
Rumen Metaproteomics Highlight the Unique Contributions of Microbe-Derived Extracellular and Intracellular Proteins for In Vitro Ruminal Fermentation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Mass Spectrometry
2.3. Sample-Specific Metagenomic Database Constructions
2.4. Metaproteomic Data Analysis
2.5. Identified Protein Function Annotation
3. Results and Discussion
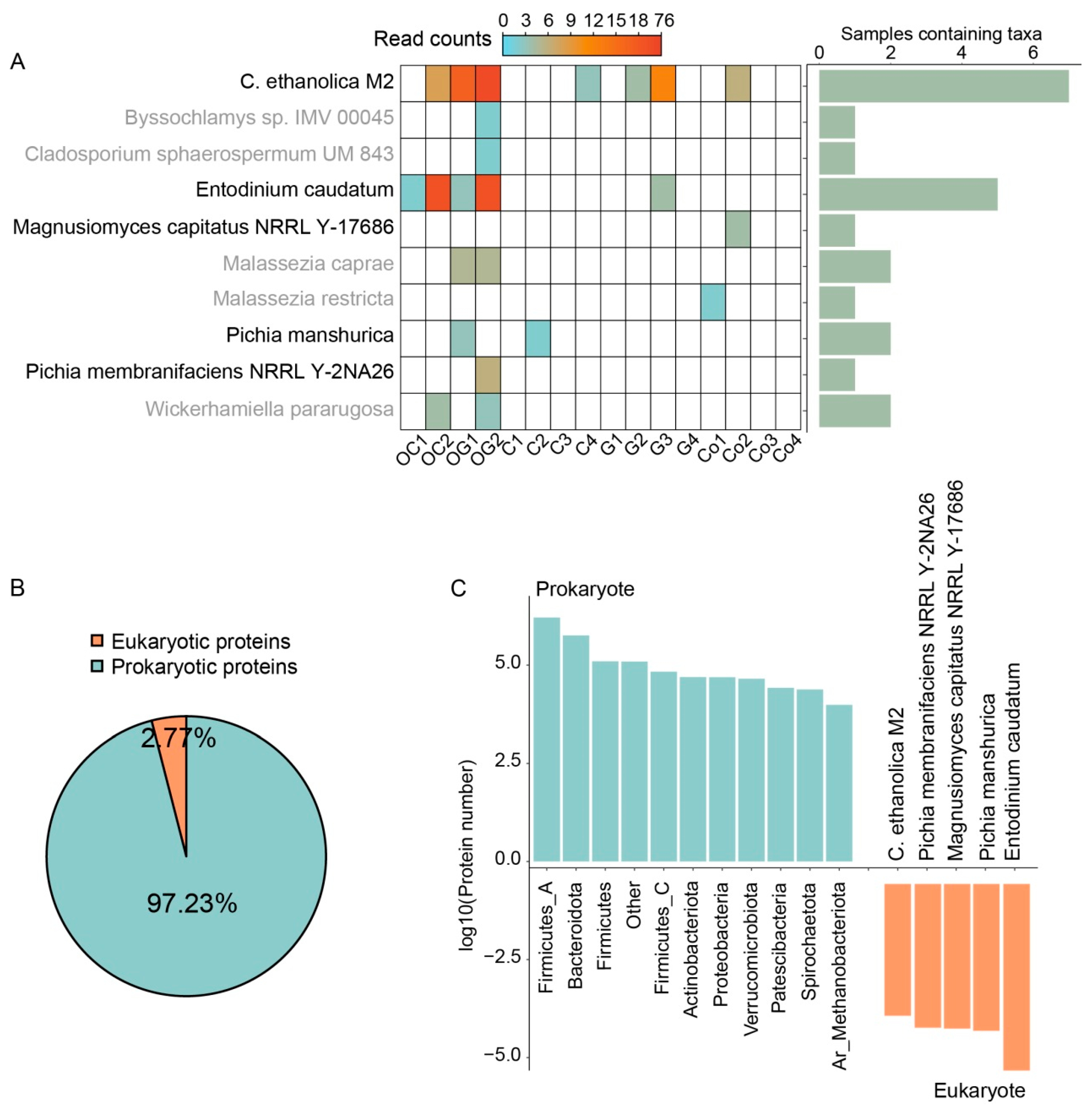
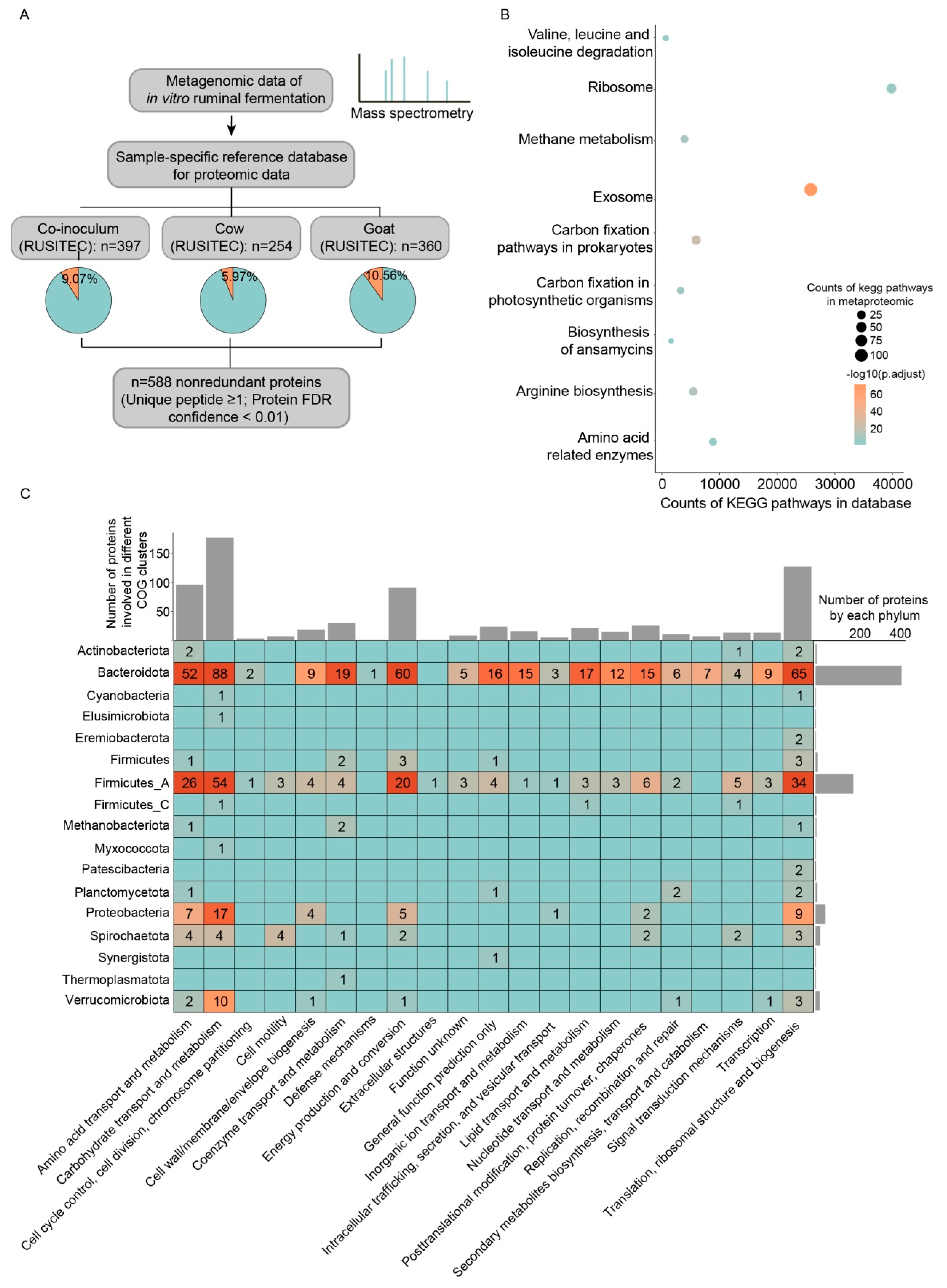
3.1. Sample-Specific Reference Database Covering Rumen Bacteria, Fungi, and Protozoa
3.2. Metaproteomic Characterization of Microorganisms in In Vitro Ruminal Fermentation Broth
3.3. Carbohydrate Active Enzymes Contained in Microbe-Derived Extracellular Proteins
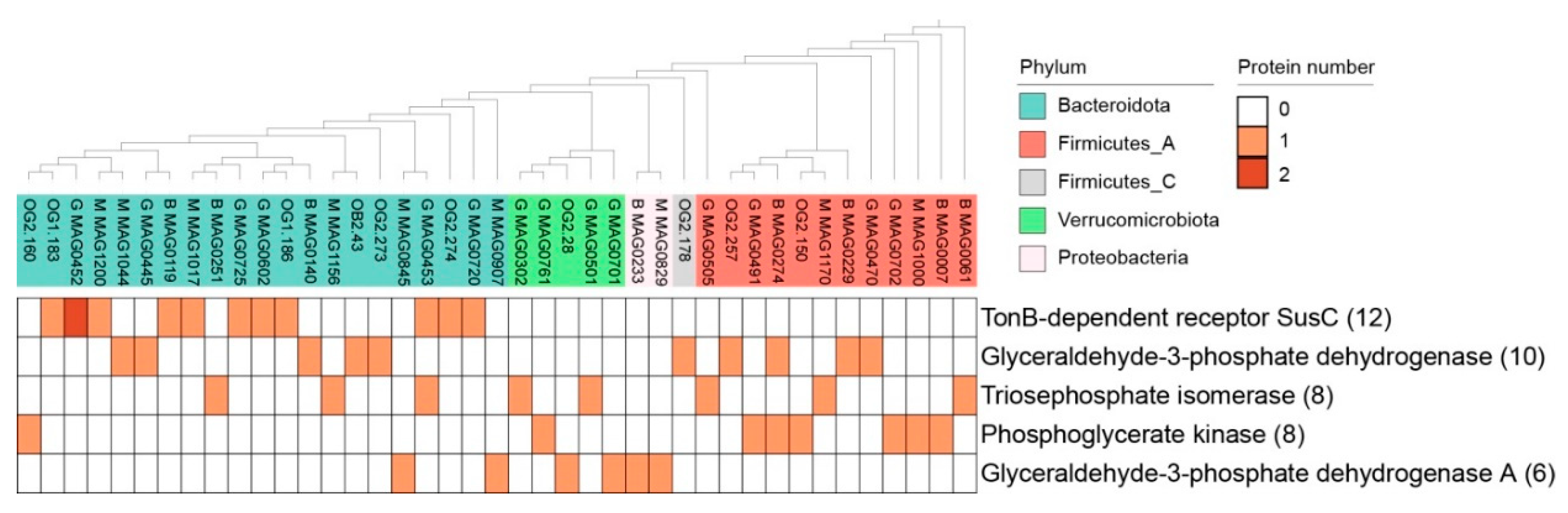
3.4. The Intracellular/Surface Moonlighting Proteins in Extracellular Proteomic of In Vitro Ruminal Fermentation Broth
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calsamiglia, S.; Busquet, M.; Cardozo, P.W.; Castillejos, L.; Ferret, A. Invited review: Essential oils as modifiers of rumen microbial fermentation. J. Dairy Sci. 2007, 90, 2580–2595. [Google Scholar] [CrossRef] [PubMed]
- Svartstrom, O.; Alneberg, J.; Terrapon, N.; Lombard, V.; de Bruijn, I.; Malmsten, J.; Dalin, A.M.; El Muller, E.; Shah, P.; Wilmes, P.; et al. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J. 2017, 11, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Walker, A.W.; Roehe, R.; Watson, M. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019, 37, 953–961. [Google Scholar] [CrossRef]
- Artzi, L.; Bayer, E.A.; Morais, S. Cellulosomes: Bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 2017, 15, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Hagen, L.H.; Brooke, C.G.; Shaw, C.A.; Norbeck, A.D.; Piao, H.; Arntzen, M.O.; Olson, H.M.; Copeland, A.; Isern, N.; Shukla, A.; et al. Proteome specialization of anaerobic fungi during ruminal degradation of recalcitrant plant fiber. ISME J. 2021, 15, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.L.; Edwards, J.E.; Wilkinson, T.J.; Allison, G.G.; McCaffrey, K.; Scott, M.B.; Rees-Stevens, P.; Kingston-Smith, A.H.; Huws, S.A. Using ‘Omic Approaches to Compare Temporal Bacterial Colonization of Lolium perenne, Lotus corniculatus, and Trifolium pratense in the Rumen. Front. Microbiol. 2018, 9, 2184. [Google Scholar] [CrossRef]
- Wei, X.; Ouyang, K.; Long, T.; Liu, Z.; Li, Y.; Qiu, Q. Dynamic Variations in Rumen Fermentation Characteristics and Bacterial Community Composition during In Vitro Fermentation. Fermentation 2022, 8, 276. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Wang, W.-K.; Wu, Q.-C.; Zhang, F.; Li, W.-J.; Yang, Z.-M.; Bo, Y.-K.; Yang, H.-J. The Effect of Different Lactic Acid Bacteria Inoculants on Silage Quality, Phenolic Acid Profiles, Bacterial Community and In Vitro Rumen Fermentation Characteristic of Whole Corn Silage. Fermentation 2022, 8, 285. [Google Scholar] [CrossRef]
- Brede, M.; Orton, T.; Pinior, B.; Roch, F.F.; Dzieciol, M.; Zwirzitz, B.; Wagner, M.; Breves, G.; Wetzels, S.U. PacBio and Illumina MiSeq Amplicon Sequencing Confirm Full Recovery of the Bacterial Community After Subacute Ruminal Acidosis Challenge in the RUSITEC System. Front. Microbiol. 2020, 11, 1813. [Google Scholar] [CrossRef]
- Baba, Y.; Matsuki, Y.; Takizawa, S.; Suyama, Y.; Tada, C.; Fukuda, Y.; Saito, M.; Nakai, Y. Pretreatment of Lignocellulosic Biomass with Cattle Rumen Fluid for Methane Production: Fate of Added Rumen Microbes and Indigenous Microbes of Methane Seed Sludge. Microbes Environ. 2019, 34, 421–428. [Google Scholar] [CrossRef]
- Oss, D.B.; Ribeiro, G.O., Jr.; Marcondes, M.I.; Yang, W.; Beauchemin, K.A.; Forster, R.J.; McAllister, T.A. Synergism of Cattle and Bison Inoculum on Ruminal Fermentation and Select Bacterial Communities in an Artificial Rumen (Rusitec) Fed a Barley Straw Based Diet. Front. Microbiol. 2016, 7, 2032. [Google Scholar] [CrossRef] [PubMed]
- Petruschke, H.; Schori, C.; Canzler, S.; Riesbeck, S.; Poehlein, A.; Daniel, R.; Frei, D.; Segessemann, T.; Zimmerman, J.; Marinos, G.; et al. Discovery of novel community-relevant small proteins in a simplified human intestinal microbiome. Microbiome 2021, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Deusch, S.; Seifert, J. Catching the tip of the iceberg—Evaluation of sample preparation protocols for metaproteomic studies of the rumen microbiota. Proteomics 2015, 15, 3590–3595. [Google Scholar] [CrossRef] [PubMed]
- Adebayo Arowolo, M.; Zhang, X.M.; Wang, M.; Wang, R.; Wen, J.N.; Hao, L.Z.; He, J.H.; Shen, W.J.; Ma, Z.Y.; Tan, Z.L. Proper motility enhances rumen fermentation and microbial protein synthesis with decreased saturation of dissolved gases in rumen simulation technique. J. Dairy Sci. 2022, 105, 231–241. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Mayne, J.; Ning, Z.; Stintzi, A.; Figeys, D. Assessing the impact of protein extraction methods for human gut metaproteomics. J. Proteom. 2018, 180, 120–127. [Google Scholar] [CrossRef]
- Adav, S.S.; Ng, C.S.; Arulmani, M.; Sze, S.K. Quantitative iTRAQ secretome analysis of cellulolytic Thermobifida fusca. J. Proteome Res. 2010, 9, 3016–3024. [Google Scholar] [CrossRef]
- Hagen, L.H.; Frank, J.A.; Zamanzadeh, M.; Eijsink, V.G.H.; Pope, P.B.; Horn, S.J.; Arntzen, M.O. Quantitative Metaproteomics Highlight the Metabolic Contributions of Uncultured Phylotypes in a Thermophilic Anaerobic Digester. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Shi, T.; Zhang, T.; Wang, X.; Wang, X.; Shen, W.; Guo, X.; Liu, Y.; Li, Z.; Jiang, Y. Copresent microbiome and short-chain fatty acids profiles of plant biomass utilization in rumen simulation technique system. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lind, A.L.; Pollard, K.S. Accurate and sensitive detection of microbial eukaryotes from whole metagenome shotgun sequencing. Microbiome 2021, 9, 58. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.; Carvalho, B.F.; Mantovani, H.C.; Schwan, R.F.; Avila, C.L.S. Identification and characterization of yeasts from bovine rumen for potential use as probiotics. J. Appl. Microbiol. 2019, 127, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Zhang, Y.; Yu, Z.; Zhang, T.; Dai, X.; Pan, X.; Jing, R.; Yan, Y.; Liu, Y.; et al. Genomic insights into the phylogeny and biomass-degrading enzymes of rumen ciliates. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hartinger, T.; Zebeli, Q. The Present Role and New Potentials of Anaerobic Fungi in Ruminant Nutrition. J. Fungi 2021, 7, 200. [Google Scholar] [CrossRef]
- Newbold, C.J.; de la Fuente, G.; Belanche, A.; Ramos-Morales, E.; McEwan, N.R. The Role of Ciliate Protozoa in the Rumen. Front. Microbiol. 2015, 6, 1313. [Google Scholar] [CrossRef]
- Honan, M.C.; Greenwood, S.L. Characterization of variations within the rumen metaproteome of Holstein dairy cattle relative to morning feed offering. Sci. Rep. 2020, 10, 3179. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, J.T.; Karnati, S.K.; Dehority, B.A.; Morrison, M.; Smith, G.L.; St-Pierre, N.R.; Firkins, J.L. Rumen ciliated protozoa decrease generation time and adjust 18S ribosomal DNA copies to adapt to decreased transfer interval, starvation, and monensin. J. Dairy Sci. 2009, 92, 256–269. [Google Scholar] [CrossRef]
- Wallace, R.J.; McPherson, C.A. Factors affecting the rate of breakdown of bacterial protein in rumen fluid. Br. J. Nutr. 1987, 58, 313–323. [Google Scholar] [CrossRef]
- Veith, P.D.; Chen, Y.Y.; Chen, D.; O’Brien-Simpson, N.M.; Cecil, J.D.; Holden, J.A.; Lenzo, J.C.; Reynolds, E.C. Tannerella forsythia Outer Membrane Vesicles Are Enriched with Substrates of the Type IX Secretion System and TonB-Dependent Receptors. J. Proteome Res. 2015, 14, 5355–5366. [Google Scholar] [CrossRef]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, C.B.; Santana, M.F. Prediction of the secretomes of endophytic and nonendophytic fungi reveals similarities in host plant infection and colonization strategies. Mycologia 2020, 112, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Kainulainen, V.; Korhonen, T.K. Dancing to another tune-adhesive moonlighting proteins in bacteria. Biology 2014, 3, 178–204. [Google Scholar] [CrossRef]
- Jeffery, C.J. Moonlighting proteins. Trends Biochem. Sci. 1999, 24, 8–11. [Google Scholar] [CrossRef]
- Jeffery, C. Intracellular proteins moonlighting as bacterial adhesion factors. AIMS Microbiol. 2018, 4, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, V.; Fischetti, V.A. A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J. Exp. Med. 1992, 176, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Dahlhamer, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease Among Adults Aged >/=18 Years—United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1166–1169. [Google Scholar] [CrossRef]
- Cho, K.H.; Salyers, A.A. Biochemical analysis of interactions between outer membrane proteins that contribute to starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 2001, 183, 7224–7230. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Accession | Classification of Microbial Origin | Unique Peptides | CAZy Family | Substrates | Enzyme |
|---|---|---|---|---|---|
| B_MAG0029_g01249 | p_Firmicutes_A; f_Lachnospiraceae;g_; s_ | 2 | GH109 a | 3-hydroxybutyryl-CoA dehydrogenase. | |
| Entodinium_caudatum_g1 | Entodinium_caudatum | 2 | GH13_1 | glycogen/starch | |
| Entodinium_caudatum_g2 | Entodinium_caudatum | 1 | CBM48 a | ||
| Entodinium_caudatum_g3 | Entodinium_caudatum | 1 | GH13_8 | glycogen/starch | |
| Entodinium_caudatum_g4 | Entodinium_caudatum | 2 | GT35 a | starch | |
| G_MAG0453_g01794 | p_Bacteroidota; g_UBA3839;s_ UBA3839 sp900313845 | 1 | GH94 | cellobiose/cellulose | Cellobiose phosphorylase. |
| M_MAG0911_g00105 | p_Firmicutes_A; g_Ruminococcus;s_ | 2 | GH48 | cellulose/chitin | Cellulose 1,4-beta-cellobiosidase (reducing end). |
| M_MAG0967_g01628 | p_Firmicutes_A; g_NK4A144;s_ | 1 | GH94 | cellobiose/cellulose | Cellobiose phosphorylase. |
| OB2.89_g00740 | p_Firmicutes_A; g_UMGS1696;s_ | 1 | CBM48=GH13_8 | glycogen/starch | 1,4-alpha-glucan branching enzyme. |
| OG2.257_g02106 | p_Firmicutes_A; g_Butyrivibrio;s_ | 1 | GH109 | Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, T.; Guo, X.; Liu, Y.; Zhang, T.; Wang, X.; Li, Z.; Jiang, Y. Rumen Metaproteomics Highlight the Unique Contributions of Microbe-Derived Extracellular and Intracellular Proteins for In Vitro Ruminal Fermentation. Fermentation 2022, 8, 394. https://doi.org/10.3390/fermentation8080394
Shi T, Guo X, Liu Y, Zhang T, Wang X, Li Z, Jiang Y. Rumen Metaproteomics Highlight the Unique Contributions of Microbe-Derived Extracellular and Intracellular Proteins for In Vitro Ruminal Fermentation. Fermentation. 2022; 8(8):394. https://doi.org/10.3390/fermentation8080394
Chicago/Turabian StyleShi, Tao, Xi Guo, Yuqin Liu, Tingting Zhang, Xiangnan Wang, Zongjun Li, and Yu Jiang. 2022. "Rumen Metaproteomics Highlight the Unique Contributions of Microbe-Derived Extracellular and Intracellular Proteins for In Vitro Ruminal Fermentation" Fermentation 8, no. 8: 394. https://doi.org/10.3390/fermentation8080394