Differences in Saprophytic Growth, Virulence, Genomes, and Secretomes of Ilyonectria robusta and I. mors-panacis Isolates from Roots of American Ginseng (Panax quinquefolius)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolates
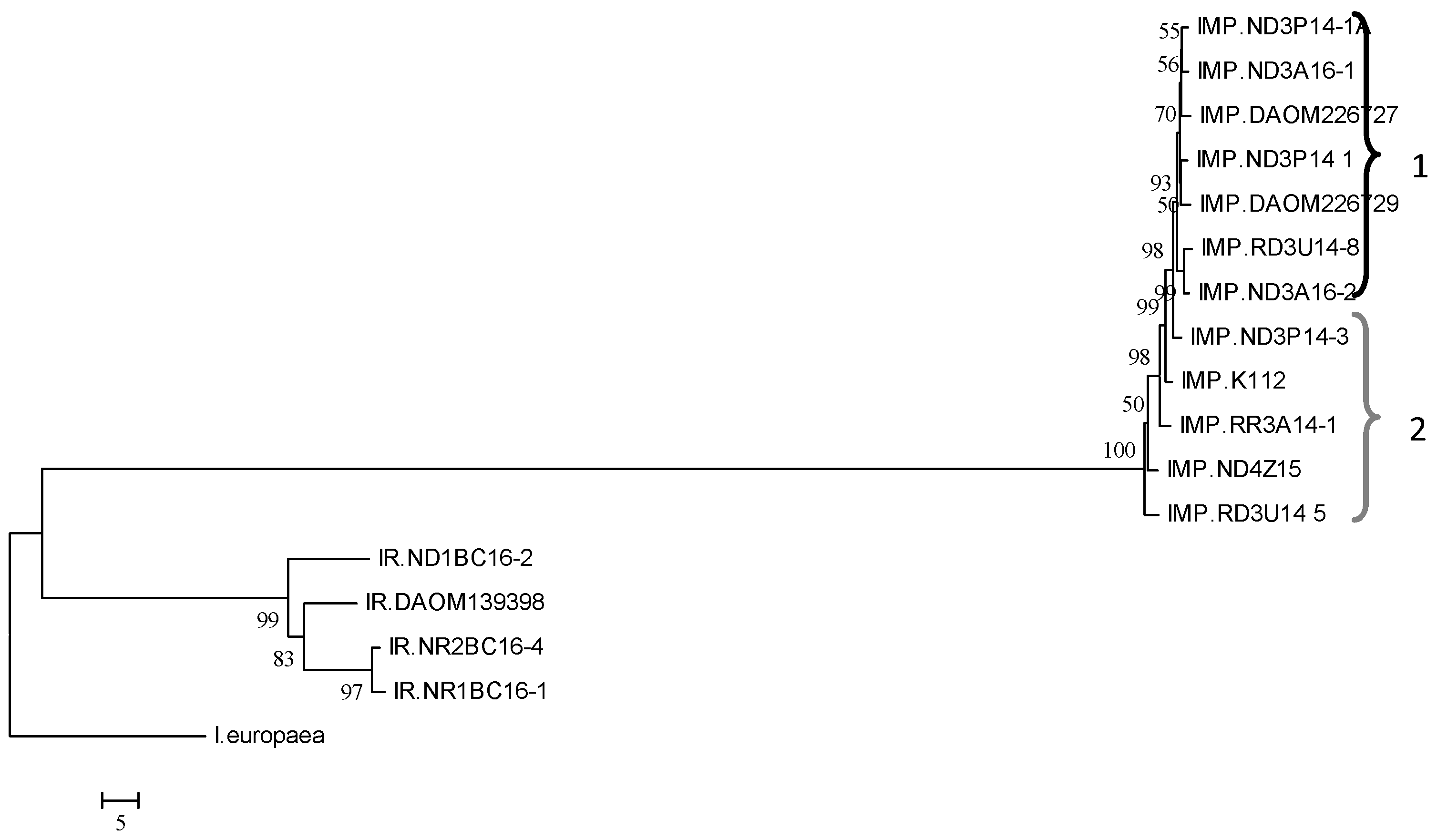
2.2. Histone H3 Sequencing
2.3. Isolate Growth Rate and Virulence
2.4. Genome Sequencing, Assembly and Gene Prediction
2.5. Total Predicted Gene Analysis
2.6. Secretome Analysis
2.7. Non-Secretome Analysis
3. Results
3.1. Histone H3 Sequencing
3.2. Growth Rate and Virulence
3.3. Genomic Sequencing and Assembly Quality
3.4. Total Predicted Genes of the Ilyonectria Isolates
3.5. Genomic Comparisons of the Ilyonectria Isolates
3.6. Secretome of the Ilyonectria Isolates
3.7. Non-Secretome of the Ilyonectria Isolates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sewell, G.F.W. Effects of Pythium species on the growth of apple and their possible causal role in apple replant disease. Ann. App. Biol. 1981, 97, 31–42. [Google Scholar] [CrossRef]
- Petit, E.; Gubler, W.D. Characterization of Cylindrocarpon species, the cause of black foot disease of grapevine in California. Plant Dis. 2005, 89, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Seifert, K.A.; Axelrood, P.E. Cylindrocarpon destructans var. destructans. Can. J. Plant Pathol. 1998, 20, 115–117. [Google Scholar] [CrossRef]
- Cabral, A.; Groenewald, J.Z.; Rego, C.; Oliveira, H.; Crous, P.W. Cylindrocarpon root rot: Multi-gene analysis reveals novel species within the Ilyonectria radicicola species complex. Mycol. Prog. 2012, 11, 655–688. [Google Scholar] [CrossRef] [Green Version]
- Matuo, T.; Miyazawa, Y. On Cylindrocarpon panacis sp. nov. causing root rot of ginseng. Trans. Mycol. Soc. Jpn. 1969, 9, 109–112. [Google Scholar]
- Reeleder, R.D.; Brammall, R.A. Pathogenicity of Pythium species, Cylindrocarpon destructans, and Rhizoctonia solani to ginseng seedlings in Ontario. Can. J. Plant Pathol. 1994, 16, 311–316. [Google Scholar] [CrossRef]
- Matuo, T.; Miyazawa, Y. Scientific name of Cylindrocarpon sp. causing root rot of ginseng. Ann. Phytopathol. Soc. Jpn. 1984, 50, 649–652. [Google Scholar] [CrossRef]
- Song, J.Y.; Seo, M.W.; Kim, S.I.; Nam, M.H.; Lim, H.S.; Kim, H.G. Genetic diversity and pathogenicity of Cylindrocarpon destructans isolates obtained from Korean Panax ginseng. Mycobiology 2014, 42, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.W.; Park, J.Y.; Kim, S. Analysis of genetic diversity and PCR assay for the detection of ginseng root rot pathogen, Ilyonectria radicicola. Res. Plant Dis. 2015, 21, 121. [Google Scholar]
- Reeleder, R.D.; Roy, R.; Capel, B. Seed and root rots of ginseng (Panax quinquefolius L.) caused by Cylindrocarpon destructans and Fusarium spp. J. Ginseng Res. 2002, 26, 151–158. [Google Scholar]
- Li, T.S.C. Asian and American ginseng—A review. HortTechnology 1995, 5, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Punja, Z.K. Factors influencing development of root rot on ginseng caused by Cylindrocarpon destructans. Phytopathology 2005, 95, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Kernaghan, G.; Reeleder, R.D.; Hoke, S.M.T. Quantification of Cylindrocarpon destructans f. sp. panacis in soils by real-time PCR. Plant Pathol. 2007, 56, 508–516. [Google Scholar] [CrossRef]
- Farh, M.E.; Kim, Y.J.; Kim, Y.J.; Yang, D.C. Cylindrocarpon destructans/Ilyonectria radicicola-species complex: Causative agent of ginseng root-rot disease and rusty symptoms. J. Ginseng Res. 2018, 42, 9–15. [Google Scholar] [CrossRef]
- Westerveld, S.M.; Shi, F. The history, etiology, and management of ginseng replant disease: A Canadian perspective in review. Can. J. Plant Sci. 2021, 101, 886–901. [Google Scholar] [CrossRef]
- Rafiqi, M.; Ellis, J.G.; Ludowici, V.A.; Hardham, A.R.; Dodds, P.N. Challenges and progress towards understanding the role of effectors in plant–fungal interactions. Curr. Opin. Plant Biol. 2012, 15, 477–482. [Google Scholar] [CrossRef]
- Plissonneau, C.; Benevenuto, J.; Mohd-Assaad, N.; Fouché, S.; Hartmann, F.E.; Croll, D. Using population and comparative genomics to understand the genetic basis of effector-driven fungal pathogen evolution. Front. Plant Sci. 2017, 8, 119. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, B.; Vandooren, J.; Gerg, M.; Ahomaa, K.; Hunger, A.; Berchtold, S.; Akbareian, S.; Schaten, S.; Knolle, P.; Edwards, D.R.; et al. Systemic ablation of MMP-9 triggers invasive growth and metastasis of pancreatic cancer via deregulation of IL6 expression in the bone marrow. Mol. Cancer Res. 2016, 14, 1147–1158. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Sun, H.; Vainio, E.J.; Raffaello, T.; Kovalchuk, A.; Morin, E.; Duplessis, S.; Asiegbu, F.O. Intraspecific comparative genomics of isolates of the Norway spruce pathogen (Heterobasidion parviporum) and identification of its potential virulence factors. BMC Genom. 2018, 19, 220. [Google Scholar] [CrossRef] [Green Version]
- Menardo, F.; Praz, C.R.; Wicker, T.; Keller, B. Rapid turnover of effectors in grass powdery mildew (Blumeria graminis). BMC Evol. Biol. 2017, 17, 223. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Chen, M.; Ma, Y.; Du, Z.; Tuan, N.; Li, Y.; Xiao, J.; Zhang, Y. Whole-genome and time-course dual RNA-Seq analyses reveal chronic pathogenicity-related gene dynamics in the ginseng rusty root rot pathogen Ilyonectria robusta. Sci. Rep. 2020, 10, 1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Wang, S.; Mi, C.Y.; Yang, R.H.; Zen, G.H.; Hu, X.F. Genome sequence resource for Ilyonectria mors-panacis, causing rusty root rot of Panax notoginseng. Mol. Plant-Microbe Interact. 2019, 32, 1468–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef]
- Taylor, A.; Vagany, A.; Jackson, A.C.; Harrison, R.J.; Rainoni, A.; Clarkson, J.P. Identification of pathogenicity-related genes in Fusarium oxysporum f. sp. cepae. Mol. Plant Pathol. 2016, 17, 1032–1047. [Google Scholar] [CrossRef] [Green Version]
- Lievens, B.; Houterman, P.M.; Rep, M. Effector gene screening allows unambiguous identification of Fusarium oxysporum f. sp. lycopersici races and discrimination from other formae specialis. FEMS Microbiol. Lett. 2009, 300, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Hissen, A.H.; Wan, A.N.; Warwas, M.L.; Pinto, L.J.; Moore, M.M. The Aspergillus fumigatus siderophore biosynthetic gene sidA, encoding L-ornithine N5-oxygenase, is required for virulence. Infect. Immun. 2005, 73, 5493–5503. [Google Scholar] [CrossRef] [Green Version]
- Wight, W.D.; Labuda, R.; Walton, J.D. Conservation of the genes for HC-toxin biosynthesis in Alternaria jesenskae. BMC Microbiol. 2013, 13, 165. [Google Scholar] [CrossRef] [Green Version]
- Quidde, T.; Buttner, P.; Tudzynski, P. Evidence for three different specific saponin-detoxifying activities in Botrytis cinerea and cloning and functional analysis of a gene coding for a putative avenacinase. Eur. J. Plant Pathol. 1999, 105, 273–283. [Google Scholar] [CrossRef]
- Yun, S.H.; Arie, T.; Kaneko, I.; Yoder, O.C.; Turgeon, G. Molecular organization of mating type loci in heterothallic, homothallic, and asexual Gibberella/Fusarium species. Fungal Genet. Biol. 2000, 31, 7–20. [Google Scholar] [CrossRef]
- Farh, M.E.; Han, J.A.; Kim, Y.J.; Kim, J.C.; Singh, P.; Yang, D.C. Discovery of a new primer set for detection and quantification of Ilyonectria mors-panacis in soils for ginseng cultivation. J. Ginseng Res. 2019, 43, 1–9. [Google Scholar] [CrossRef]
- Seifert, K.A.; McMullen, C.R.; Yee, D.; Reeleder, R.D.; Dobinson, K.F. Molecular differentiation and detection of ginseng adapted isolates of the root rot fungus C. destructans. Popul. Biol. 2003, 93, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Raffaele, S.; Kamoun, S. Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat. Rev. Microbiol. 2012, 10, 417–430. [Google Scholar] [CrossRef]
- Spanu, P.D.; Abbott, J.C.; Amselem, J.; Burgis, T.A.; Soanes, D.M.; Stüber, K.; Van Themaat, E.V.L.; Brown, J.K.M.; Butcher, S.A.; Gurr, S.I.; et al. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 2010, 330, 1543–1546. [Google Scholar] [CrossRef]
- Noble, L.M.; Andrianopoulos, A. Fungal genes in context: Genome architecture reflects regulatory complexity and function. Genome Biol. Evol. 2013, 5, 1336–1352. [Google Scholar] [CrossRef] [Green Version]
- Vogelgsang, S.; Widmer, F.; Jenny, E.; Enkerl, J. Characterisation of novel Fusarium graminearum microsatellite markers in different Fusarium species from various countries. Eur. J. Plant Pathol. 2008, 123, 477–482. [Google Scholar] [CrossRef]
- Van der burgt, A.; Karimi Jashni, M.; Bahkali, A.H.; De Wit, P.J.G.M. Pseudogenization in pathogenic fungi with different host plants and lifestyles might reflect their evolutionary past. Mol. Plant Pathol. 2014, 15, 133–144. [Google Scholar] [CrossRef]
- Lafontaine, I.; Dujon, B. Origin and fate of pseudogenes in Hemiascomycetes: A comparative analysis. BMC Genom. 2010, 11, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Notley-McRobb, L.; Lim, M.; Carter, D.A. A comparison of the nature and abundance of microsatellites in 14 fungal genomes. Fungal Genet. Biol. 2004, 41, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Testa, A.C.; Oliver, R.P.; James, K.; Hane, J.K. OcculterCut: A comprehensive survey of AT-rich regions in fungal genomes. Genome Biol. Evol. 2016, 8, 2044–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, H.; Eisendle, M.; Turgeon, B.G. Siderophores in fungal physiology and virulence. Annu. Rev. Phytopathol. 2008, 46, 149–187. [Google Scholar] [CrossRef]
- Walsh, J.; DesRochers, N.; Renaud, J.B.; Seifert, K.A.; Yeung, K.K.C.; Sumarah, M.W. Identification of N,N′,N″-triacetylfusarinine C as a key metabolite for root rot disease virulence in American ginseng. J. Ginseng Res. 2019, 45, 156–162. [Google Scholar] [CrossRef]
- Brosch, G.; Ransom, R.; Lechner, T.; Walton, J.D.; Loidl, P. Inhibition of maize histone deacetylases by HC toxin, the host-selective toxin of Cochliobolus carbonum. Plant Cell 1995, 7, 1941–1950. [Google Scholar]
- Bernards, M.A.; Ivanov, D.A.; Neculai, M.A.; Nicol, R.W. Ginsenosides: Phytoanticipins or host recognition factors? In The Biological Activity of Phytochemicals; Gang, D.R., Ed.; Springer: New York, NY, USA, 2011; pp. 13–46. [Google Scholar]
- Searels, J.M.; Keen, K.D.; Horton, J.L.; Clarke, H.D.; Ward, J.R. Comparing ginsenoside production in leaves and roots of wild American ginseng (Panax quinquefolius). Am. J. Plant Sci. 2013, 4, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Farh, M.E.; Kim, Y.J.; Abbai, R.; Singh, P.; Jung, K.H.; Kim, Y.J.; Yang, D.C. Pathogenesis strategies and regulation of ginsenosides by two species of Ilyonectria in Panax ginseng: Power of speciation. J. Ginseng Res. 2020, 44, 332–340. [Google Scholar] [CrossRef]
- Klix, V.; Nowrousian, M.; Ringelberg, C.; Loros, J.J.; Dunlap, J.C.; Pöggeler, S. Functional characterization of MAT1-1-specific mating-type genes in the homothallic ascomycete Sordaria macrospora provides new insights into essential and nonessential sexual regulators. Eukaryot. Cell 2010, 9, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.H.; Wingfield, B.D.; Wingfield, M.J.; Steenkamp, E.T. Causes and consequences of variability in peptide mating pheromones of Ascomycete fungi. Mol. Biol. Evol. 2011, 28, 1987–2003. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, G.; Yoder, O.C. Proposed nomenclature for mating type genes of filamentous Ascomycetes. Fungal Genet. Biol. 2000, 31, 1–5. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef]
- Jashni, M.K.; Dols, I.H.; Iida, Y.; Boeren, S.; Beenen, H.G.; Mehrabi, R.; Collemare, J.; de Wit, P.J.G.M. Synergistic action of a metalloprotease and a serine protease from Fusarium oxysporum f. sp. lycopersici cleaves chitin-binding tomato chitinases, reduces their antifungal activity, and enhances fungal virulence. Mol. Plant-Microbe Interact. 2015, 28, 996–1008. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Code | Field Type B | Symptoms | Location | Year Isolated |
|---|---|---|---|---|
| IR.NR1BC16-1 C | N | Rusty root | Summerland, BC | 2016 |
| IR.ND1BC16-2 C | N | Black-brown lesion | Summerland, BC | 2016 |
| IR.NR2BC16-4 C | N | Rusty root | Summerland, BC | 2016 |
| IR.DAOM139398 E | Cherry orchard | ND | Georgian Bay, ON | - |
| IMP.ND3P14-1A | N | Black-brown lesion | St. Williams, ON | 2016 |
| IMP.ND3P14-3 D | N | Black-brown lesion | Lynedoch, ON | 2014 |
| IMP.ND3A16-1 D | N | Black-brown lesion | Delhi, ON | 2016 |
| IMP.ND4Z15 D | N | Black-brown lesion | Simcoe, ON | 2015 |
| IMP.ND3A16-2 D | N | Black-brown lesion | Delhi, ON | 2016 |
| IMP.ND3P14-1 D | N | Black-brown lesion | Lynedoch, ON | 2014 |
| IMP.RD3U14-8 D | R | Black-brown lesion | Scotland, ON | 2014 |
| IMP.RR3A14-1 D | R | Black-brown lesion and rusty root | Delhi, ON | 2014 |
| IMP.RD3U14-5 D | R | Black-brown lesion | Scotland, ON | 2014 |
| IMP.K112 F | ND | ND | Kamloops, BC | 2006 |
| IMP.DAOM226727 G | ND | ND | Delhi, ON | 1996 |
| IMP.DAOM226729 | ND | ND | Delhi, ON | 1935 |
| I. europaea PMI-82 A | Poplar forest | ND | ND | - |
| Isolate | Growth Rate (cm/day) A | Lesion Size (cm2) A |
|---|---|---|
| IR.NR1BC16-1 | 0.63 a | 0.18 d |
| IR.ND1BC16-2 | 0.61 a | 0.14 ef |
| IR.NR2BC16-4 | 0.58 ab | 0.13 ef |
| IR.DAOM139398 | 0.52 c | 0.18 d |
| IMP.ND3P14-1A | 0.41 e | 0.23 b |
| IMP.ND3P14-3 | 0.43 de | 0.09 g |
| IMP.ND3A16-1 | 0.25 h | 0.30 a |
| IMP.ND4Z15 | 0.30 fg | 0.20 cd |
| IMP.ND3A16-2 | 0.25 h | 0.22 b |
| IMP.ND3P14-1 | 0.48 cd | 0.18 d |
| IMP.RD3U14-8 | 0.27 gh | 0.09 g |
| IMP.RR3A14-1 | 0.27 gh | 0.17 d |
| IMP.RD3U14-5 | 0.26 gh | 0.21 bc |
| IMP.K112 | 0.53 bc | 0.14 e |
| IMP.DAOM226727 | 0.34 f | 0.14 ef |
| IMP.DAOM226729 | 0.43 de | 0.12 f |
| BUSCO (3817 Groups in Sardariomycetes_odb10) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Isolate A | WGS Accession | Scaffolds | N50 | L50 | Complete | Single-Copy | Duplicated | Fragmented | Missing |
| IR.NR1BC16-1 | JAQYTY000000000 | 2652 | 1,630,377 | 67 | 3764 | 3730 | 34 | 7 | 46 |
| IR.ND1BC16-2 | JAQYTZ000000000 | 10,371 | 899,178 | 67 | 3766 | 3713 | 53 | 6 | 45 |
| IR.NR2BC16-4 | JAQYTX000000000 | 1549 | 1,566,478 | 67 | 3763 | 3728 | 35 | 6 | 48 |
| IR.DAOM139398 | JAQYUA000000000 | 1690 | 1,976,210 | 67 | 3767 | 3737 | 30 | 6 | 44 |
| IMP.ND3P14-1A | JAQYUG000000000 | 8384 | 270,784 | 67 | 3760 | 3731 | 37 | 11 | 38 |
| IMP.ND3P14-3 | JAQYUF000000000 | 4472 | 303,308 | 64 | 3768 | 3729 | 39 | 12 | 37 |
| IMP.ND3A16-1 | JAQYUJ000000000 | 7456 | 371,676 | 54 | 3769 | 3732 | 37 | 12 | 36 |
| IMP.ND4Z15 | JAQYUE000000000 | 4256 | 179,452 | 67 | 3766 | 3730 | 36 | 13 | 38 |
| IMP.ND3A16-2 | JAQYUI000000000 | 8029 | 285,004 | 66 | 3768 | 3729 | 39 | 12 | 37 |
| IMP.ND3P14-1 | JAQYUH000000000 | 8662 | 291,290 | 62 | 3768 | 3730 | 38 | 11 | 38 |
| IMP.RD3U14-8 | JAQYUC000000000 | 7651 | 334,621 | 67 | 3767 | 3730 | 37 | 13 | 37 |
| IMP.RR3A14-1 | JAQYUB000000000 | 3907 | 178,193 | 67 | 3766 | 3727 | 39 | 14 | 37 |
| IMP.RD3U14-5 | JAQYUD000000000 | 3372 | 178,475 | 67 | 3767 | 3728 | 39 | 13 | 37 |
| IMP.K112 | JAQYUK000000000 | 4268 | 278,127 | 61 | 3768 | 3728 | 40 | 13 | 36 |
| IMP. DAOM226727 | JAQYUM000000000 | 8002 | 284,075 | 63 | 3769 | 3731 | 38 | 12 | 36 |
| IMP. DAOM226729 | JAQYUL000000000 | 7918 | 353,126 | 58 | 3769 | 3730 | 39 | 13 | 35 |
| I. europaea PMI-82 | N/A | 325 | 327,392 | 9 | 3763 | 3703 | 60 | 6 | 48 |
| Isolate A | Genome Size (Mbp) B | Predicted Genes B | GC Content B |
|---|---|---|---|
| I. robusta | 59.2 b | 17,603.5 b | 50.1% a |
| I. mors-panacis type 1 | 65.02 a | 18,183.4 ab | 48.92% b |
| I. mors-panacis type 2 | 65.04 a | 18,370.2 a | 48.98% b |
| I. europaea PMI-82 | 63.7 | 19,307 | 51.13% |
| Isolate | Total Non-Secreted Proteins | Total Secreted Proteins | Secreted SSNPs | Secreted SSCPs | Secreted CAZymes | Secreted Proteases | Secreted Lipases | Other Secreted Annotated Proteins | Secreted Hypothetical/Unknown Proteins |
|---|---|---|---|---|---|---|---|---|---|
| I. robusta | 8869 a | 8736 a | 1269 b | 677 b | 673 a | 585 a | 414 a | 3013 a | 1917 a |
| I. mors-panacis type 1 | 8945 a | 9240 a | 1370 a | 698 a | 669 a | 595 a | 409 a | 2622 a | 1876 a |
| I. mors-panacis type 2 | 9180 a | 9191 a | 1372 a | 705 a | 666 a | 595 a | 410 a | 3075 a | 2196 a |
| I. europaea PMI-82 | 9474 | 9834 | 2386 | 1159 | 782 | 654 | 504 | 3077 | 1613 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behdarvandi, B.; Hsiang, T.; Valliani, M.; Goodwin, P.H. Differences in Saprophytic Growth, Virulence, Genomes, and Secretomes of Ilyonectria robusta and I. mors-panacis Isolates from Roots of American Ginseng (Panax quinquefolius). Horticulturae 2023, 9, 713. https://doi.org/10.3390/horticulturae9060713
Behdarvandi B, Hsiang T, Valliani M, Goodwin PH. Differences in Saprophytic Growth, Virulence, Genomes, and Secretomes of Ilyonectria robusta and I. mors-panacis Isolates from Roots of American Ginseng (Panax quinquefolius). Horticulturae. 2023; 9(6):713. https://doi.org/10.3390/horticulturae9060713
Chicago/Turabian StyleBehdarvandi, Behrang, Tom Hsiang, Moez Valliani, and Paul H. Goodwin. 2023. "Differences in Saprophytic Growth, Virulence, Genomes, and Secretomes of Ilyonectria robusta and I. mors-panacis Isolates from Roots of American Ginseng (Panax quinquefolius)" Horticulturae 9, no. 6: 713. https://doi.org/10.3390/horticulturae9060713