster. The maximum value is close to the expected value for an
S = 4 ground spin-state with a
g-value of 2.2. However, this g-value is quite high, in agreement with ground spin-state anisotropy. At 2 K, magnetization increases continuously with the field (
Figure 2b) but does not reach saturation at 5 T.
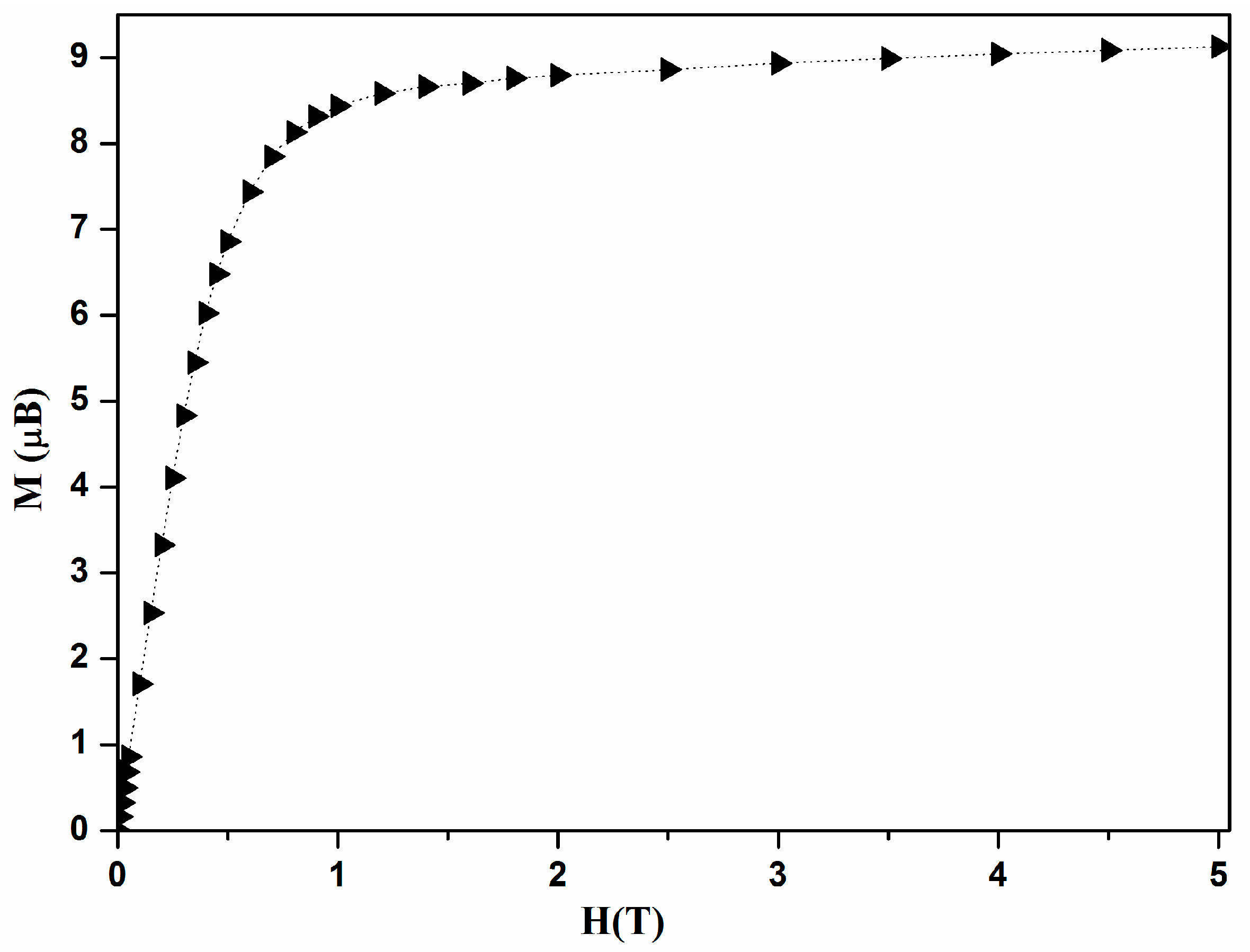
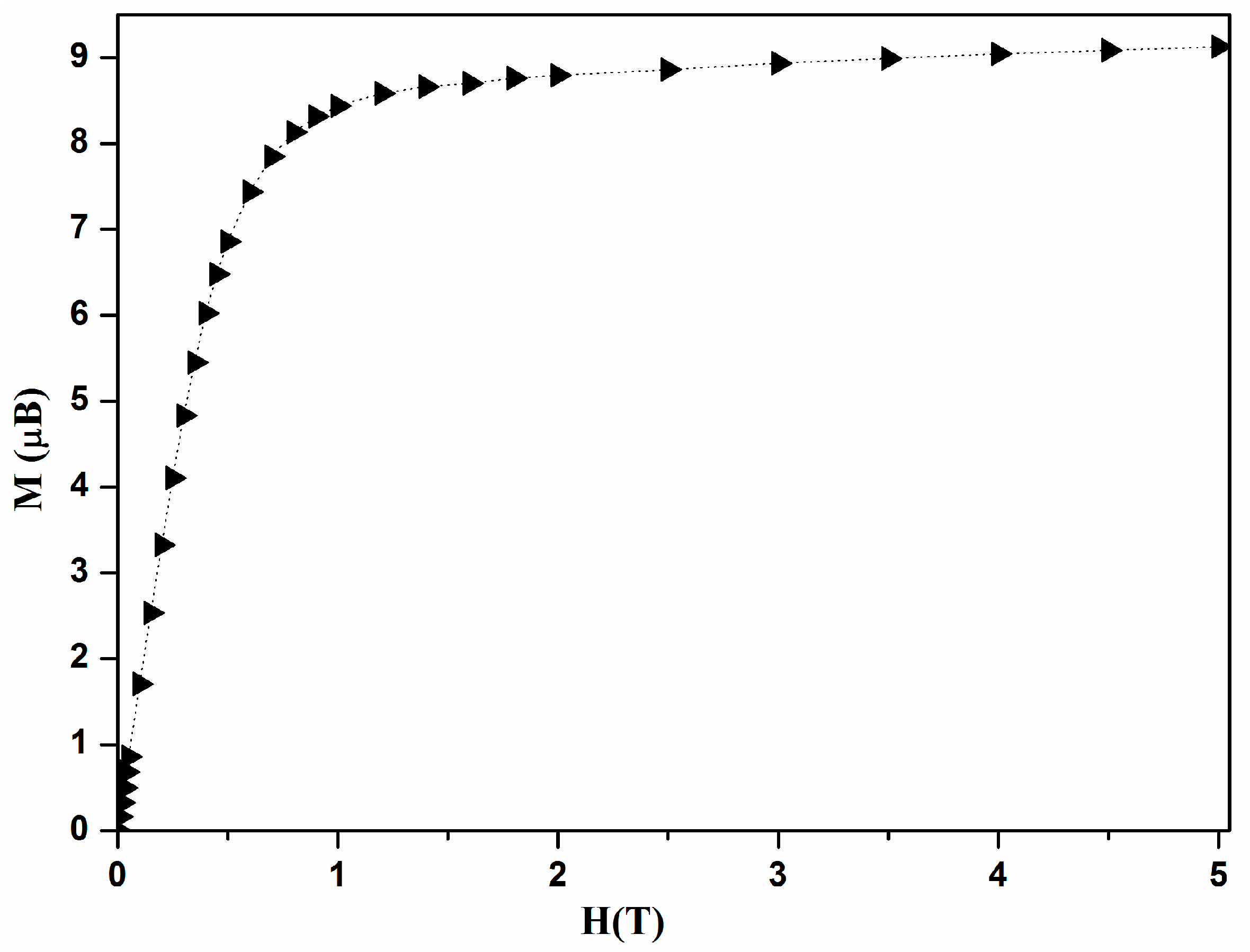
We report in
Figure 3 the field dependence of magnetization at 2 K for a single crystal of [Ni
4], with its easy axis directed along the applied field. It shows a rapid increase and reaches a saturation value of 9 µ
B which corresponds to a ground spin-state
S = 4 with
g = 2.2.
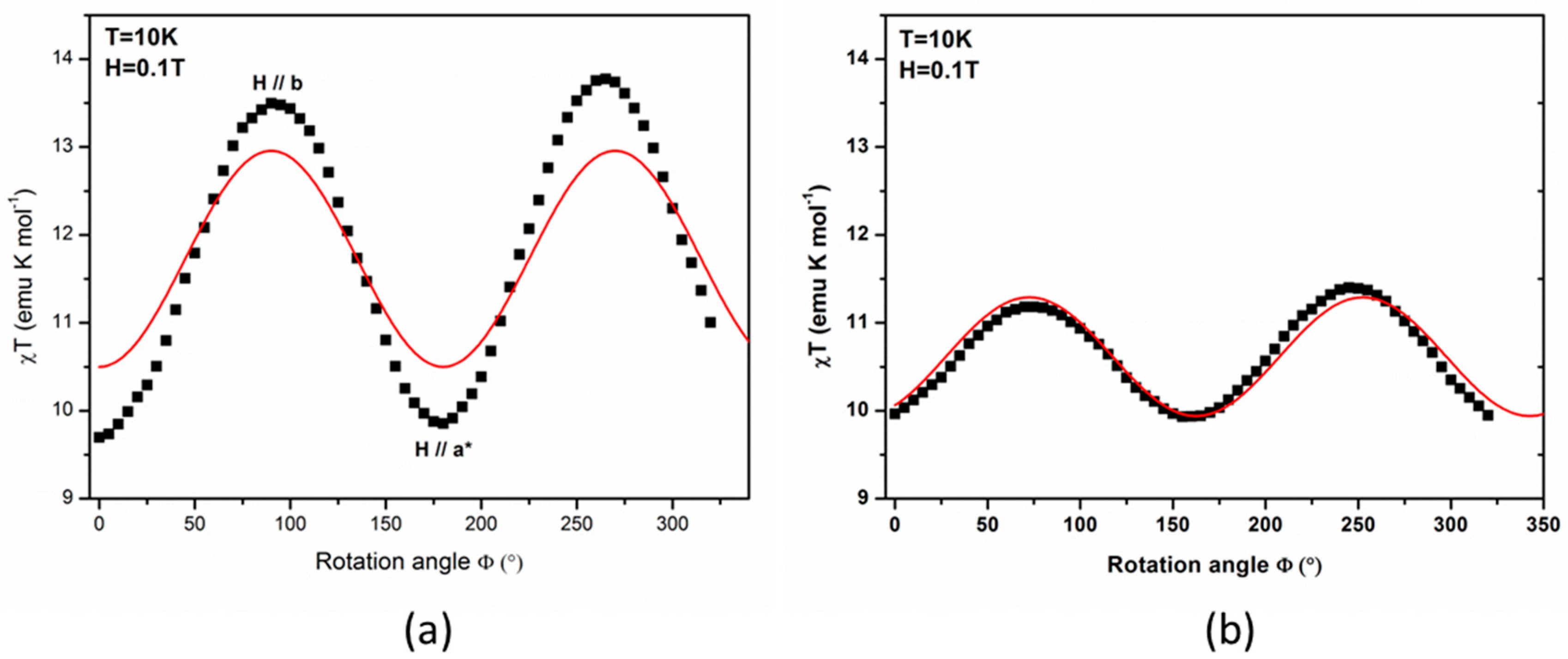
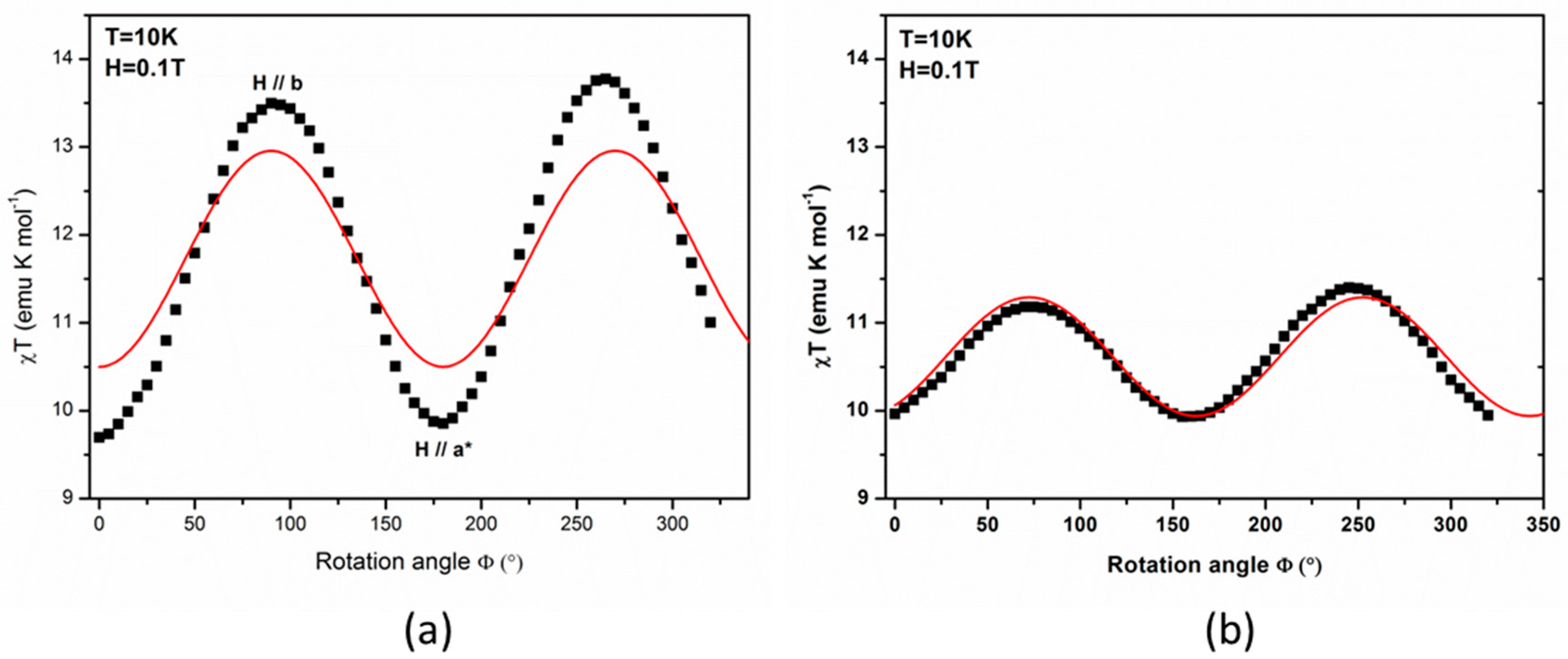
The results of angle-resolved susceptibility measurements on single crystal are displayed in
Figure 4, in the form of the product of the magnetic susceptibility with temperature (χ
T) versus the rotation angle (Φ). The crystal was successively rotated around the
and
crystal axes with, in both cases, the
axis initially set along the applied magnetic field (rotation angle Φ = 0). As may be expected, the presence of minima and maxima clearly reveals the existence of an appreciable magnetic anisotropy. It can be noticed that the maximal χ
T values for the rotation around the
axis are well above the ones observed for the rotation around the
axis, while the minima are similar for both experiments.
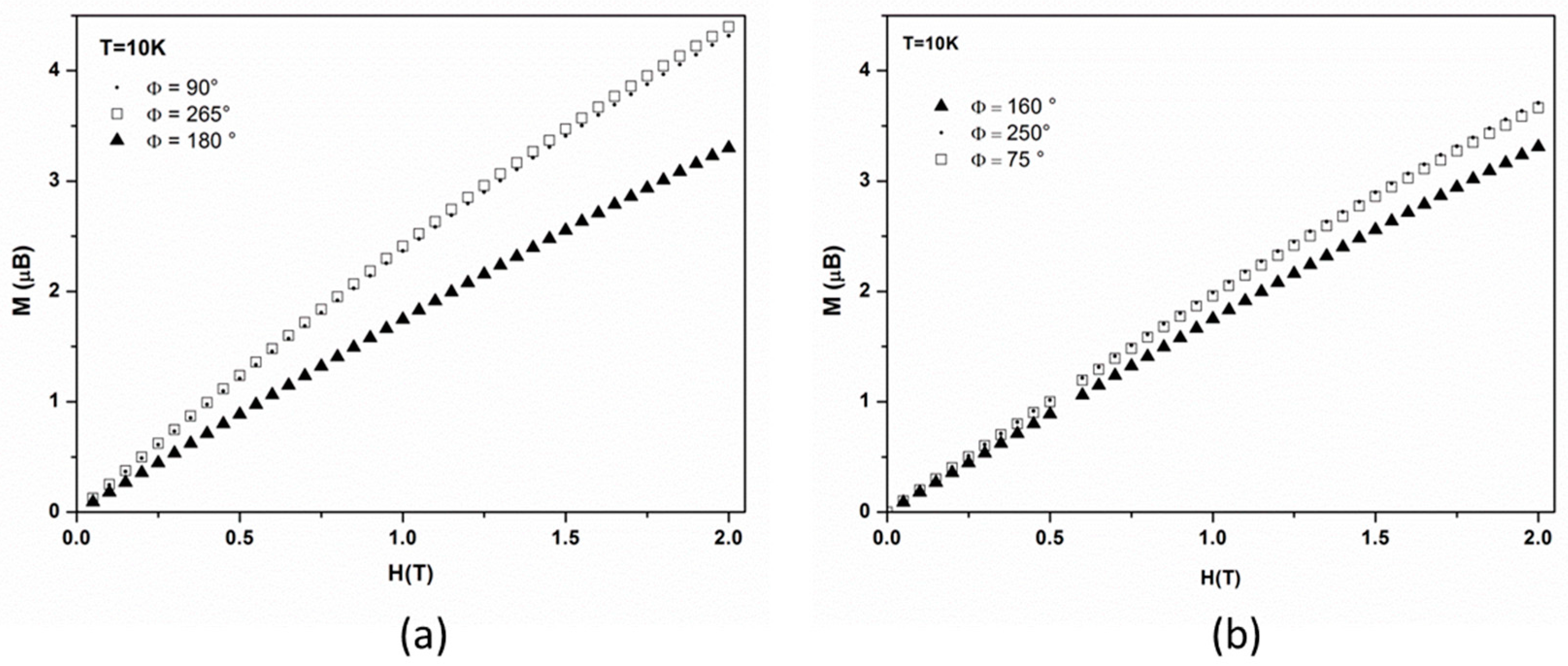
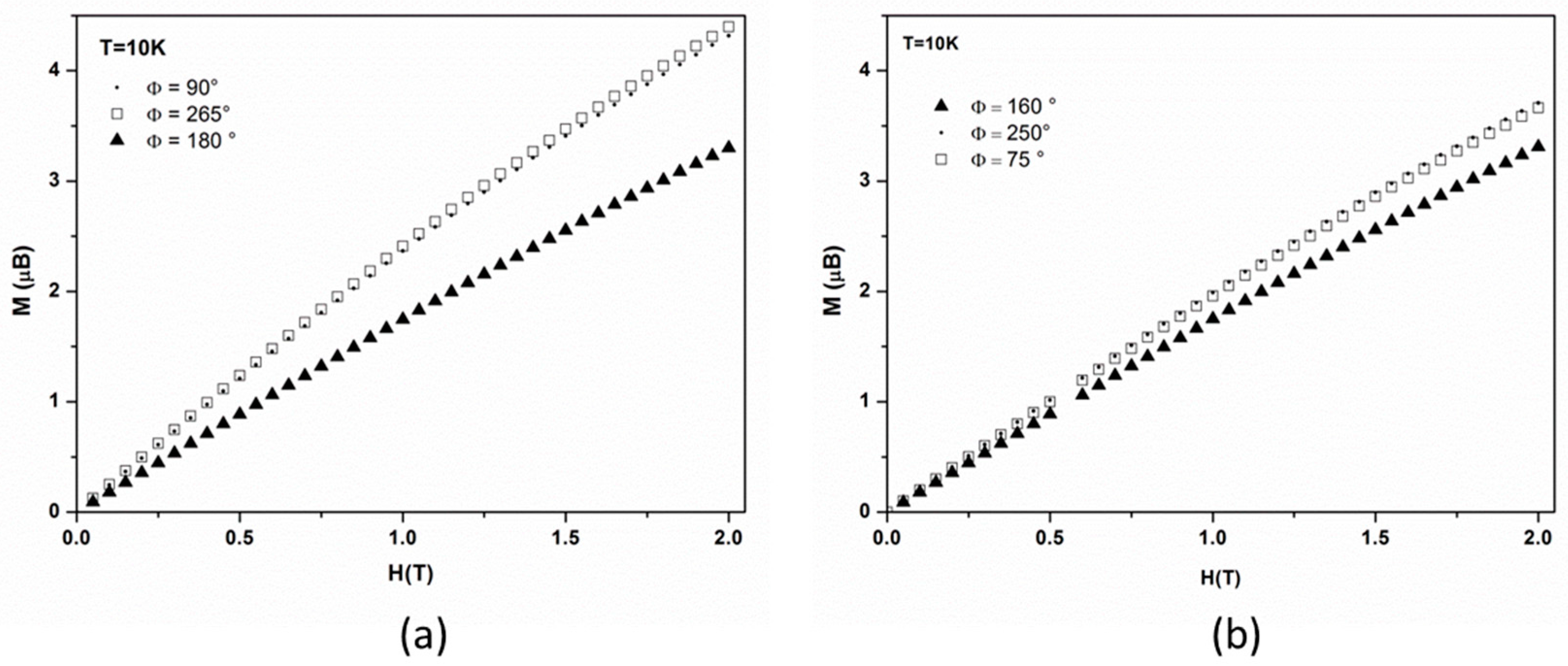
For each set of data we also measured the dependence of magnetization with the magnetic field at each maximum and minimum, at 10 K and up to 2 T, which is the magnetic field used during the PND study. For rotation around the
axis (
Figure 5a), the field dependence of magnetization for the two maxima (
//
and
//
) superimpose as may be expected, and magnetization continuously increases up to 4.4 µ
B at 2 T, while, for the minimum (
//
), it increases up to 3.4 µ
B. For the
axis rotation (
Figure 5b), close values (3.3 and 3.7 µ
B) are observed at 2 T for both the minimum and two maxima of
Figure 4b.
3.2. Local Susceptibility Tensor from PND
The paramagnetic susceptibility tensor of the crystal results from the sum of the local susceptibility tensors of each molecular cluster in the lattice, which are related by the symmetry operations of the space group. There are four clusters with two different molecular orientations in the cell and, therefore, the magnetic proper axes of the crystal (bulk) may not coincide with the molecular cluster proper axes:
By the successive application of a magnetic field on a single crystal of Ni4, along three orthogonal directions, we managed to collect three experimental flipping ratio datasets. This enables the refinement of the six components of the local (molecular) cluster tensors,
, which are reported in the first row of
Table 3. However, due to the low number of accurate flipping ratios (see
Table 4), the
and
components provided by refinement are below the limit of significance (less than 1.5σ) and, therefore, have been set to zero in the final refinement. The local susceptibility tensor expressed in the (
,
,
) basis set, i.e., a Cartesian basis set with
//
,
//
,
//
, is then the following:
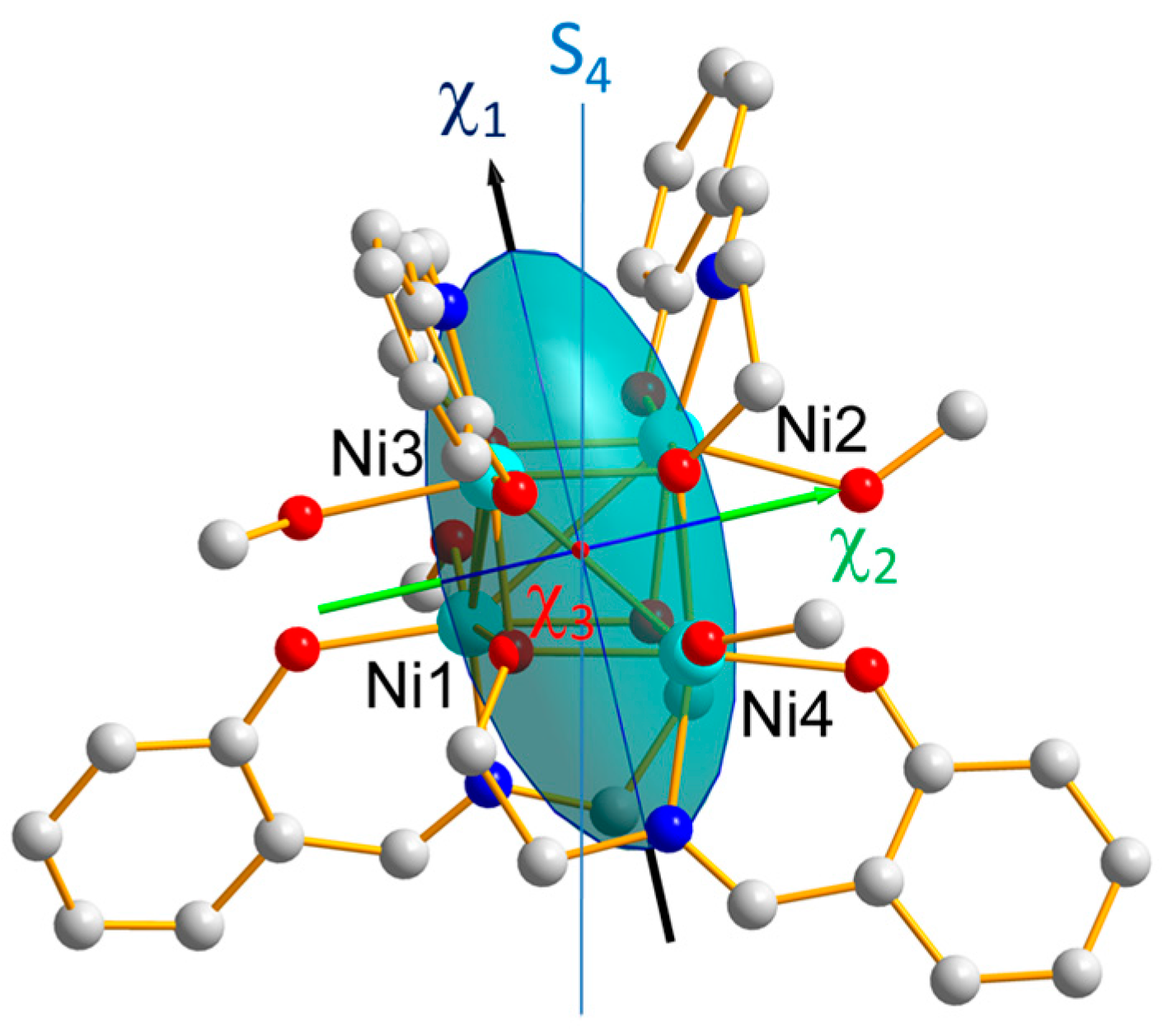
Diagonalization of this tensor provides its eigenvectors and eigenvalues, i.e., the directions of the cluster magnetic axes and the associated magnetic susceptibilities. The latter are gathered in
Table 4 and the components of the eigenvectors in the (
,
,
) frame are provided in
Table S1. The directions of the cluster magnetic axes are displayed in
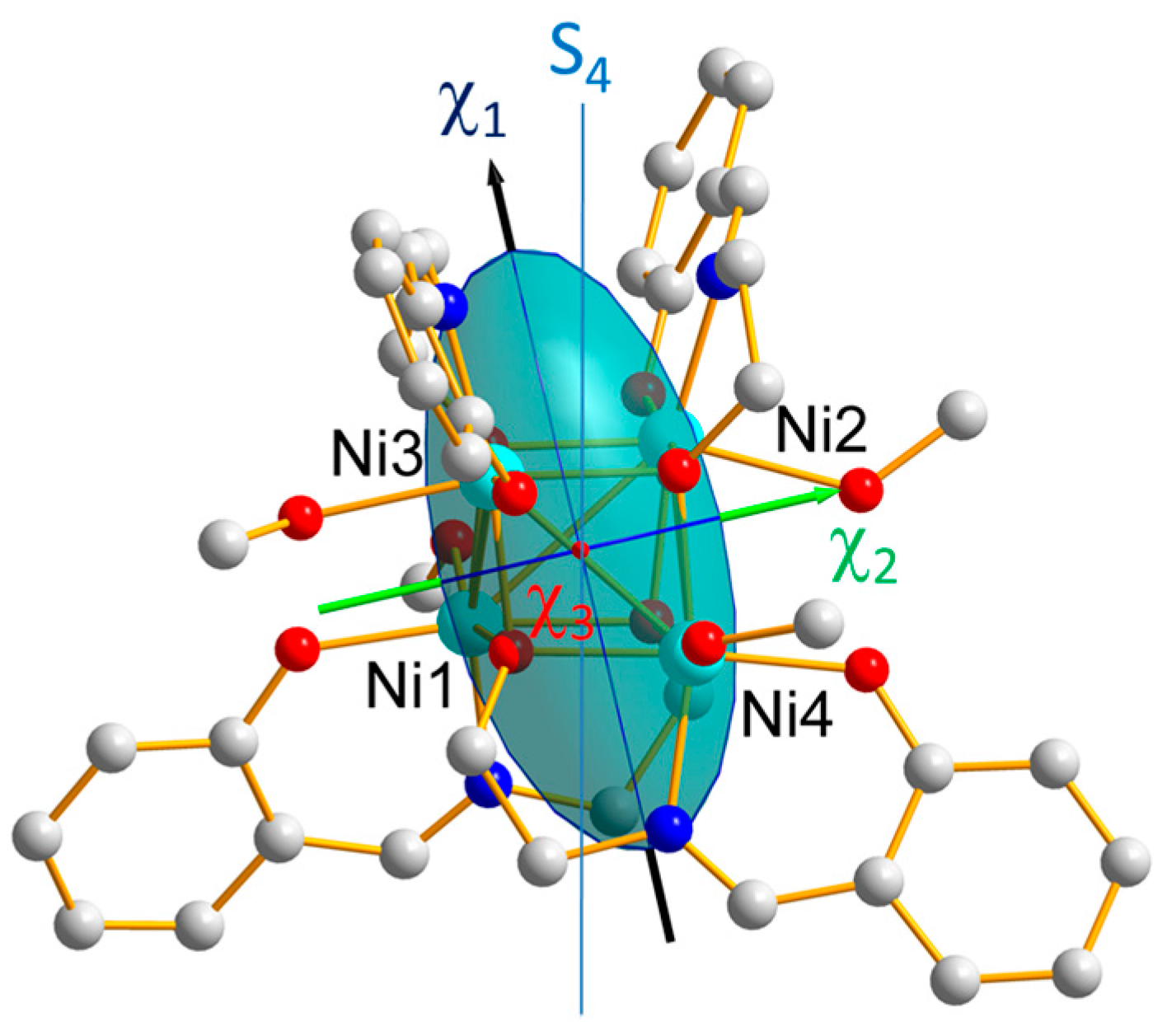
Figure 6, altogether with the magnetic ellipsoid of the cluster.
As we already stated, the susceptibility tensor of the cluster results from the sum of individual Ni tensors involving single ion paramagnetic contributions (
) and additional terms due to ferro- (W
F) and antiferromagnetic (W
AF) exchange between the Ni(II) ions:
This tensor can be rewritten as a sum of “effective tensors”
which include the perturbation due to the antiferromagnetic exchange between Ni(II) ions belonging to Ni–Ni pairs and the ferromagnetic exchange interactions between the Ni atoms along the diagonal of the Ni
4O
4 cube. This approximation is justified by the weakness of the magnetic exchange interactions in the cluster, as shown by the coupling constants deduced from magnetic measurements,
JF = 8(1) cm
−1 and
JAF = −3(1) cm
−1 from [
11]:
The six components χ
ij for each Ni ion were first refined, on the joint basis of the three datasets, but only those for which the obtained value was larger than 1.5 times the error bar were included in the final refinement. The refinement of the local susceptibility tensors
(with
l = 1, 4) evidences a strong similarity between the Ni1 and Ni4 susceptibilities on one side and between Ni2 and Ni3 on the other side as showed by the reported components in
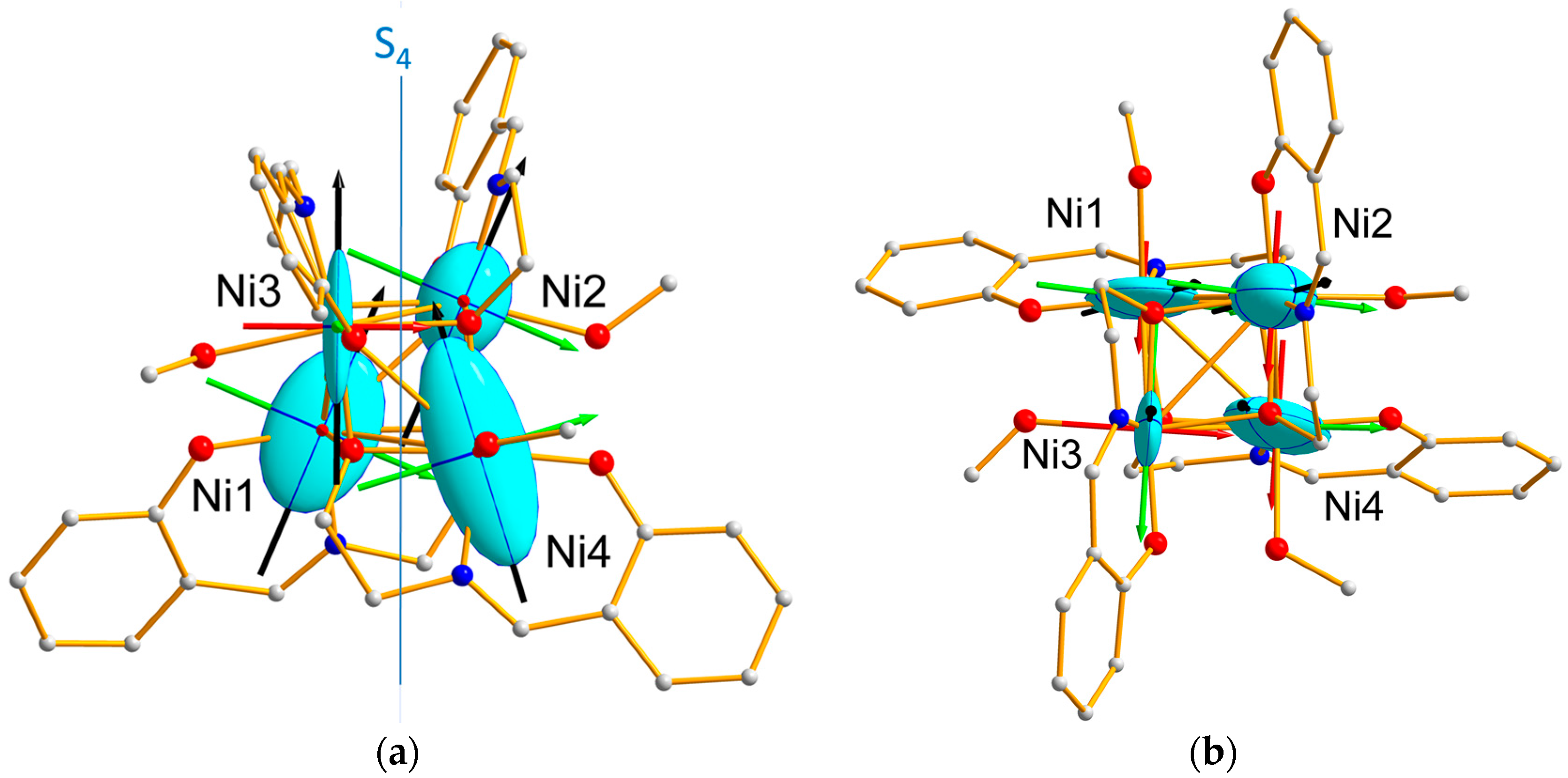
Table 1. This suggests that the two Ni atoms belonging to a same antiferromagnetic (AF) pair, (Ni1, Ni4) and (Ni2, Ni3), respectively, have similar magnetic behaviors with respect to the applied magnetic field. Furthermore, the refined eigenvalues of each Ni local tensor, reported in
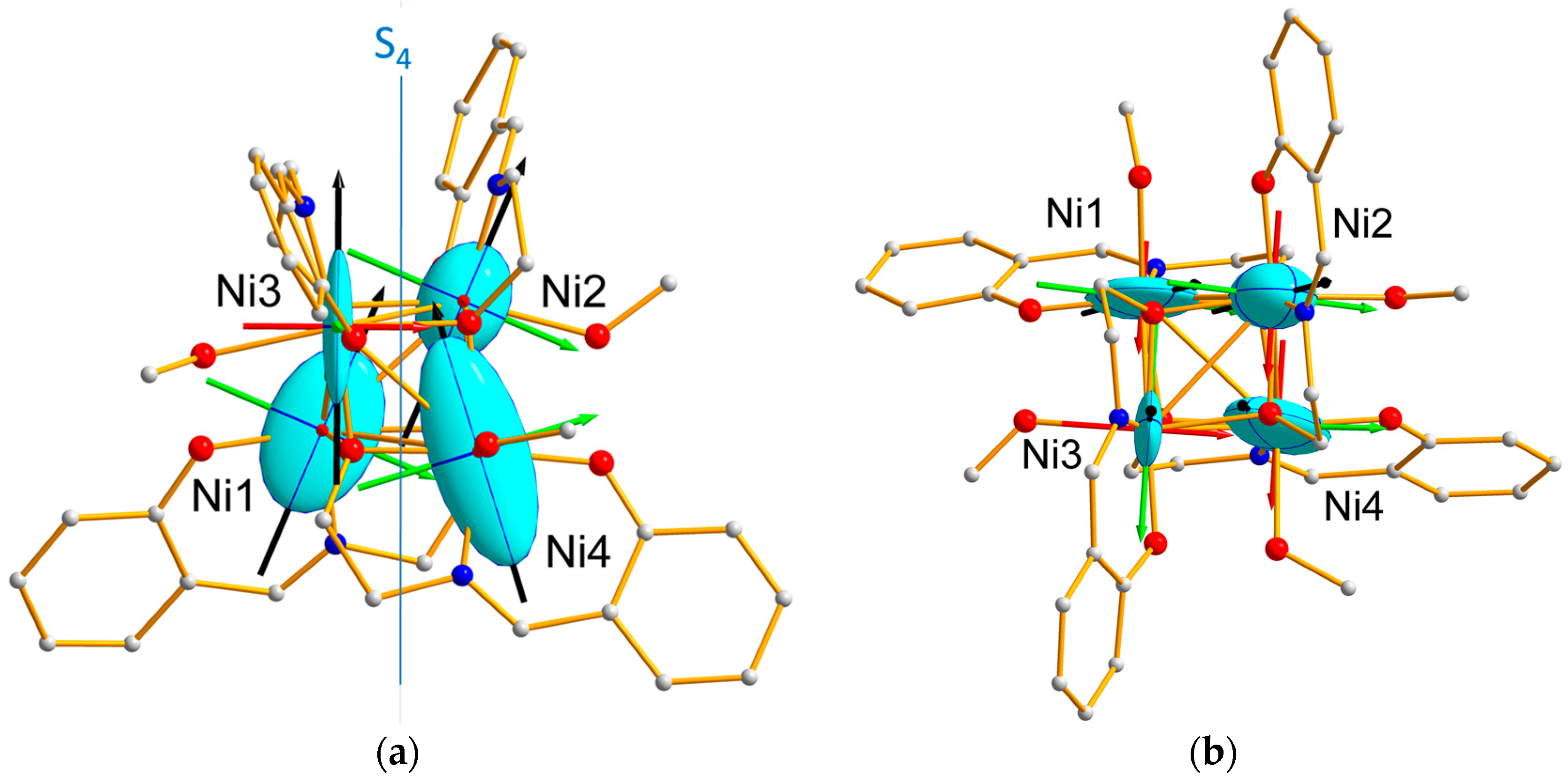
Table 2, show that the Ni1 and Ni4 tensors are more anisotropic than those of Ni2 and Ni3. This feature is reflected by the magnetic ellipsoids drawn in
Figure 7, together with the directions of the eigenvectors, whose components are reported in
Table S1.
Therefore we introduced two “effective tensors” (with k = 1, 2) associated with each of the AF pairs, which include the perturbation by ferromagnetic exchange interactions between the Ni atoms of the pairs.
The components of the two tensors associated to each AF pair are reported in
Table 1 and the corresponding eigenvalues in
Table 2. The components of the eigenvectors can be found in
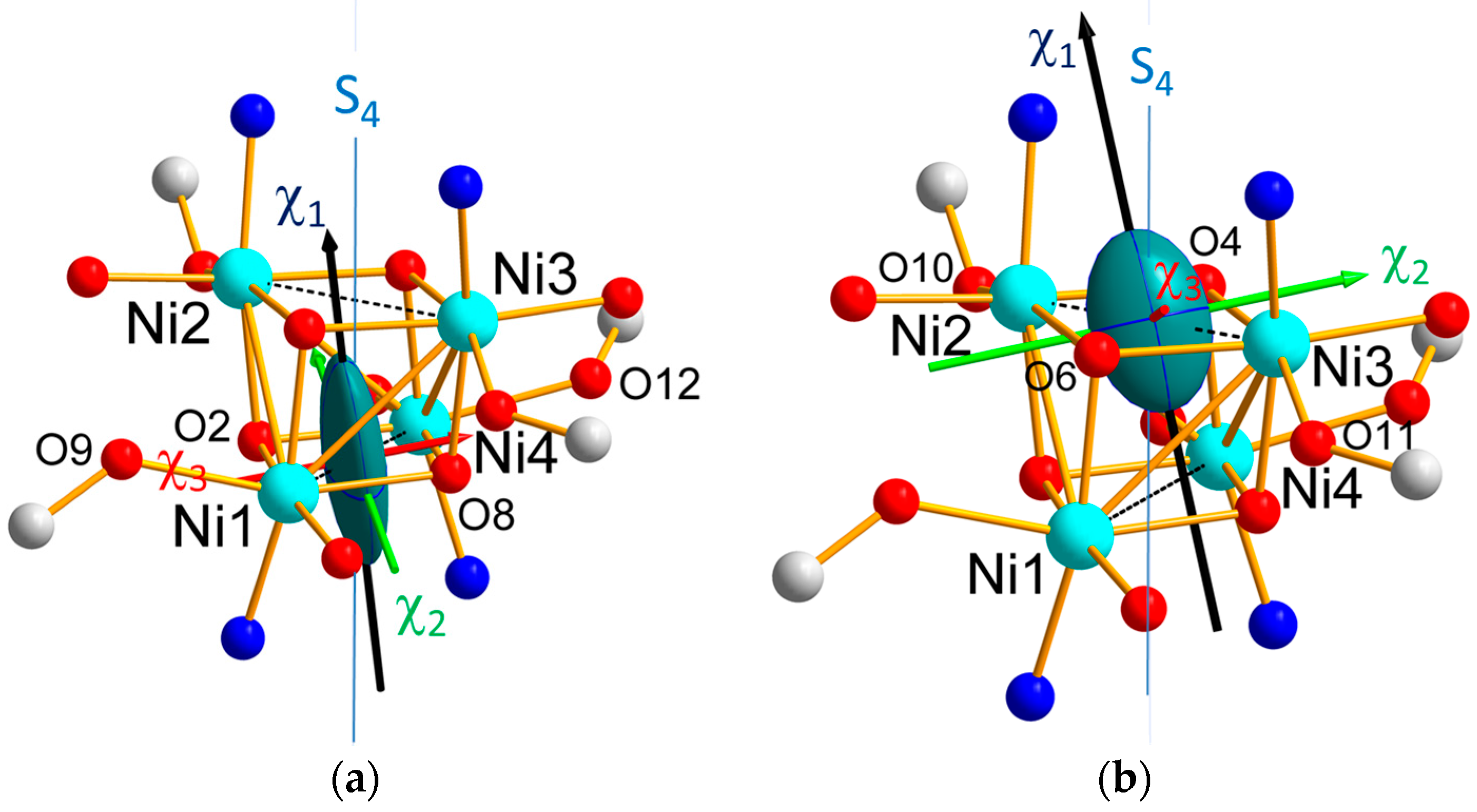
Table S1. The obtained values indicate the tensor associated to the Ni1-Ni4 pair is more anisotropic than the tensor relative to the Ni2–Ni3 pair, in agreement with the previous findings on the single ion tensors. The magnetic ellipsoids and principal magnetic axes are represented in
Figure 8. The easy axes (
) of the pairs are both close to the S
4 pseudo-symmetry axis, but the hard (
) and intermediate (
axes are found nearly perpendicular to each other axes, as well.
3.3. Discussion
The main features of the local geometry around the Ni atoms in [Ni
4] are reported in
Table 5. The deformation of the coordination octahedron around each Ni atom of this cluster is due a tetragonal elongation along the O–Ni–O direction involving the coordinated methanol and the bridging alkoxide [
15]. The local geometries around Ni1 and Ni4 (Ni2 and Ni3, respectively) are very similar, as exemplified by the longest distance between opposite edge atoms in the octahedron (which reflects the distortion of the octahedron): 4.246 Å for Ni1, and 4.254 Å for Ni4. Noteworthy, this distance is slightly larger (4.275 Å) for both Ni2 and Ni3.
It has to be noticed that the directions of tetragonal elongation are roughly parallel for Ni1 and Ni4 on one side and for Ni2 and Ni3 on the other side, i.e., for atoms which are AF coupled. In contrast, the elongation direction for one AF pair is orthogonal to the elongation direction of the other pair, i.e., for atoms which are ferromagnetically coupled. The structural characteristics of the six Ni
2O
2 bridges of the cubane are summarized in
Table 6. The AF bridges present longer Ni…Ni distances (3.19 Å) with two short and two long Ni–O bonds and bridging angles of 100–101 deg., while the ferromagnetic bridges display shorter Ni…Ni distances (3.04 Å) with three short and one long Ni–O bonds and bridging angles of 91 to 97 deg, as shown in
Figures S1 and S2 in the Supplementary Information.
The similarity between the Ni “effective” single-ion susceptibility tensors obtained from PND analysis, for (Ni1, Ni4) on one hand, and (Ni2, Ni3) on the other hand, can be correlated to the two corresponding local geometries. Although the local tetragonal distortion is slightly larger for (Ni2, Ni3) than for (Ni1, Ni4), with a O-Ni-O distance of 4.275 Å compared to 4.25 Å, the (Ni1, Ni4) tensors are more anisotropic than the (Ni2, Ni3) ones. This suggests that the axial distortion of the coordination octahedron is not the only source of magnetic anisotropy. Actually, another feature of the octahedral environment distortion for the Ni1 and Ni4 ions may be related to the more anisotropic character of their single-ion magnetic behavior: the deformation of the triangular faces of the octahedron. The sum of the deviations from 60 deg (in absolute magnitude) of the inner angles of the eight faces is indeed equal to 115.6 deg for (Ni1, Ni4) and 112.6 deg for (Ni2, Ni3). This would in turn mean that deformation in the equatorial directions of the coordination octahedron plays an important role.
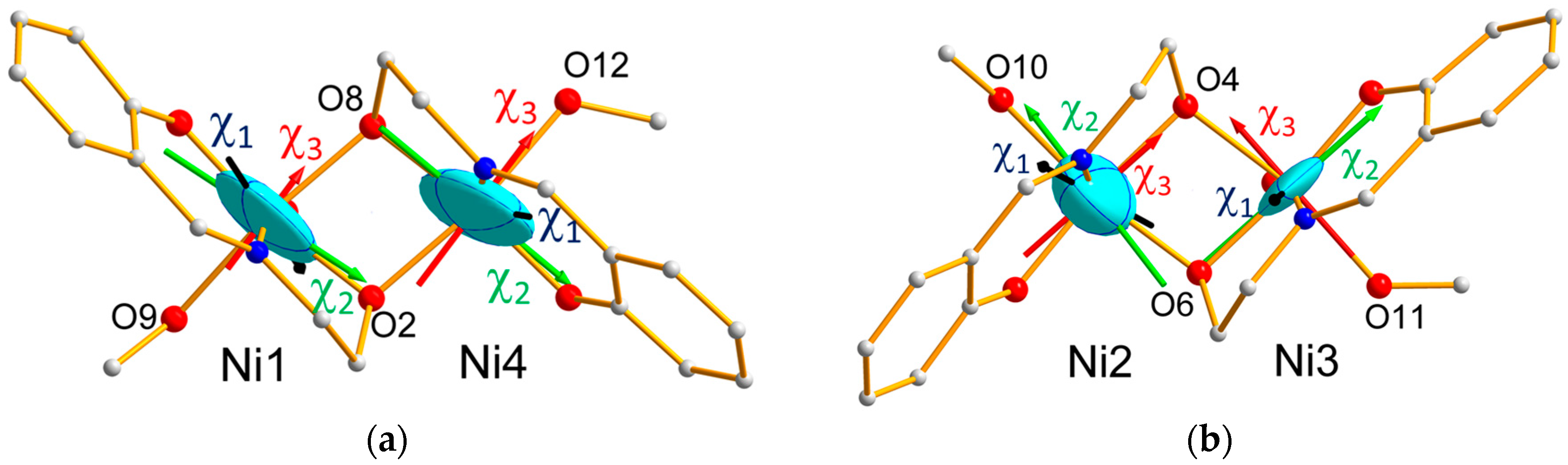
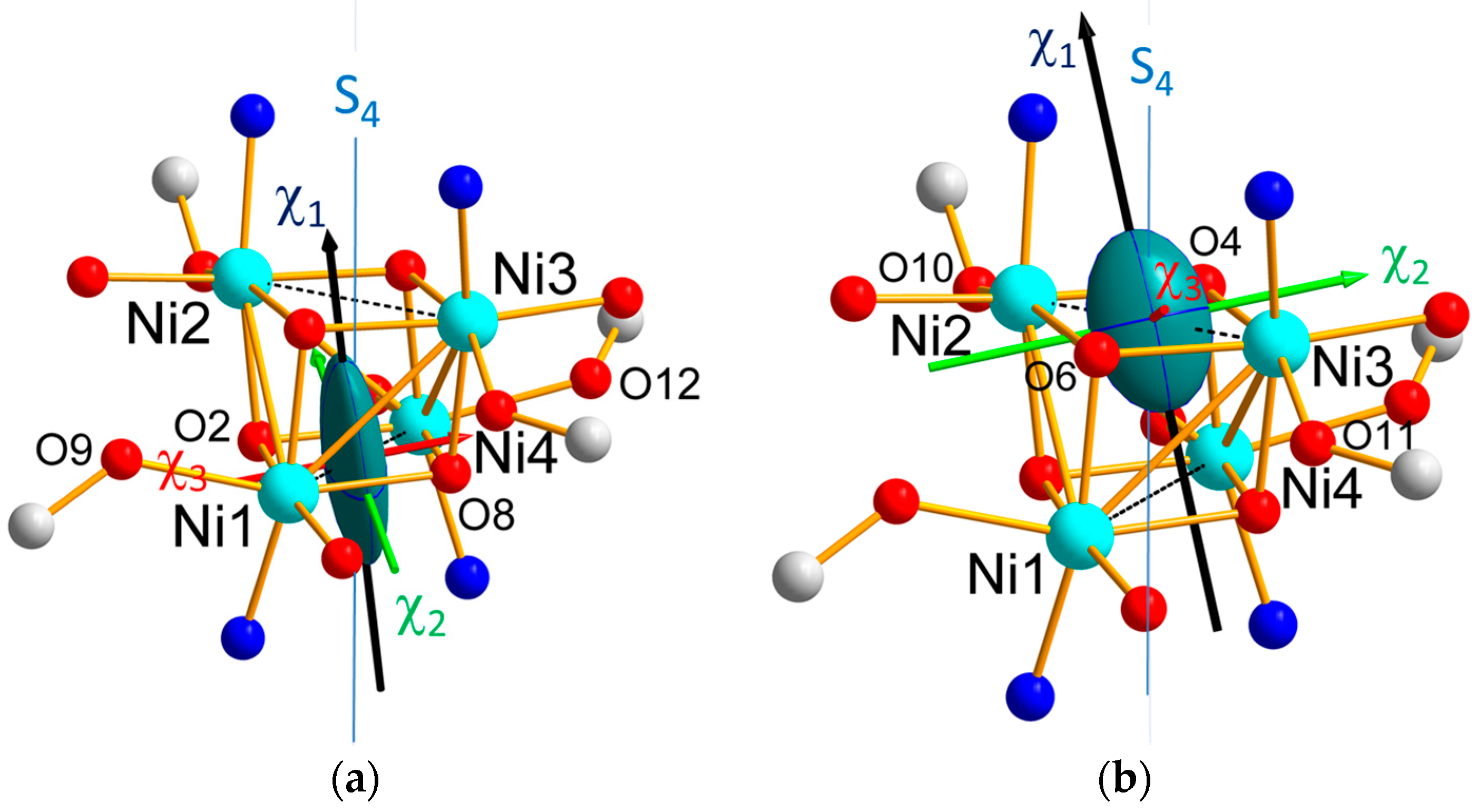
As it can be seen in
Figure 4a,b, which display the ellipsoids relative to the Ni1O
2Ni4 and Ni2O
2Ni3 bridges, the directions of the
hard magnetic axes for Ni1, Ni3 and Ni4 are close to the local distortion axes, i.e., along the O–Ni–O direction involving methanol and bridging alkoxide, as speculated in [
15]. This is not verified for the Ni2 ion. However, from
Table 4, the eigenvalues of the Ni2 local tensor corresponding to the mean and hard axes are close to each other (χ
2 = 0.42(20)μ
B/T and χ
3 = 0.33(6)μ
B/T) with overlapping error bars, showing that it is difficult to discriminate between these two directions and that the exact direction of the hard axis in the plane perpendicular to the easy axis cannot be determined from the Ni2 local tensor.
The effective magnetic tensors associated to each antiferromagnetic pair (
Figure 8) reflect the resultant of the effective single ion tensors of the Ni ions involved in the pair (
Figure 9). Noticeably, the hard magnetic axes of the both pairs are perpendicular to each other, in agreement with the local directions of tetragonal distortion.
The cluster experimental local susceptibility tensor (Equation (11)) at 10 K obtained from the PND data analysis may be used to calculate the bulk magnetization induced by a magnetic field, applied in any direction of space, for a more direct comparison with the angular-resolved measurements. Taking into account the symmetry operations of the P2
1/c space group, the crystal susceptibility tensor can be deduced from Equations (10) and (11), which leads to the bulk susceptibility tensor:
This tensor is diagonal in the (,,) basis set and, therefore, its eigenvectors, i.e., the bulk magnetic proper axes, coincide with the , , and axes, which fully agrees with the SQUID data analysis.
In order to further compare the PND results with the angular-resolved magnetic data, the single crystal SQUID data were simulated using the experimental cluster susceptibility tensor from PND and a homemade program taking into account the magnetic field orientations and the lattice symmetry. For the first set of data, given as rotations around the crystal
axis, the simulation reproduces well the positions of maxima at 90° and 180° and the minimum at 265° as shown in
Figure 4a (solid curves). Thus, the maxima at 90° and 180° can be ascribed to the
axis and the minimum at 265° to the
axis.
For the second set, given as rotations around the
axis, the simulation of the maxima at 75° and 160° and minimum at 250° was not possible as such. Moreover in
Figure 5a, the magnetization value at 2 T for the minimum (3.3 µ
B) differs from the expected one (2µ
B) from the PND bulk tensor (Equation (15)) for
//
, while for the maximum (3.7 µ
B) it is larger than the expected value (3.8 µ
B) from Equation (15) for the intermediate axis
//
. This is very likely due to a tilt of the rotation axis in the (
,
) plane, as the precise orientation of the
axis proved to be very hard to find because of the crystal shape. The simulation of the data reported in
Figure 4b only proved possible when considering the true rotation axis was tilted from the
axis in the (
,
) plane, with the magnetic field initially close to the
axis, as already proposed. The best agreement between our simulation and the experimental data, displayed on
Figure 4b, was obtained for a tilt angle ω of −65°, suggesting the rotation axis was actually closer to
than to
as first expected. As one can note, with such a hypothesis a very good agreement is reached with the experimental data, both on the locations and magnitudes of the extrema of the curve. Alas, this implies that the orientations in the lattice corresponding to these extrema are rather ill-defined and, thus, prevents a proper assignment of the maxima or the minima.
As shown in
Figure 1, the local easy magnetic axis
of the cluster is close to the pseudo-symmetry S
4 axis, in agreement with the conclusion of the previous INS investigation [
3]. The following Hamiltonian was used for the full description of the eight transitions in the INS spectra (with selection rules ΔM
S = ±1) [
3]:
where
is the zero-field splitting (ZFS) Hamiltonian:
and
are high order terms (Stevens operators) corresponding to crystal field effects.
The axial and rhomboedral ZFS parameters of the cluster were found to be equal to D = −0.93(2) cm−1 and E =0.023(8) cm−1(due to deviations from S4 cluster symmetry). The observed axial anisotropy of the cluster magnetic ellipsoid (χ1 >> χ2, χ3) confirms the easy axis-type magnetic anisotropy of the cluster predicted by the negative sign of D.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}