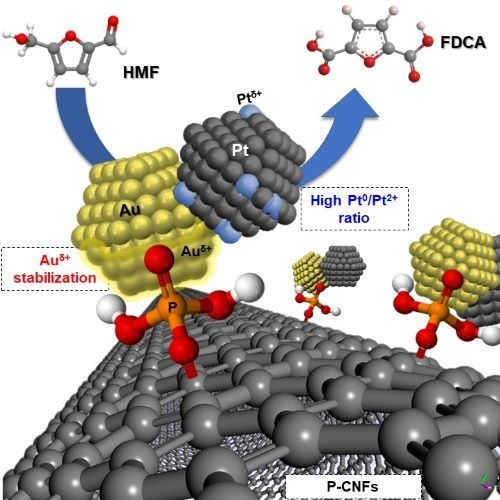
Catalytic Performances of Au–Pt Nanoparticles on Phosphorous Functionalized Carbon Nanofibers towards HMF Oxidation

 ,
,  ,
, 
 ,
, 

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalytic Tests
2.3. Characterization of Catalysts
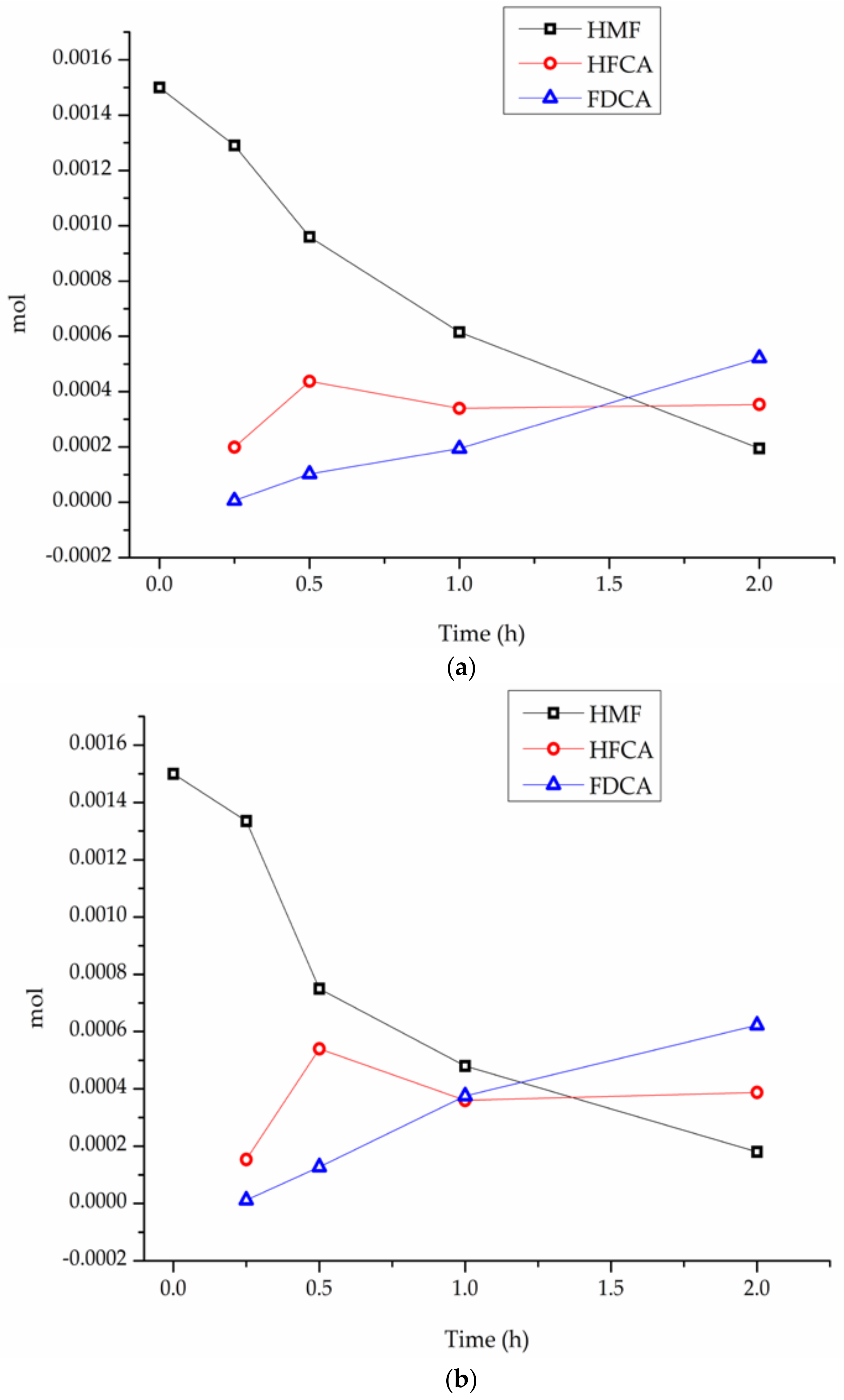
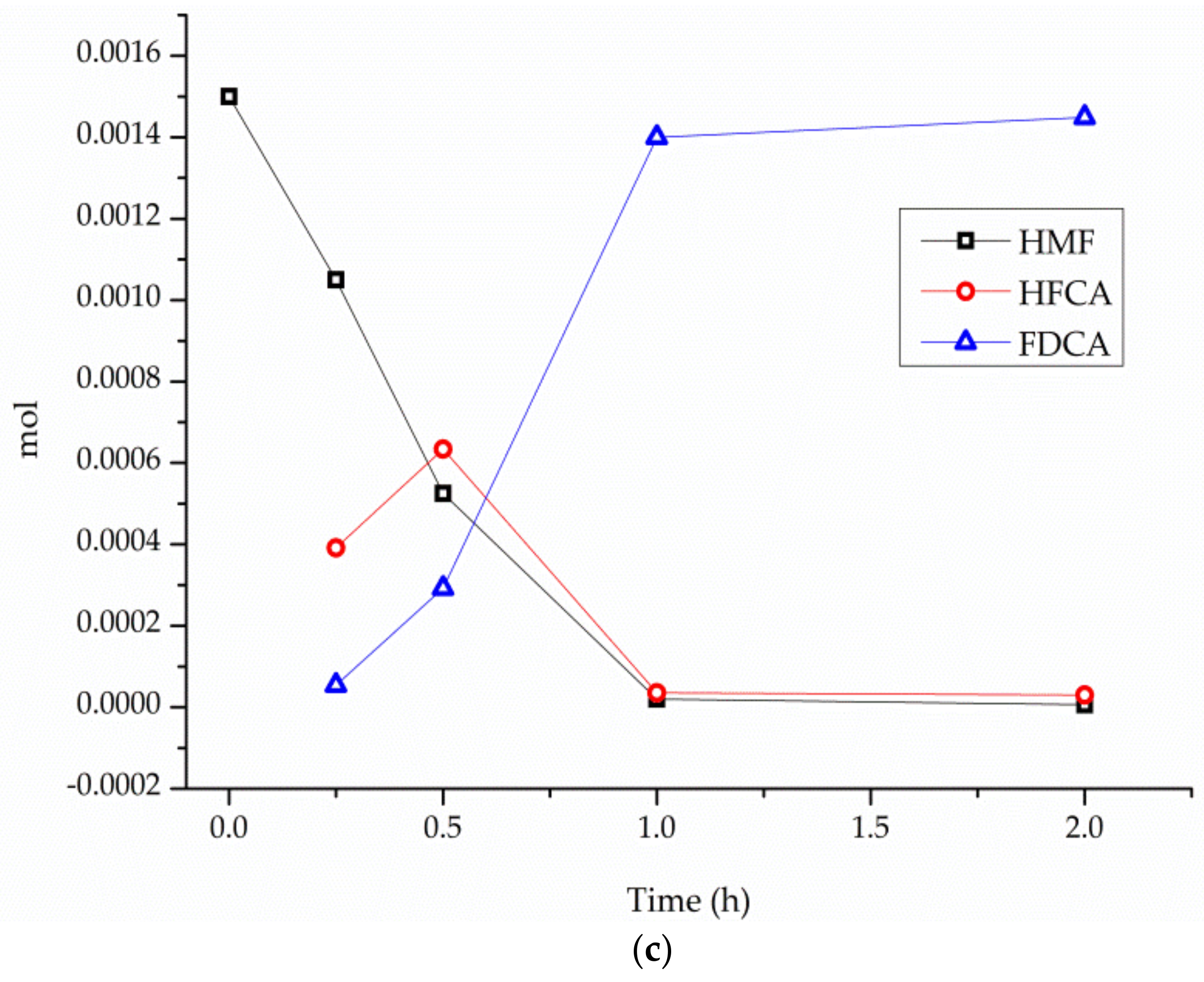
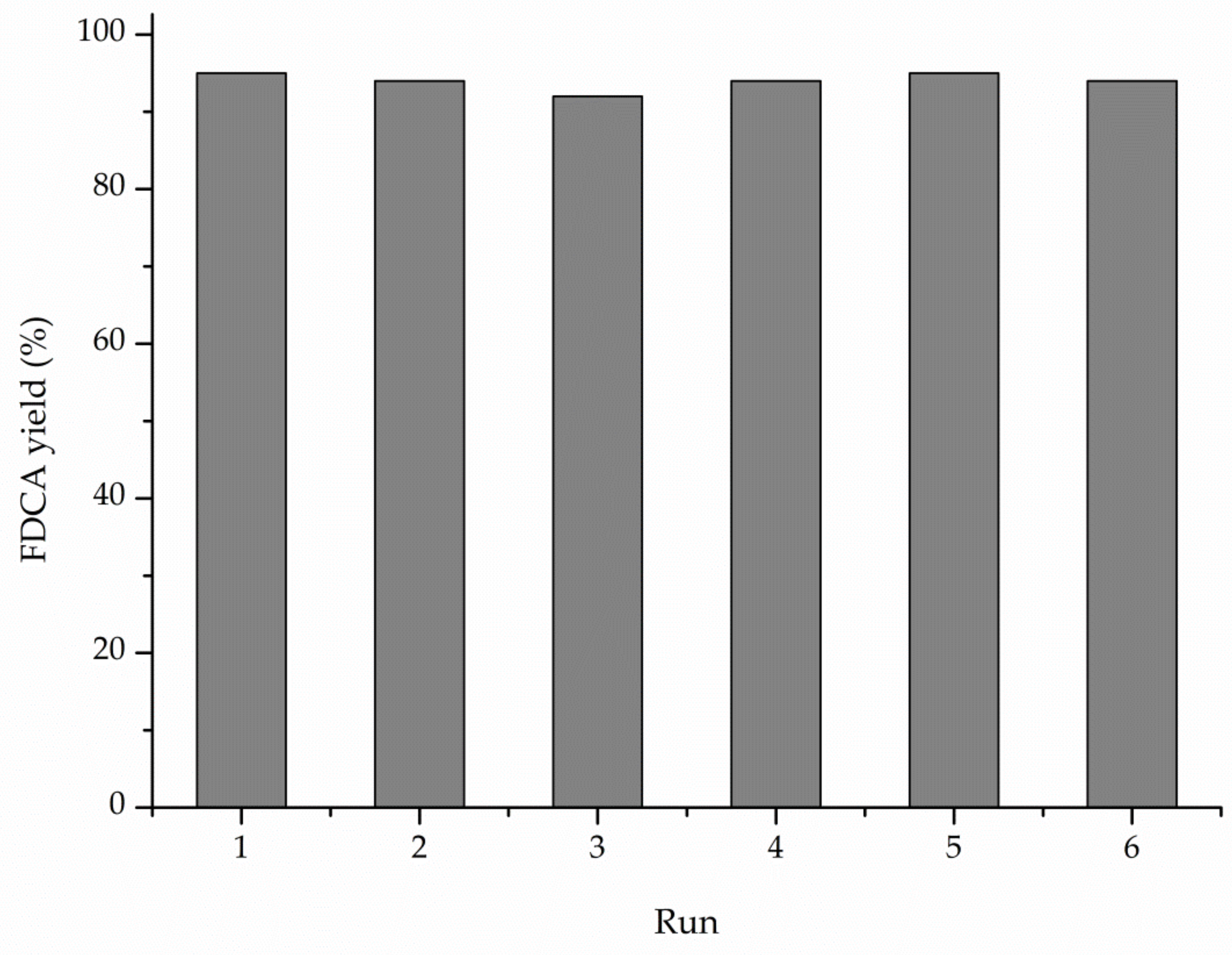
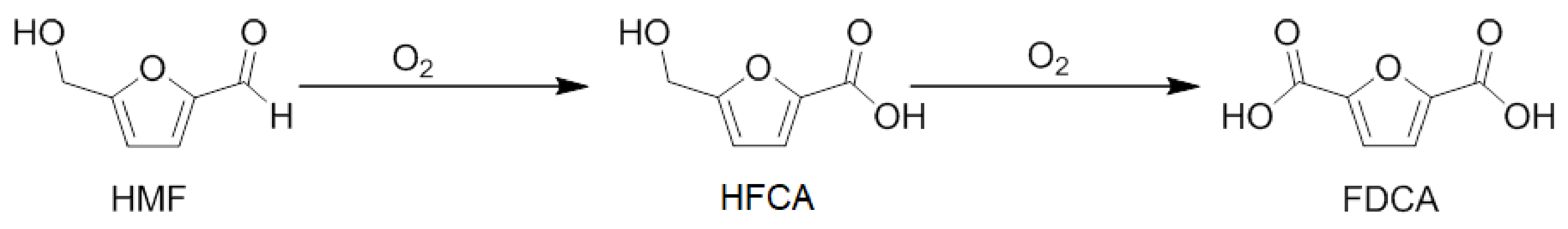
3. Results
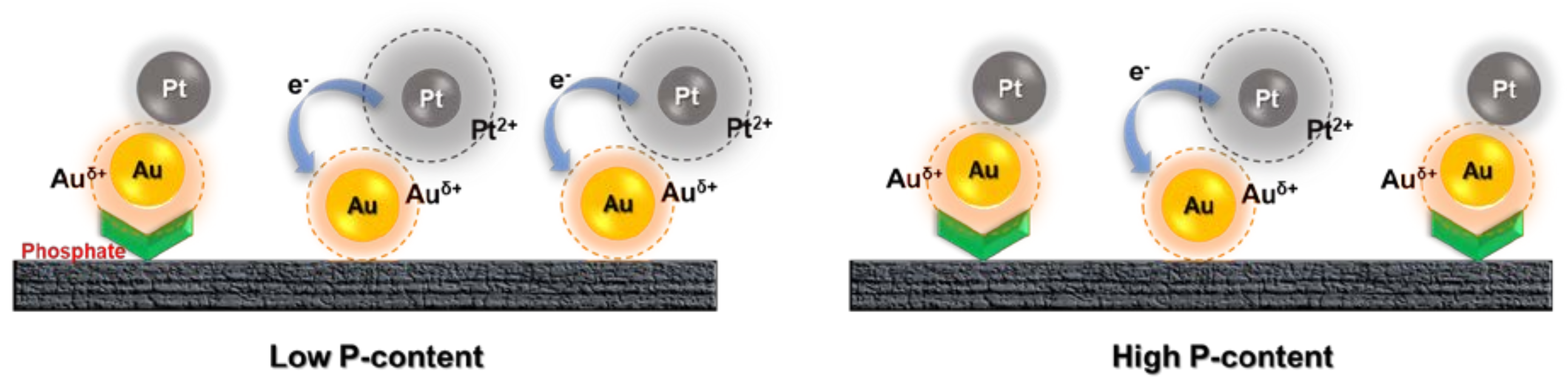
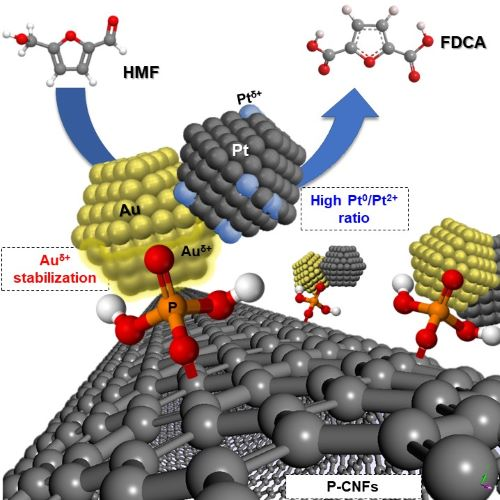
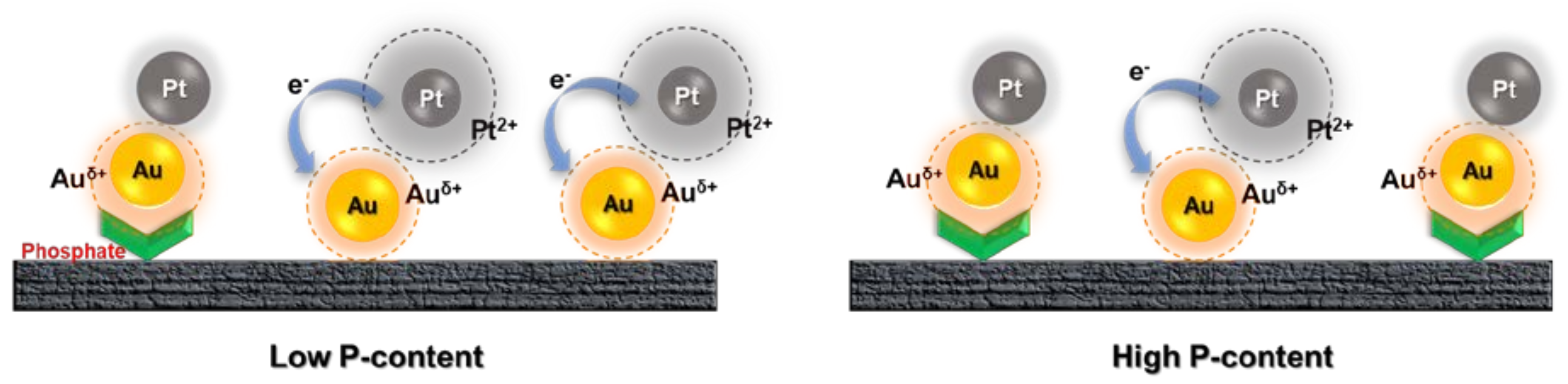
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rodríguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Serp, P.; Figueiredo, J.L. Carbon Materials for Catalysis; John Wiley & Sons: New Jersey, NJ, USA, 2009; ISBN 9780470178850. [Google Scholar]
- Bitter, J.H. Nanostructured carbons in catalysis a Janus material industrial applicability and fundamental insights. J. Mater. Chem. 2010, 20, 7312–7321. [Google Scholar] [CrossRef]
- Su, D.S.; Perathoner, S.; Centi, G. Nanocarbons for the development of advanced catalysts. Chem. Rev. 2013, 113, 5782–5816. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.; Luong, J.H.T. Carbon materials as catalyst supports and catalysts in the transformation of biomass to fuels and chemicals. ACS Catal. 2014, 4, 3393–3410. [Google Scholar] [CrossRef]
- Prati, L.; Villa, A.; Lupini, A.R.; Veith, G.M. Gold on carbon: One billion catalysts under a single label. Phys. Chem. Chem. Phys. 2012, 14, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodriguez, S.; Rillo, N.; Lazaro, M.J.; Pastor, E. Pd catalysts supported onto nanostructured carbon materials for CO2 valorization by electrochemical reduction. Appl. Catal. B Environ. 2015, 163, 83–95. [Google Scholar] [CrossRef]
- Tasis, D.; Tagmatarchis, N.; Bianco, A.; Prato, M. Chemistry of carbon nanotubes. Chem. Rev. 2006, 106, 1105–1136. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Dimitratos, N. (Eds.) Catalysis Series. In Metal-Free Functionalized Carbons in Catalysis; Royal Society of Chemistry: Cambridge, UK, 2018; ISBN 978-1-78262-863-7. [Google Scholar]
- Balasubramanian, K.; Burghard, M. Chemically functionalized carbon nanotubes. Small 2005, 1, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Maiti, U.N.; Lee, J.M.; Lim, J.; Han, T.H.; Kim, S.O. Nitrogen-doped carbon nanotubes and graphene composite structures for energy and catalytic applications. Chem. Commun. 2014, 50, 6818–6830. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.A.; Jordão, E.; Mendes, M.J.; Freitas, M.M.A.; Faria, J.L.; Figueiredo, J.L. Properties of carbon-supported platinum catalysts: Role of carbon surface sites. J. Catal. 2002, 209, 355–364. [Google Scholar] [CrossRef]
- Campisi, S.; Chan-Thaw, C.; Villa, A. Understanding heteroatom-mediated metal-support interactions in functionalized carbons: A. perspective review. Appl. Sci. 2018, 8, 1159. [Google Scholar] [CrossRef]
- Rocha, R.; Soares, O.; Figueiredo, J.; Pereira, M. Tuning CNT properties for metal-free environmental catalytic applications. C 2016, 2, 17. [Google Scholar] [CrossRef]
- Ovejero, G.; Sotelo, J.L.; Romero, M.D.; Rodríguez, A.; Ocaña, M.A.; Rodríguez, G.; García, J. Multiwalled carbon nanotubes for liquid-phase oxidation functionalization, characterization, and catalytic activity. Ind. Eng. Chem. Res. 2006, 45, 2206–2212. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. The role of surface chemistry in catalysis with carbons. Catal. Today 2010, 150, 2–7. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Li, J.; Tian, Q.; Jiang, S.; Zhang, Y.; Wu, Y. Electrocatalytic performances of phosphorus doped carbon supported Pd towards formic acid oxidation. Electrochim. Acta 2016, 213, 21–30. [Google Scholar] [CrossRef]
- Silva, J.C.M.; de Freitas, I.C.; Neto, A.O.; Spinacé, E.V.; Ribeiro, V.A. Palladium nanoparticles supported on phosphorus-doped carbon for ethanol electro-oxidation in alkaline media. Ionics 2018, 24, 1111–1119. [Google Scholar] [CrossRef]
- Higgins, D.C.; Meza, D.; Chen, Z. Nitrogen-doped carbon nanotubes as platinum catalyst supports for oxygen reduction reaction in proton exchange membrane fuel cells. J. Phys. Chem. C 2010, 114, 21982–21988. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, Q.; Zhang, R.; Wang, Q.; Kang, G.; Peng, F. Phosphorus-doped carbon nanotubes supported low Pt loading catalyst for the oxygen reduction reaction in acidic fuel cells. J. Power Sources 2014, 268, 171–175. [Google Scholar] [CrossRef]
- Shiva, K.S.; Ramakrishna, S.U.B.; Rama, D.B.; Himabindu, V. Phosphorus-doped carbon nanoparticles supported palladium electrocatalyst for the hydrogen evolution reaction (HER) in PEM water electrolysis. Ionics 2018, 1–9. [Google Scholar] [CrossRef]
- Yin, M.; Huang, Y.; Li, Q.; Jensen, J.O.; Cleemann, L.N.; Zhang, W.; Bjerrum, N.J.; Xing, W. Phosphate-Doped carbon black as Pt catalyst support: Co-catalytic functionality for dimethyl ether and methanol electro-oxidation. ChemElectroChem 2014, 1, 448–454. [Google Scholar] [CrossRef]
- Zhu, J.; He, G.; Liang, L.; Wan, Q.; Shen, P.K. Direct anchoring of platinum nanoparticles on nitrogen and phosphorus-dual-doped carbon nanotube arrays for oxygen reduction reaction. Electrochim. Acta 2015, 158, 374–382. [Google Scholar] [CrossRef]
- Wang, B.; Yu, L.; Zhang, J.; Pu, Y.; Zhang, H.; Li, W. Phosphorus-doped carbon supports enhance gold-based catalysts for acetylene hydrochlorination. RSC Adv. 2014, 4, 15877–15885. [Google Scholar] [CrossRef]
- Lu, C.; Wang, M.; Feng, Z.; Qi, Y.; Feng, F.; Ma, L.; Zhang, Q.; Li, X. A phosphorus-carbon framework over activated carbon supported palladium nanoparticles for the chemoselective hydrogenation of para-chloronitrobenzene. Catal. Sci. Technol. 2017, 7, 1581–1589. [Google Scholar] [CrossRef]
- Guo, W.; Niu, S.; Shi, W.; Zhang, B.; Yu, W.; Xie, Y.; Ji, X.; Wu, Y.; Su, D.; Shao, L. Pd–P nanoalloys supported on a porous carbon frame as an efficient catalyst for benzyl alcohol oxidation. Catal. Sci. Technol. 2018, 8, 2333–2339. [Google Scholar] [CrossRef]
- Chen, C.; Li, X.; Wang, L.; Liang, T.; Wang, L.; Zhang, Y.; Zhang, J. Highly porous nitrogen- and phosphorus-codoped graphene: An outstanding support for Pd catalysts to oxidize 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid. ACS Sustain. Chem. Eng. 2017, 5, 11300–11306. [Google Scholar] [CrossRef]
- Gandini, A.; Silvestre, A.J.D.; Neto, C.P.; Sousa, A.F.; Gomes, M. The furan counterpart of polyethylene terephthalate: An alternative material based on renewable resources. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 295–298. [Google Scholar] [CrossRef]
- Nakajima, H.; Dijkstra, P.; Loos, K. The recent developments in biobased polymers toward general and engineering applications: Polymers that are upgraded from biodegradable polymers, analogous to petroleum-derived polymers, and newly developed. Polymers 2017, 9, 523. [Google Scholar] [CrossRef]
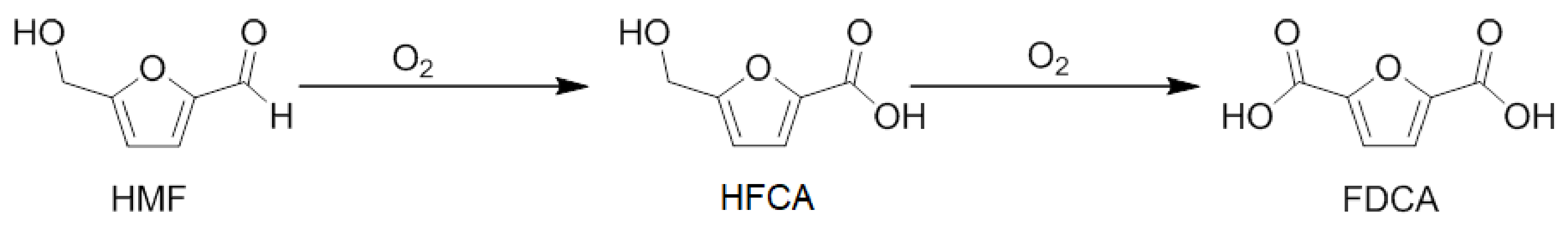
- Rosatella, A.A.; Simeonov, S.P.; Frade, R.F.M.; Afonso, C.A.M. 5-Hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754–793. [Google Scholar] [CrossRef]
- Corma Canos, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into chemicals: Aerobic oxidation of 5-hydroxymethyl-2-furfural into 2,5-furandicarboxylic acid with gold nanoparticle catalysts. ChemSusChem Chem. Sustain. Energy Mater. 2009, 2, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.E.; Houk, L.R.; Tamargo, E.C.; Datye, A.K.; Davis, R.J. Oxidation of 5-hydroxymethylfurfural over supported Pt, Pd and Au catalysts. Catal. Today 2011, 160, 55–60. [Google Scholar] [CrossRef]
- Zheng, L.; Zhao, J.; Du, Z.; Zong, B.; Liu, H. Efficient aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on Ru/C. catalysts. Sci. China Chem. 2017, 60, 950–957. [Google Scholar] [CrossRef]
- Pasini, T.; Piccinini, M.; Blosi, M.; Bonelli, R.; Albonetti, S.; Dimitratos, N.; Lopez-Sanchez, J.A.; Sankar, M.; He, Q.; Kiely, C.J.; et al. Selective oxidation of 5-hydroxymethyl-2-furfural using supported gold-copper nanoparticles. Green Chem. 2011, 13, 2091–2099. [Google Scholar] [CrossRef]
- Artz, J.; Palkovits, R. Base-Free aqueous-phase oxidation of 5-hydroxymethylfurfural over ruthenium catalysts supported on covalent triazine frameworks. ChemSusChem. 2015, 8, 3832–3838. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Teong, S.P.; Zhang, Y. Base-free conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Ru/C catalyst. Green Chem. 2016, 18, 979–983. [Google Scholar] [CrossRef]
- Davis, S.E.; Zope, B.N.; Davis, R.J. On the mechanism of selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over supported Pt and Au catalysts. Green Chem. 2012, 14, 143–147. [Google Scholar] [CrossRef]
- Zope, B.N.; Davis, S.E.; Davis, R.J. Influence of reaction conditions on diacid formation during Au-catalyzed oxidation of glycerol and hydroxymethylfurfural. Top. Catal. 2012, 55, 24–32. [Google Scholar] [CrossRef]
- Zhou, C.; Deng, W.; Wan, X.; Zhang, Q.; Yang, Y.; Wang, Y. Functionalized carbon nanotubes for biomass conversion: The base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over platinum supported on a carbon nanotube catalyst. ChemCatChem 2015, 7, 2853–2863. [Google Scholar] [CrossRef]
- Wan, X.; Zhou, C.; Chen, J.; Deng, W.; Zhang, Q.; Yang, Y.; Wang, Y. Base-free aerobic oxidation of 5-hydroxymethyl-furfural to 2,5-furandicarboxylic acid in water catalyzed by functionalized carbon nanotube-supported Au-Pd alloy nanoparticles. ACS Catal. 2014, 4, 2175–2185. [Google Scholar] [CrossRef]
- Villa, A.; Schiavoni, M.; Campisi, S.; Veith, G.M.; Prati, L. Pd-modified Au on carbon as an effective and durable catalyst for the direct oxidation of HMF to 2,5-furandicarboxylic acid. ChemSusChem 2013, 6, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Campisi, S.; Sanchez Trujillo, F.; Motta, D.; Davies, T.; Dimitratos, N.; Villa, A. Controlling the incorporation of phosphorus functionalities on carbon nanofibers: Effects on the catalytic performance of fructose dehydration. C 2018, 4, 9. [Google Scholar] [CrossRef]
- Tessonnier, J.P.; Rosenthal, D.; Hansen, T.W.; Hess, C.; Schuster, M.E.; Blume, R.; Girgsdies, F.; Pfaender, N.; Timpe, O.; Su, D.S.; et al. Analysis of the structure and chemical properties of some commercial carbon nanostructures. Carbon 2009, 47, 1779–1798. [Google Scholar] [CrossRef] [Green Version]
- Torres, D.; Pinilla, J.L.; Moliner, R.; Suelves, I. On the oxidation degree of few-layer graphene oxide sheets obtained from chemically oxidized multiwall carbon nanotubes. Carbon 2015, 81, 405–417. [Google Scholar] [CrossRef]
- Villa, A.; Manzoli, M.; Vindigni, F.; Chinchilla, L.E.; Botton, G.A.; Prati, L. Diols production from glycerol over Pt-Based catalysts: On the role played by the acid sites of the support. Catal. Lett. 2017, 147, 2523–2533. [Google Scholar] [CrossRef]
- Jouve, A.; Stucchi, M.; Barlocco, I.; Evangelisti, C.; Somodic, F.; Villa, A.; Prati, L. Carbon-Supported au nanoparticles: Catalytic activity ruled out by carbon support. Top. Catal. 2018, 1–11. [Google Scholar] [CrossRef]
- Gil, S.; Muñoz, L.; Sánchez-Silva, L.; Romero, A.; Valverde, J.L. Synthesis and characterization of Au supported on carbonaceous material-based catalysts for the selective oxidation of glycerol. Chem. Eng. J. 2011, 172, 418–429. [Google Scholar] [CrossRef]
- Han, X.; Geng, L.; Guo, Y.; Jia, R.; Liu, X.; Zhang, Y.; Wang, Y. Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Pt/C–O–Mg catalyst. Green Chem. 2016, 18, 1597–1604. [Google Scholar] [CrossRef]
- Ait, R.H.; Essayem, N.; Besson, M. Selective aqueous phase oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Pt/C catalysts: Influence of the base and effect of bismuth promotion. Green Chem. 2013, 15, 2240–2251. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Graphitization Degree (ID/IG) 1 | |
|---|---|---|
| Pristine | P–CNFs | |
| PS-CNFs | 0.69 | 0.37 |
| LHT-CNFs | 0.93 | 1.18 |
| HHT-CNFs | 0.12 | 0.14 |
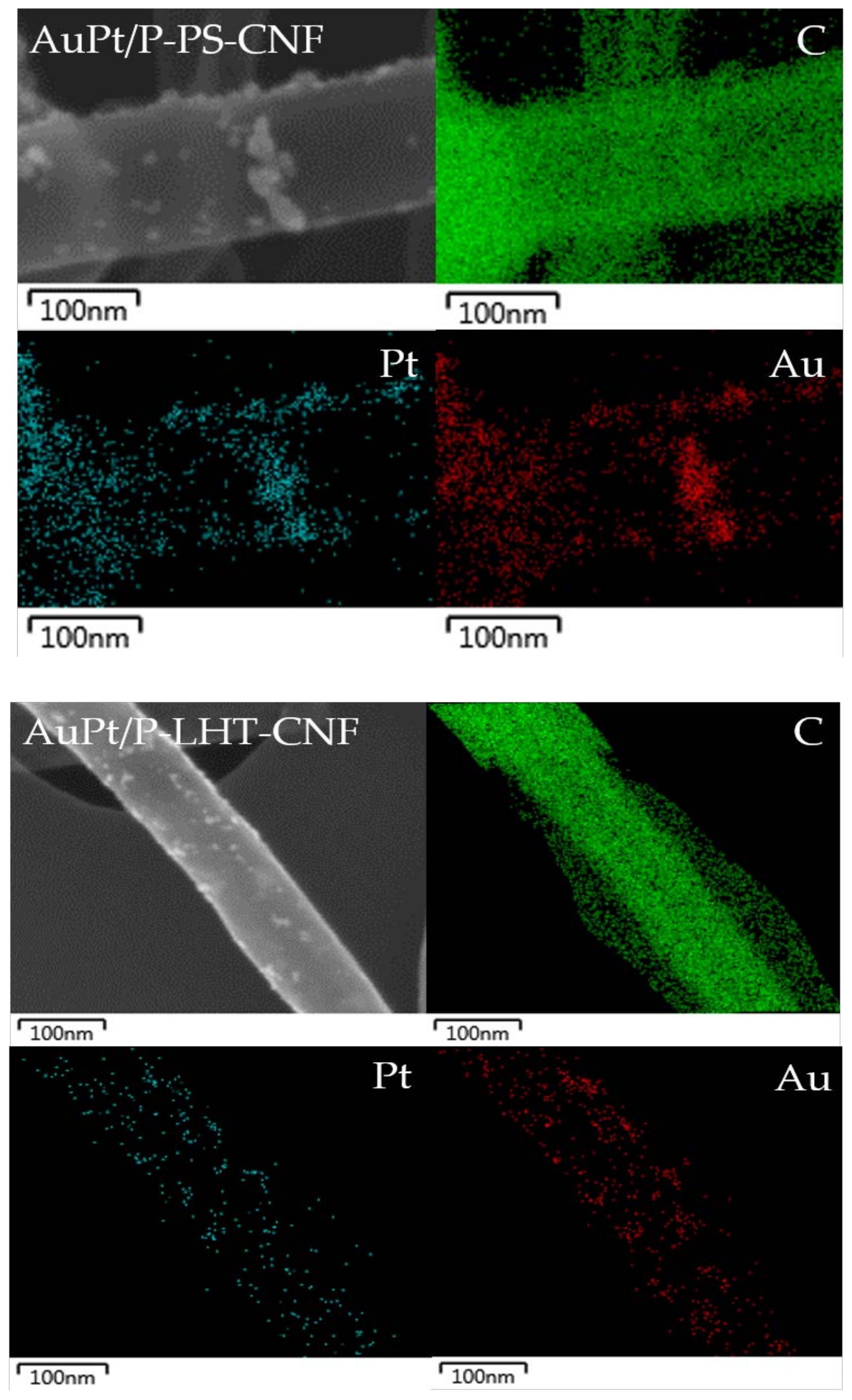
| Sample | Atomic Ratio % C:O:P |
|---|---|
| P–PS-CNFs | 87.12:12.65:0.23 |
| P–LHT-CNFs | 96.06:3.70:0.24 |
| P–HHT-CNFs | 90.89:7.92:1.19 |
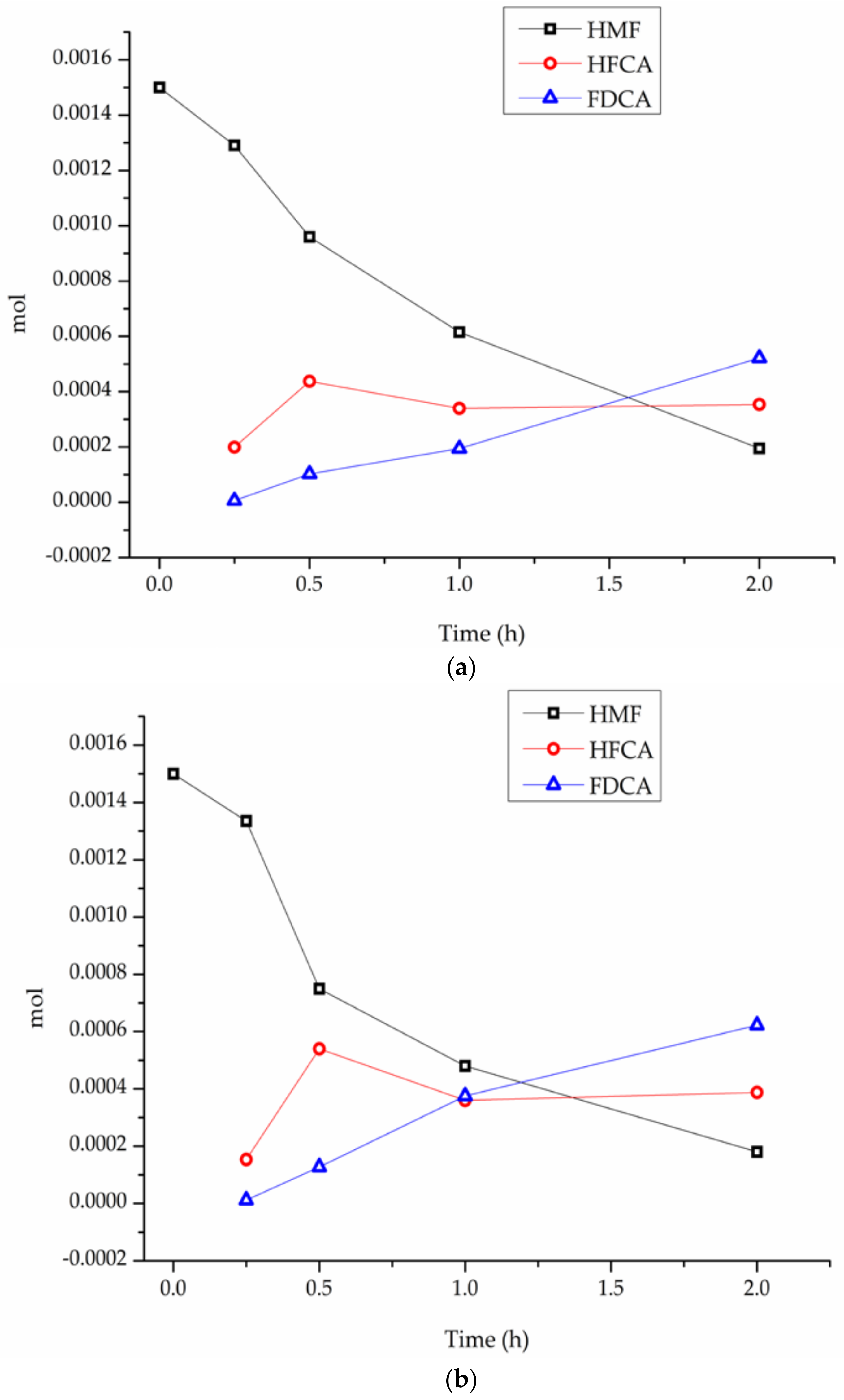
| Catalyst | Activity (h−1) b | Conversion% After 2 h | Selectivity d | ||
|---|---|---|---|---|---|
| HFCA | FDCA | Others | |||
| Au/PS-CNF | 92 | 54 | 53 e | 43 e | 4 e |
| Pt/PS-CNF | 58 | 43 | 73 e | 22 e | 5 e |
| Au–Pt/PS-CNF | 136 | 83 | 47 | 51 | 2 |
| Au–Pt/LHT-CNF | 140 | 86 | 45 | 54 | 1 |
| Au–Pt/HHT-CNF | 185 | 88 | 30 | 68 | 2 |
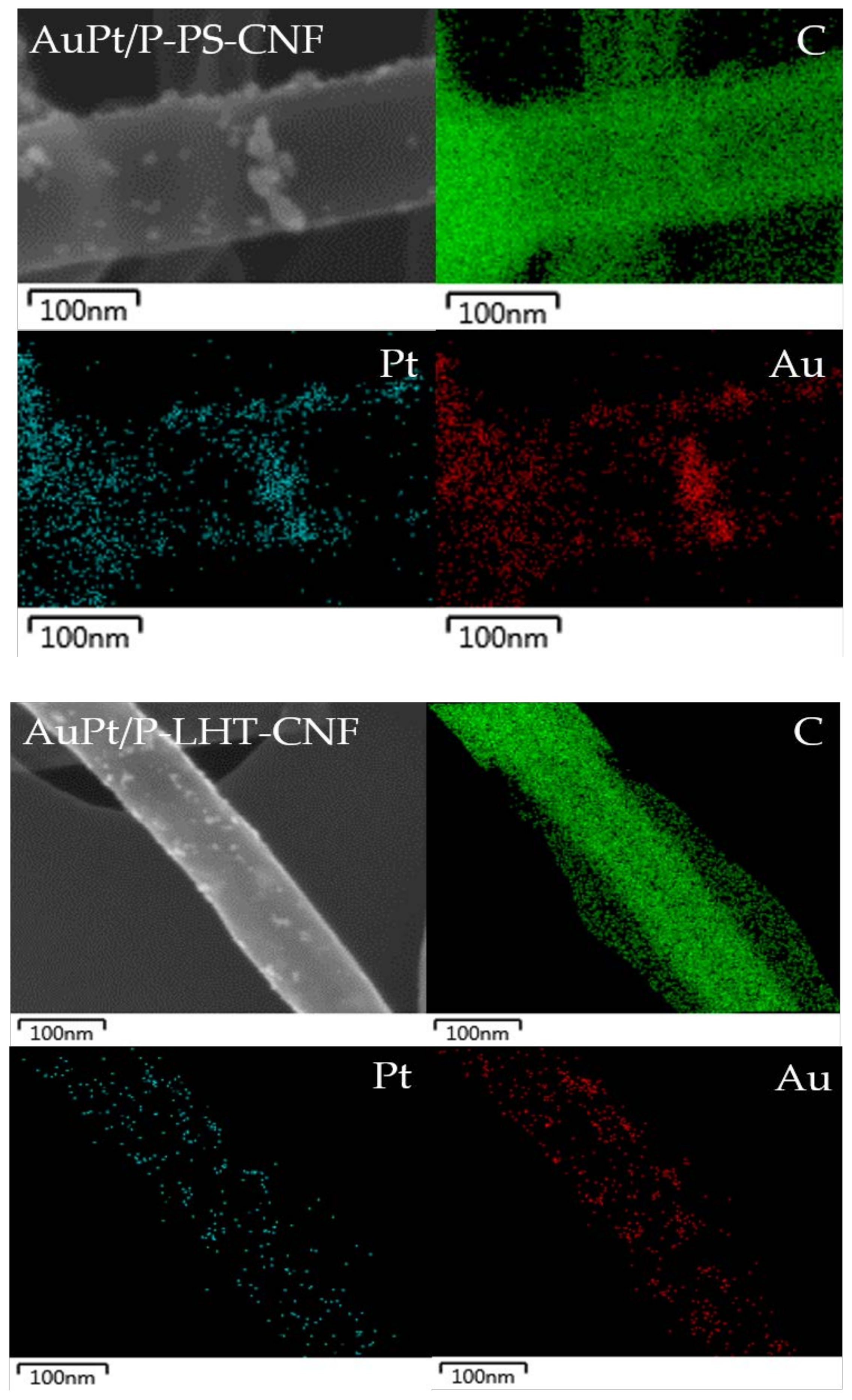
| Au–Pt/P–PS-CNF | 144 | 90 | 41 | 56 | 3 |
| Au–Pt/P–LHT-CNF | 171 | 92 | 36 | 61 | 3 |
| Au–Pt/P–HHT-CNF | 368 | 98 c | 2 | 96 | 2 |
| Catalyst | Pt 4f7/2 | Au 4f7/2 | P 1s | ||
|---|---|---|---|---|---|
| Pt0 BE (eV) | Pt2+ BE (eV) | Pt0/Pt2+ Ratio | BE (eV) | C–O–PO3 C–P BE (eV) | |
| Au–Pt/P–PS-CNF | 71.28 | 72.60 | 4.7 | 84.07 | 133.38 |
| Au–Pt/P–LHT-CNF | 71.28 | 72.49 | 6.4 | 84.06 | 133.68 |
| Au–Pt/P–HHT-CNF | 71.28 | 72.40 | 7.6 | 84.12 | 134.08 |
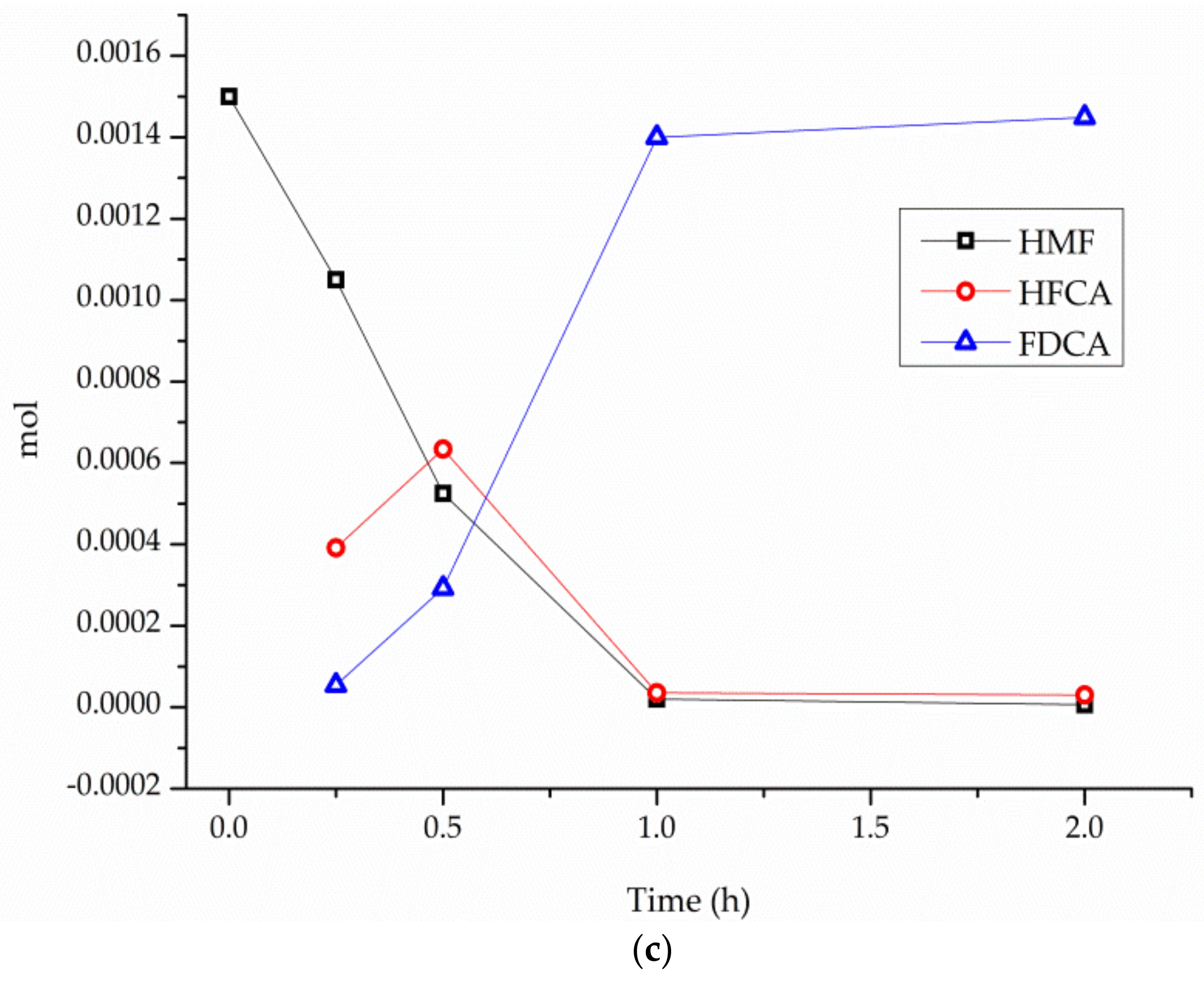
| NaOH Equiv. | Activity (h−1) b | Conversion% After 1 h | Selectivity c | ||
|---|---|---|---|---|---|
| HFCA | FDCA | Others | |||
| 0 | 89 | 15 | 68 d | 2 d | 30 d |
| 1 | 301 | 80 | 15 | 84 | 1 |
| 2 | 368 | 98 | 2 | 96 | 2 |
| 4 | 403 | >99 | - | 92 | 5 |
| Temperature (°C) | Activity (h−1) b | Conversion% After 1 h | Selectivity c | ||
|---|---|---|---|---|---|
| HFCA | FDCA | Others | |||
| 30 | 243 | 84 | 22 | 78 | - |
| 50 | 321 | 97 | 6 | 93 | 1 |
| 60 | 368 | 98 | 2 | 96 | 2 |
| 80 | 393 | >99 | - | 89 | 11 |
| Oxygen Pressure Atm | Activity (h−1) b | Conversion% After 1 h | Selectivity c | ||
|---|---|---|---|---|---|
| HFCA | FDCA | Others | |||
| 1 | 325 | 92 | 53 | 33 | 14 |
| 2 | 358 | 95 | 21 | 76 | 3 |
| 3 | 368 | 98 | 2 | 96 | 2 |
| 5 | 426 | >99 | - | 98 | 1 |
| Catalyst | Catalyst Structural Properties | Catalytic Results | |||||
|---|---|---|---|---|---|---|---|
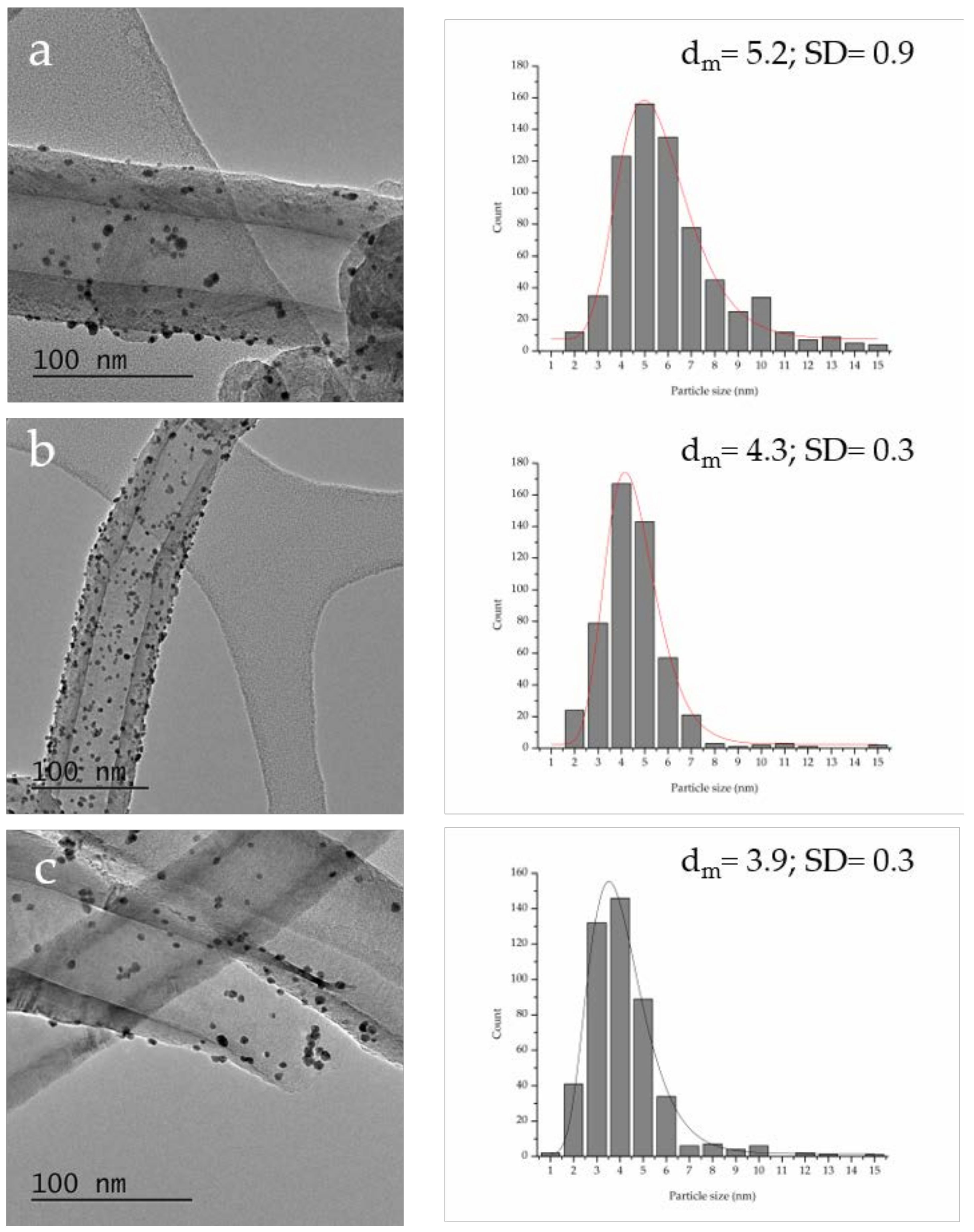
| P Content in Bare Support (%at.) a | Graphitization Degree (ID/IG) b | Particle Size (nm) c | Pt0/Pt2+ Ratio a | Au 4f7/2 Peak Position (eV) a | Activity (h−1) d | FDCA/HFCA Molar Ratio e | |
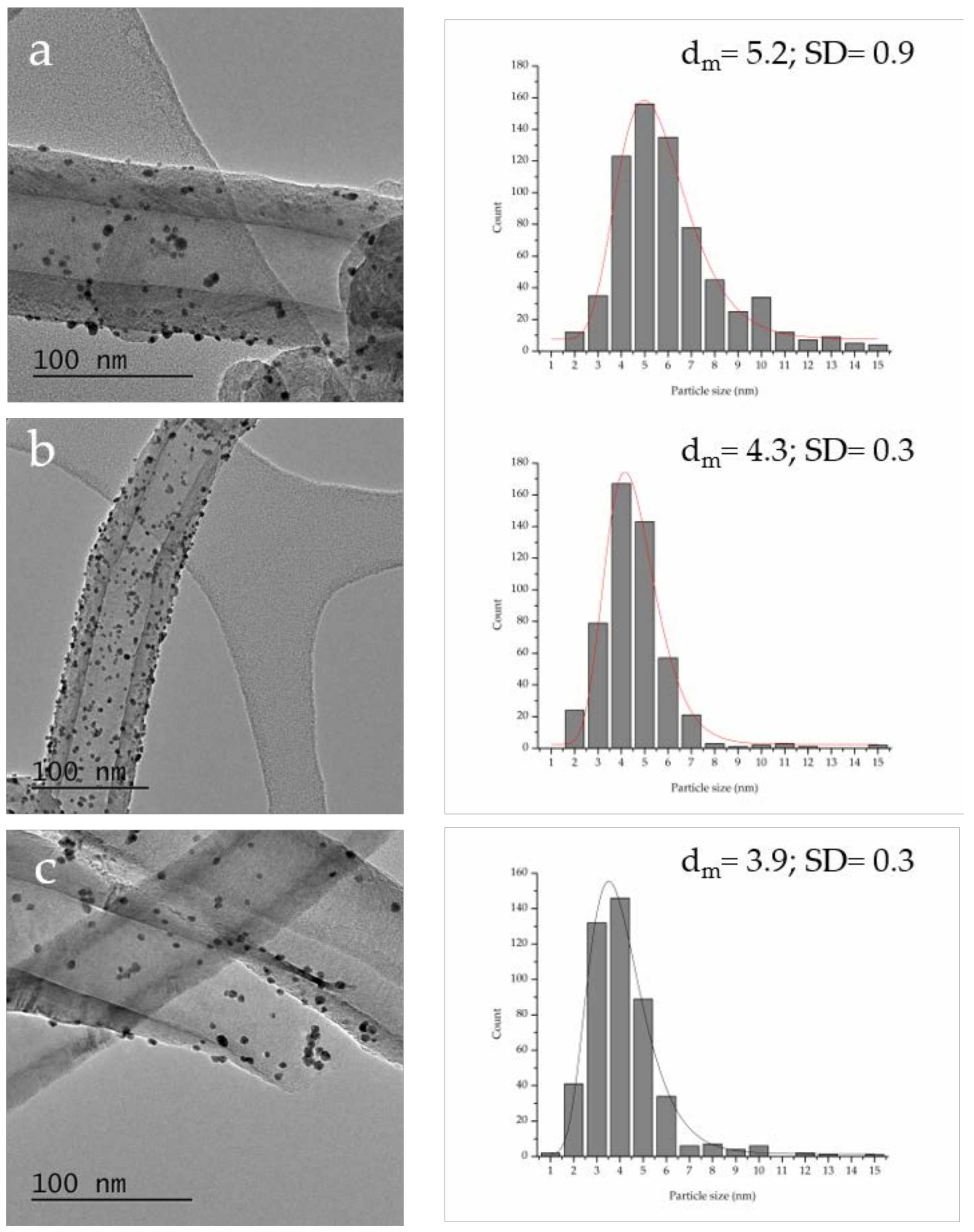
| Au–Pt/P–PS-CNF | 0.23 | 0.63 | 5.2 ± 0.3 | 4.7 | 84.07 | 144 | 1.4 |
| Au–Pt/P–LHT-CNF | 0.24 | 0.53 | 4.3 ± 0.3 | 6.4 | 84.06 | 171 | 1.7 |
| Au–Pt/P–HHT-CNF | 1.19 | 0.50 | 3.9 ± 0.3 | 7.6 | 84.12 | 368 | 48 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campisi, S.; Capelli, S.; Motta, D.; Trujillo, F.J.S.; Davies, T.E.; Prati, L.; Dimitratos, N.; Villa, A. Catalytic Performances of Au–Pt Nanoparticles on Phosphorous Functionalized Carbon Nanofibers towards HMF Oxidation. C 2018, 4, 48. https://doi.org/10.3390/c4030048
Campisi S, Capelli S, Motta D, Trujillo FJS, Davies TE, Prati L, Dimitratos N, Villa A. Catalytic Performances of Au–Pt Nanoparticles on Phosphorous Functionalized Carbon Nanofibers towards HMF Oxidation. C. 2018; 4(3):48. https://doi.org/10.3390/c4030048
Chicago/Turabian StyleCampisi, Sebastiano, Sofia Capelli, Davide Motta, Felipe J Sanchez Trujillo, Thomas E. Davies, Laura Prati, Nikolaos Dimitratos, and Alberto Villa. 2018. "Catalytic Performances of Au–Pt Nanoparticles on Phosphorous Functionalized Carbon Nanofibers towards HMF Oxidation" C 4, no. 3: 48. https://doi.org/10.3390/c4030048
APA StyleCampisi, S., Capelli, S., Motta, D., Trujillo, F. J. S., Davies, T. E., Prati, L., Dimitratos, N., & Villa, A. (2018). Catalytic Performances of Au–Pt Nanoparticles on Phosphorous Functionalized Carbon Nanofibers towards HMF Oxidation. C, 4(3), 48. https://doi.org/10.3390/c4030048