1. Introduction
Ordered mesoporous carbons (OMC) received tremendous attention during the last decade mainly due to their tunable and uniform pore size/geometry, pore connectivity and adjustable surface functionalities. All these properties made them valuable materials in many fields of applications such as the catalysis, gas adsorption and separation, energy storage, drug delivery, and gas sensors [
1,
2,
3,
4,
5,
6,
7]. The design of ordered carbon materials can be achieved only via two specific synthesis pathways, i.e., the hard and soft-template. Herein, the soft template has been chosen taking into consideration the advantages such as simplicity, time efficiency and convenient removal of the template by simple thermal annealing. This approach consists in supramolecular self-assembly organization of organic species. In general, cross-linked phenolic resins are used as carbon-yielding components, which are able to co-assembly with an amphiphilic block co-polymer acting as a pore-forming component. Thermopolymerization of such assemblies allows the formation of a thermosetting phenolic resin and further thermal annealing induces the decomposition of the phenolic resin and of the soft template resulting in a mesoporous carbon formation. Due to the flexibility of this synthesis, the incorporation of other species is very simple allowing to obtain heteroatom’s doped or hybrid mesoporous carbons.
In the recent years, nitrogen became the most studied hetoroatom since it allows to enhance carbon electronic conductivity, surface polarity and electron-donor tendency, improving the carbon performances in different applications. N-doped porous carbons show great potential in energy storage and they are particularly used in supercapacitors [
8] which are known to be environmentally friendly and a high safety system combining high power density [
9] and long cycling life [
10]. Depending on the charge storage mechanism, two types of supercapacitors are known [
11], i.e., electrical double layer capacitors (EDLC) where the charge is stored at the carbon/electrolyte interface and redox-based electrochemical capacitors [
12,
13,
14,
15] where the charge is stored via redox reactions promoted by the material in the presence of the electrolyte. While in the first case, the performances are related to the carbon porosity [
16,
17,
18], in the second case they are affected by the carbon surface chemistry [
19], namely, the nature and amount of functional groups. Therefore, it appears that the combination of an optimal carbon porosity and surface chemistry must be achieved in order to improve the electrochemical performances [
12,
15,
16,
20].
N-doped mesoporous carbons can be synthesized by post-synthesis routs involving the impregnation of a carbon with urea, melamine or polypyrrole, followed by thermal annealing under inert atmosphere or by direct exposure to ammonia gas at high temperatures [
8]. However, these methods are generally time consuming and costly for practical application and for this reason the direct thermal transformation of nitrogen containing polymers into N-doped carbon was also explored. Wei et al. [
21] reported the synthesis of N-doped mesoporous carbon using the evaporation-induced self-assembly (EISA) process and phenol-formaldehyde derived resol as carbon source, Pluronic F-127 as template and dicyandiamide as nitrogen source. The carbon resol and dicyandiamide were able to assemble with the micelles of pluronic template via hydrogen bonding and electrostatic interactions giving rise after thermal annealing to N-doped mesoporous carbon presenting tunable mesostructures, pore size and high nitrogen content (13.1 wt %). Wang et al. [
22] proposed the use of aminophenol as carbon and nitrogen source, formaldehyde as cross-linker and pluronic F-127 as pore agent. The co-assembly of these molecules resulted in N-doped mesoporous carbon with a hexagonal structure and nitrogen amount of 3.3 at %. By employing aminophenol, hexamethylenetetramine (HMTA), formaldehyde and Pluronic F-127, Chen et al. [
23] demonstrated successful synthesis of single crystals of N-doped mesoporous carbon through a soft-template approach. Yu et al. [
24] used urea as nitrogen source, hexamethylenetetramine, resorcinol–formaldehyde and pluronic F-127 co-assembled in an aqueous ammonia solution in a soft-template route under hydrothermal conditions. N-doped mesoporous carbon exhibiting a cubic structure and ~2.5 wt % of nitrogen was obtained. Using the same soft-template approach assisted by hydrothermal conditions, nitrogen-doped carbon could be obtained by self-assembly of poly(benzoxazine) with resorcinol–formaldehyde resin and lysine as precursors [
25,
26]. Melamine–formaldehyde resins [
27,
28] and aniline [
29] were reported as well as nitrogen source for the preparation of N-doped mesoporous carbons.
The main inconvenience presented by the above mentioned synthesis pathways are related either to the use of toxic precursors (phenol, formaldehyde, aniline …), or strong acids/bases for the polymerization of phenolic resin and hydrothermal conditions for rigidifying the resin. Therefore, developing more environmentally friendly synthesis routs involving limited synthesis steps and that can be cost-effective for large-scale applications are of great need. In this aim, significant progress was made by our group in the recent years to improve the soft-template route. Firstly, the glyoxylic acid as a green precursor extracted from plants was proposed for the first time as cross-linker alternative instead of formaldehyde with no requirements of supplementary base/acid catalyst in the synthesis. Such a green synthesis pathway was successfully used to prepare mesoporous carbon powders and films with tuned pore size and geometries, various graphitization levels via classical approaches like EISA and phase separation [
30], but also by novel more unconventional routes assisted by light [
31,
32]. Recently two green approaches have been developed to prepare N-doped porous carbon. Phloroglucinol/glyoxylic acid resin was cross-linked with triethylenediamine (TEDA) as nitrogen source in water at room temperature and in the absence of a template, resulting in the formation of microporous carbon spheres [
33]. Chitosan, a biocompatible and green precursor was as well proposed as a simultaneous carbon and nitrogen precursor which was able to assemble with Pluronic F-127 template to obtain N-doped carbon beads with hierarchical porosity by a soft-template assisted by a freezing-drying technique [
34].
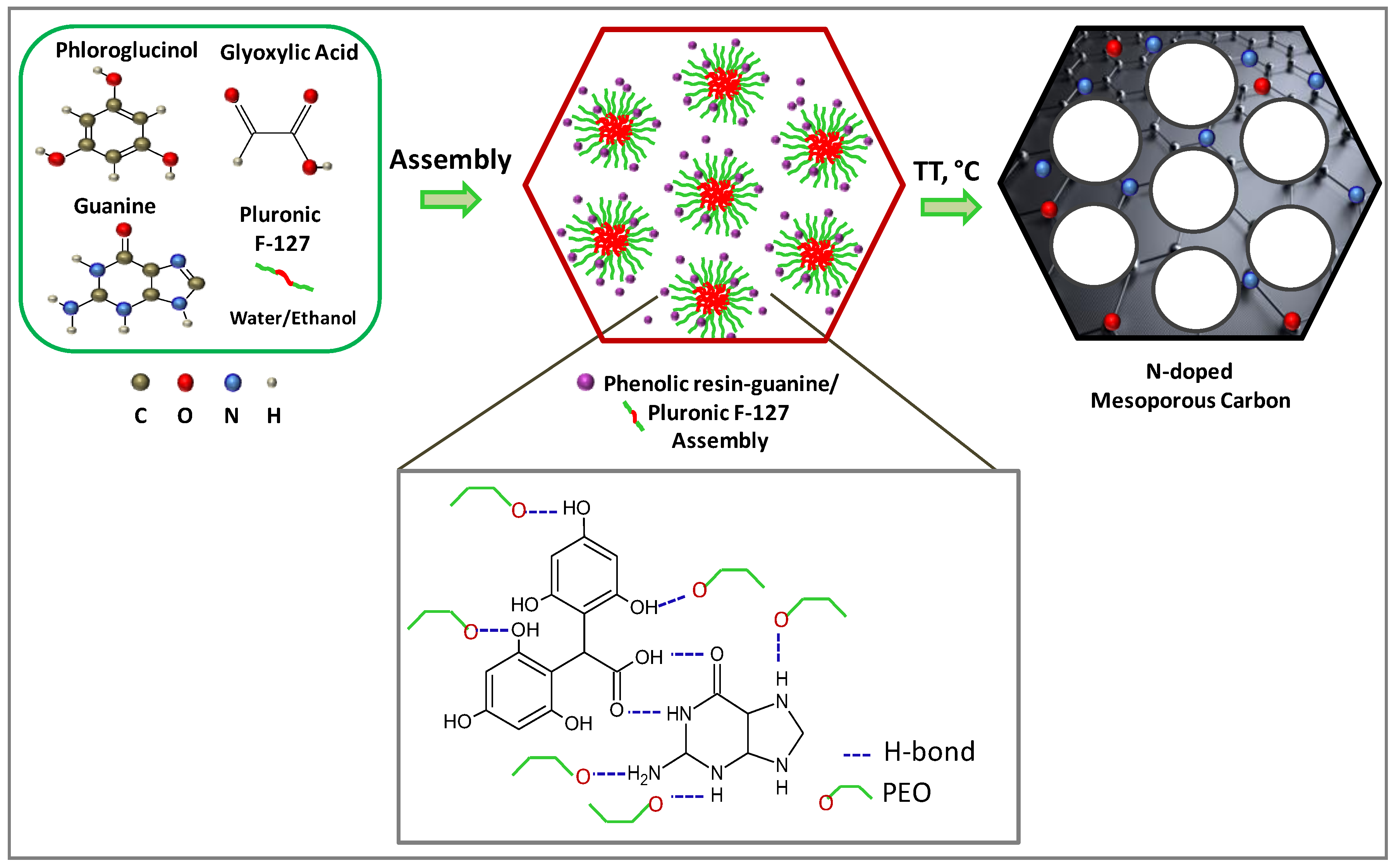
Inspired by our recent works, herein, we report a direct synthesis of N-doped ordered mesoporous carbons with a high nitrogen content and tuned porosity by co-assembly of phloroglucinol–glyoxylic acid resin and guanine with Pluronic F-127 template in water/ethanol mixture at room temperature. Guanine is proposed as a new green precursor which contains high nitrogen content (46.3%) coming from one nitrile group and two amine groups in its composition. Several synthesis parameters were investigated (synthesis procedure, template amount and annealing temperature) and their impact on the final material texture and structure finely characterized. Thus, insight on the formation mechanism of the N-doped mesoporous carbon using guanine is proposed based on several techniques. Selected materials were tested as supercapacitors and the performances are discussed in terms of materials characteristics.
3. Results and Discussion
The synthesis of N-doped carbons was performed via the soft-template approach involving the organic-organic self-assembly of phenolic resins molecules with a soft-template (triblock polymer, Pluronic F-127), which is able to create the mesoporosity and to organize it in different shapes and size depending of several factors (
Figure 1).
Herein, few important parameters have been explored in order to evaluate their influence on the final carbon characteristics. The phase separation, even if less used compared to the EISA method, was selected for this study taking into consideration the following advantages (a) allows to prepare high quantities of material per batch of synthesis, therefore possible to up-scale and (b) easy to perform and to recover the polymer for carbonization (scratching of polymer film from petri dishes being necessary for EISA method). However, the control of porosity (surface area, pore size and architecture) is of great importance, particularly when new precursors are incorporated in the synthesis mixture. This is the case of guanine which was used for the first time in order to introduce nitrogen in the carbon framework.
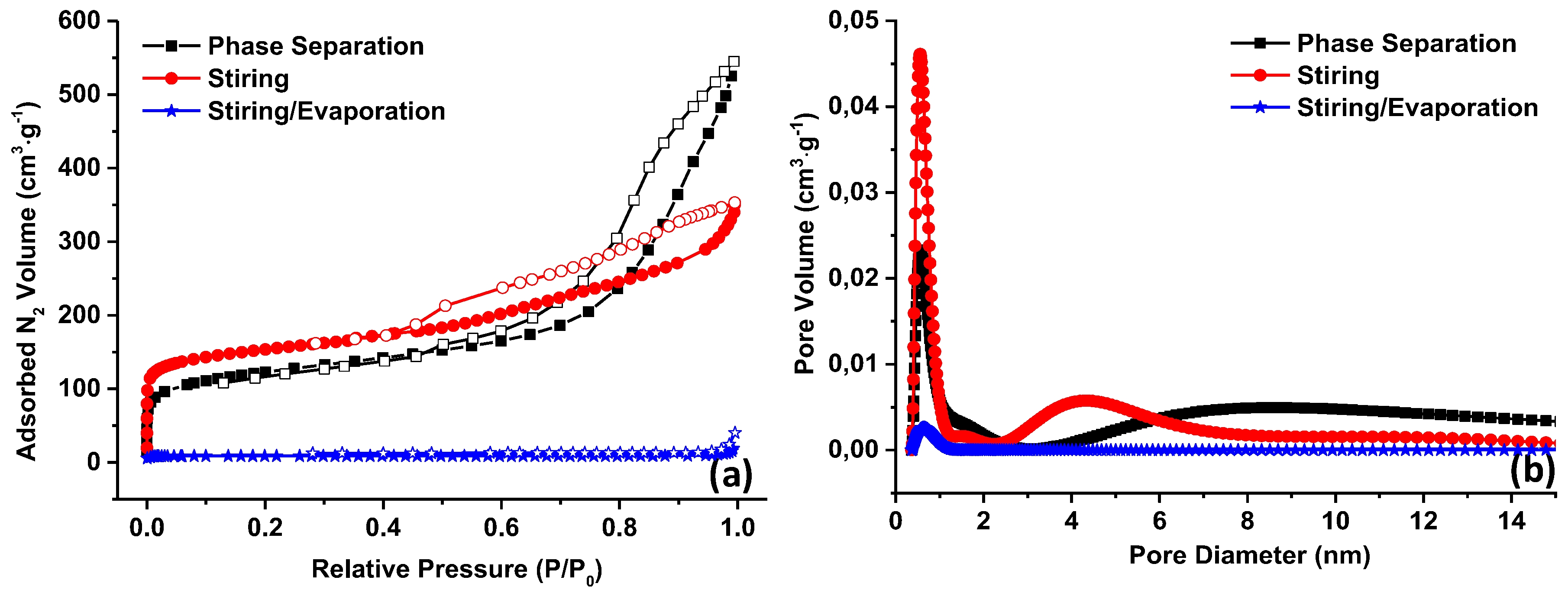
Firstly, the impact of the synthesis type on the carbon porosity was investigated and three different procedures were evaluated: (a) the phase separation, involving macroscopic phase separation of polymeric resin and the solvent; (b) phase separation assisted by stirring, herein “stirring”; and (c) stirring/evaporation method, a similar approach as method (b) but being conducted in an uncovered vessel to allow complete evaporation of the solvent. The as-obtained gels after the synthesis were thermopolymerized and pyrolyzed and the obtained carbons analyzed by transmission electron microscopy (
Figure 2). The carbon obtained by phase separation (
Figure 2a) present regular parallel channels indicating an ordered porous structure. When the synthesis is assisted by stirring (
Figure 2b), the morphology became less ordered or even completely disordered when the synthesis is performed under evaporation (
Figure 2c). This result indicates that the way of synthesis preparation has a great impact on the mesoporosity formation.
To obtain further details about the materials porosity, nitrogen adsorption measurements were performed and the obtained nitrogen adsorption/desorption isotherms are shown in
Figure 3a. For the materials prepared by phase separation and stirring, the isotherms present a type IV shape with a hysteresis loop of type 2 between 0.4 and 1,
P/P0 relative pressure, confirming the mesoporous character of the materials. An increase of the adsorbed nitrogen volume in the low relative pressure region is observed as well suggesting the presence of micropores. Comparing the two materials, it can be noticed that the microporous part is rather similar, slightly higher in the case of carbon obtained by stirring which translates in the same tendency for specific surface areas (445 and 570 m
2∙g
−1 for phase separation and stirring, respectively) and microporous volume (0.19 vs. 0.25 cm
3∙g
−1,
Table 1). If the mesoporous part of the isotherms are compared (
P/P0 > 0.4), it can be clearly seen a much defined hysteresis with higher adsorbed nitrogen volumes for phase separation derived carbon. This implies that this carbon presents a more developed mesoporosity as confirmed by the determination of mesoporous volume which is 0.50 cm
3∙g
−1, therefore five times higher than in the case of stirring derived carbon, 0.11 cm
3∙g
−1. The mesopore size is different as well (
Figure 3b), ~4 nm and 8 nm for phase separation and stirring, respectively. The micropores are visible on the pore size distribution, with sizes less than 1 nm for both materials. As the mesoporosity is much affected by the stirring procedure compared to microporosity, we can believe that this had an effect on the self-assembly of the phenolic resin with the template rather than on the formation of the resin. As a reminder, the thermal decomposition of phenolic resin (cross-linked phloroglucinol with glyoxylic acid) is responsible for the micropore creation while the decomposition of the template is related to the mesoporous formation [
38]. Surprisingly, if the stirring synthesis is assisted by evaporation, the resulting carbon material is not porous, the surface being very small (36 m
2∙g
−1). Therefore, the cross-linking of the phloroglucinol with glyoxylic acid and the self-assembly of the resulting resin with the template is strongly affected by the evaporation of the solvent. Taking into consideration the higher evaporation rate of ethanol over water, we can expect that the removal of ethanol is done before the water and that the polymer solution will be enriched in water solvent. As some precursors, such as phloroglucinol have limited solubility in water, it can be supposed that its cross-linking with glyoxylic acid and further self-assembly with the template is not promoted. However, at this point the mechanism behind the porosity loss is not yet clear, but it is evident that this approach is detrimental for the preparation of carbons with controlled porosity. Therefore, we select the stirring procedure to investigate further the template amount influence.
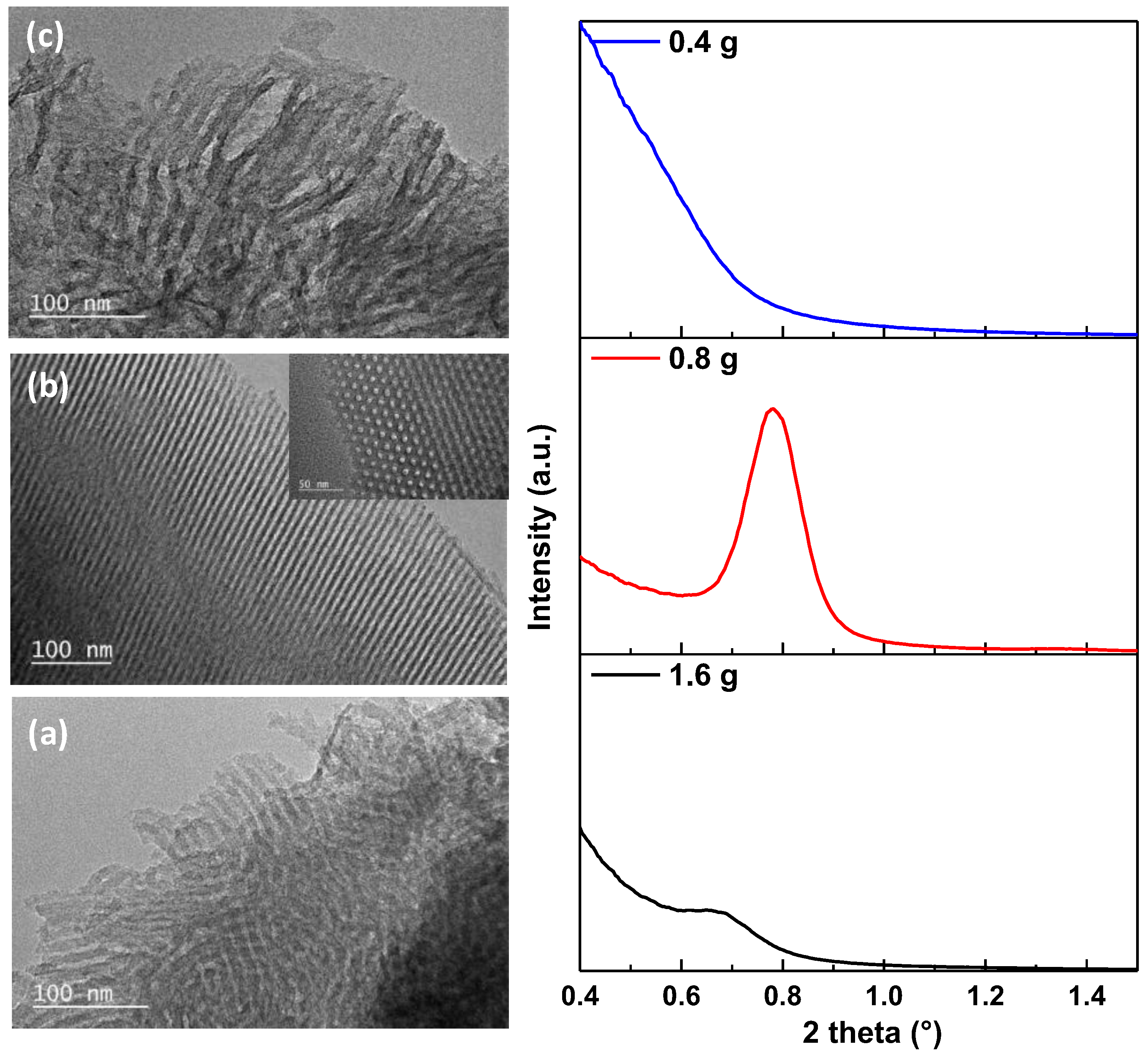
Figure 4 presents the TEM images and the small-angle X-ray scattering of carbon materials resulting by changing the initial amount of template from 1.6 g to 0.8 g and 0.4 g, respectively. It can be observed in the TEM pictures that diminishing the quantity from 1.6 g (
Figure 4a, left) to 0.8 g (
Figure 4b, left), the morphology changes, an ordered material presenting uniform parallel channels with ordered distributed porosity (in-set) being obtained. However, if the quantity is further decreased to 0.4 g, the obtained material is more disorganized (
Figure 4c, left) compared the other two materials. These results are sustained by the SAXS measurements, showing a well-defined diffraction peak at around 0.8° 2theta for 0.8 g materials. This peak corresponds to the 10 diffraction planes of a 2D hexagonal ordered mesostructure having
p6
m symmetry [
30]. On the contrary, only a small peak is seen for 1.6 g material and no peak for 0.4 g material indicating low ordering in line with the TEM observations.
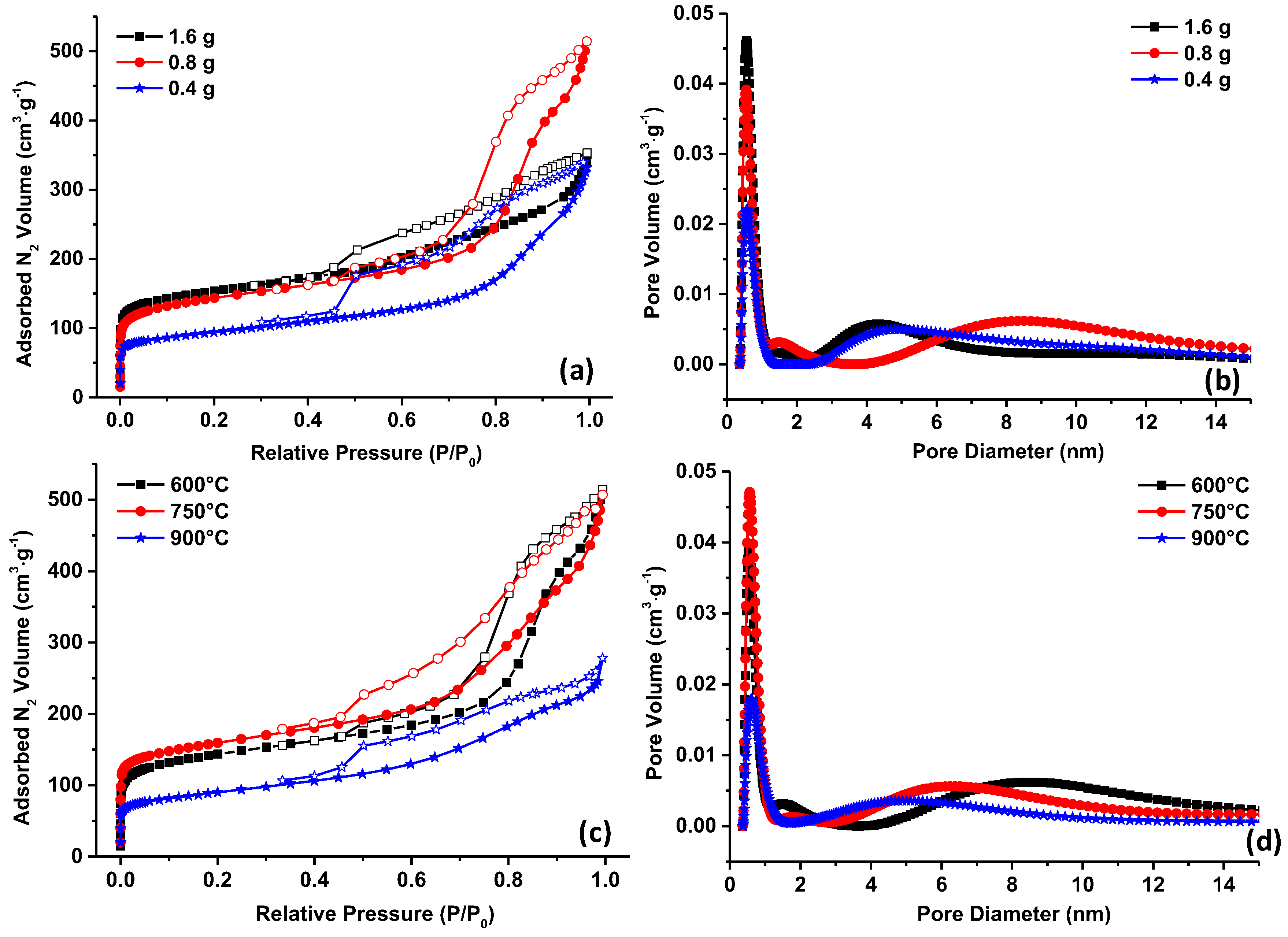
The nitrogen adsorption/desorption isotherms show for all materials type IV isotherms specific to micro/mesoporous materials (
Figure 5a). For 1.6 and 0.8 g, the low-pressure curves are almost overlapped, inducing similar microporosity (
Vmicro,
Table 1) and SSA (570 vs. 527 m
2·g
−1, respectively), while the high pressure region corresponding to the mesopores is much developed for 0.8 g material (0.11 vs. 0.40 cm
3·g
−1 for 1.6 and 0.8 g material, respectively). For 0.4 g material, the mesoporosity is similar with 1.6 g material while the microporosity and the SSA is the smallest one among the studied materials (341 m
2·g
−1,
Table 1). The pore size is also different depending on the template amount as demonstrated in
Figure 5b. This suggests that a certain quantity of template is required to obtain high surface area materials with ordered porosity. This is a balance between the formations of ordered template micelles able to interact with the phenolic resin through H-bondings. The 0.8 g material is therefore the most interesting one to investigate further the impact of annealing temperature on the porosity but also on the surface chemistry.
The nitrogen adsorption/desorption measurements for carbon materials were prepared at standard 600 °C but also at higher temperatures 750 °C and 900 °C, depicted in
Figure 5c. For 600 and 750 °C, the shapes of the curves are similar, slightly higher for 750 °C resulting in similar textural values as seen in
Table 1. Increasing the temperature to 900 °C has a negative impact on the porosity, the SSA being reduced from 587 (750 °C) to 322 m
2∙g
−1 (900 °C). The pore size is affected as well by the temperature as shown in
Figure 5d. The micropore size is increasing from 0.54 nm for 600 °C to 0.62 nm for 900 °C, while for the mesopores an opposite behavior is seen, i.e., shrinkage from ~ 9 nm to 7 nm and 5 nm for 600, 750 and 900 °C materials, respectively. The modification of pore size can be in some extent related to the collapse of the micropores with the formation of larger one and to the densification of the carbon matrix due to the removal of hetoroatoms. This last aspect was analyzed in more detail since it may also influence the electrochemical performances of the supercapacitors. The heteroatoms may change the wettability behavior of the material with the electrolyte, the electronic conductivity and may induce redox reactions with the electrolyte.
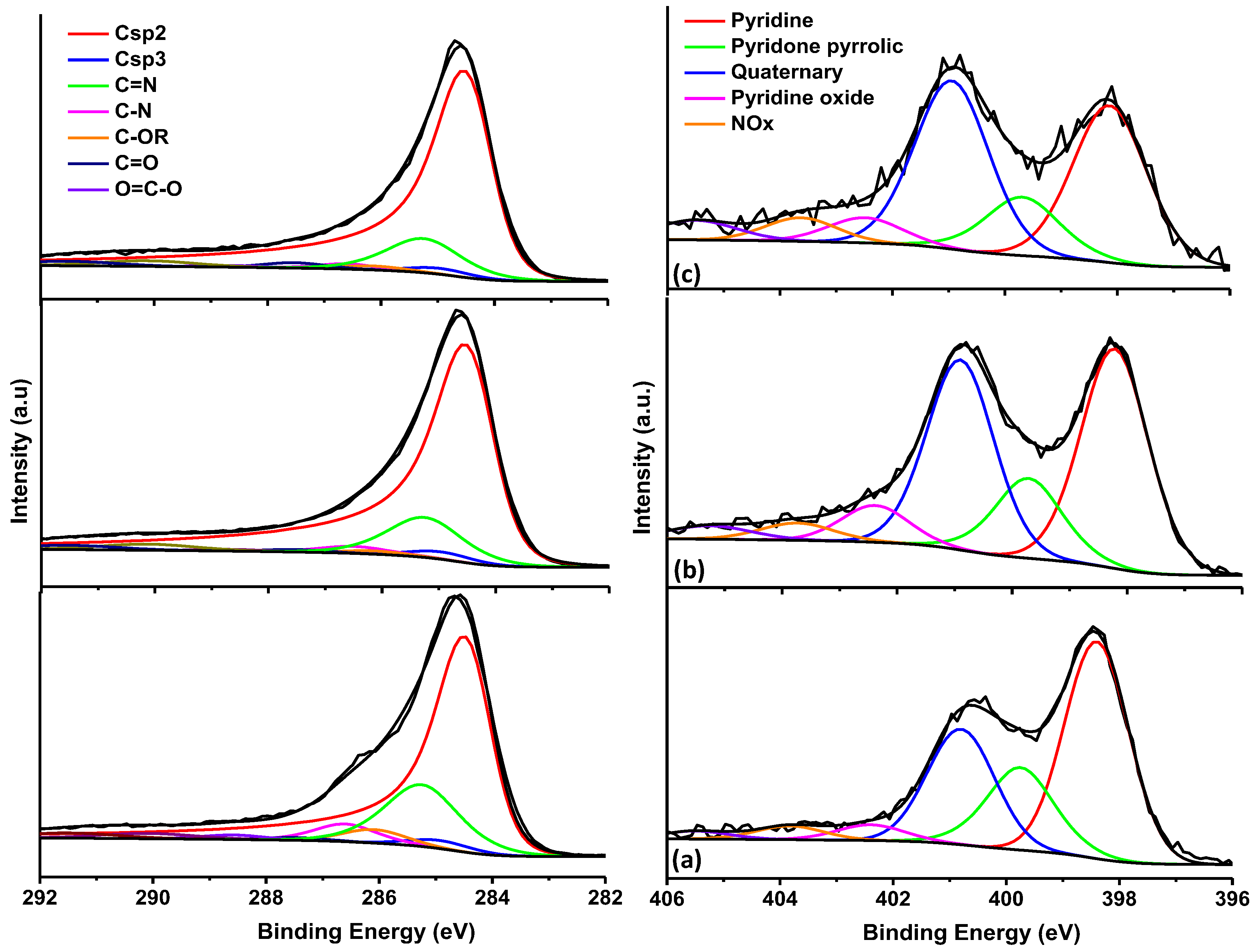
In this aim, the XPS analyses were performed for the carbon treated at different temperatures. The survey spectra were recorded and besides the carbon presence, oxygen and nitrogen were detected. For 600 °C material, the C, O and N contents are 83.4, 6.2 and 10.4 at % respectively. It is worth noting, the high quantity of nitrogen that could be introduced by using the guanine. Increasing the temperature to 900 °C, both the oxygen and nitrogen amount are reduced to ~4 and 6 at %, respectively (
Table 2). The high-resolution spectra of C1s were deconvoluted in several components (
Figure 6, left). The most intense one is related to Csp
2 followed by C=N and C–N bonds [
39,
40]. The increase of the full width at half maximum (FWMH) of the C1s peak at 600 °C compared to 700 °C and 900 °C, and the appearance of a peak towards 286.5 eV, is related to the presence of oxygen functional groups such as ethers, carbonyl, carboxyl (–C–OR, –C=O and O=C–O) and nitrogen groups (C=N, C–N), which are located at higher binding energy compared to the Csp
2.
Increasing the temperature, the FWHM of C1s peak decreases due to the removal of some oxygen and nitrogen groups. In addition, the N1s peaks exhibit five contributions located at 398.1, 399.6, 400.9, 402.5 and 403.6 eV (
Figure 6, right) corresponding to pyridine, pyridine pyrrolic, quaternary, pyridine oxide and NO
x nitrogen functional groups [
40,
41]. At 600 °C, the pyridine groups are predominant while increasing the temperature they are partly removed leaving place to quaternary groups which become predominant at 900 °C.
The distribution of N and O atoms in the carbon framework was evaluated by EDX mapping (
Figure 7) and it can be observed that the heteroatoms are very well distributed in the carbon matrix at atomic level. This indicates a good repartition of the guanine in the phenolic resin resulting in a uniform N-doped carbon.
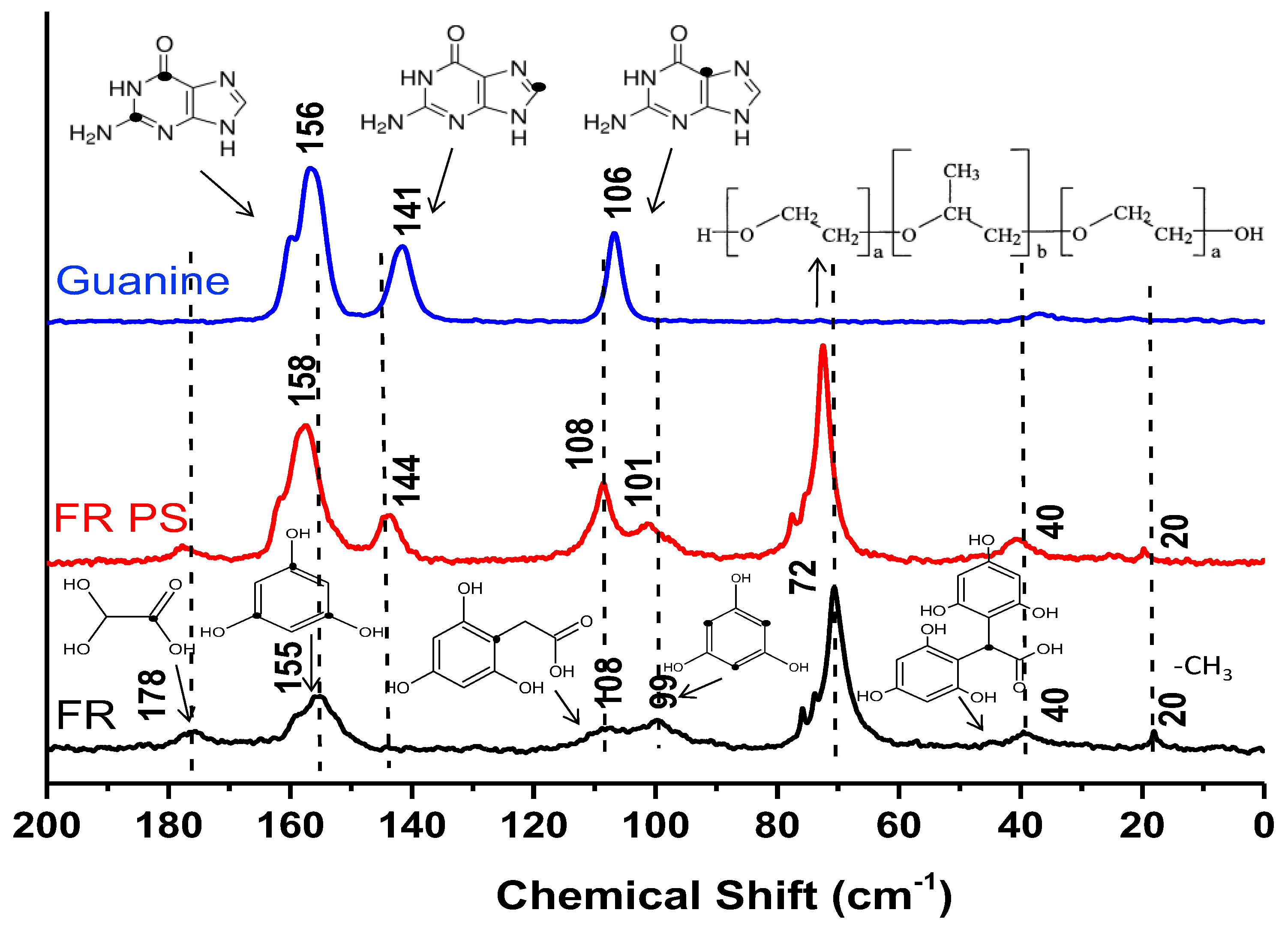
To get further insights on the formation of these materials,
13C NMR was performed on a phenolic resin containing guanine and in addition for comparison purposes on a phenolic resin which is free of guanine and on guanine precursor (
Figure 8). The
13C NMR of guanine exhibit three main peaks placed at 156, 141 and 106 ppm, respectively. The first peak (156 ppm) is the most intense one and correspond to carbon atoms bonded to oxygen (=O) in the pyrimidine cycle. The peak presents a shoulder which assigned to saturated and unsaturated carbon atoms bonded to nitrogen pyrimidine cycle (in-set
Figure 8).
The peak for 141 ppm is related to the carbon atoms bonded to nitrogen in the imodazole cycle, while the peak placed at 106 ppm is assigned to carbon atoms bonded simultaneous with carbonyl groups and nitrogen atoms. The NMR conducted on phenolic resin containing guanine (FR PS) confirms the presence of guanine by the appearance of the three peaks mentioned before. In addition, other peaks are seen at 178, 158 and 101 ppm, which are mainly related to the carbon atoms present in the initial precursors (phloroglucinol and glyoxylic acid) suggesting still some un-reacted precursors. Another two peaks are detected at 108 and 40 ppm indicating cross-linked phloroglucinol with glyoxylic acid via covalent bondings. The detailed mechanism of cross-linking was previously reported in our works [
30,
32,
38]. An intense contribution is seen at ~72 ppm and a small one at 20 ppm corresponding to the carbons atoms in –CH, –CH
2 and –CH
3 groups present in the PPO (polypropylene oxide) and PEO (polyethylene oxide) moieties of the Pluronic F-127 template. It is worth noting that if we compare all three NMR spectra, the spectra of phenolic resin containing the guanine is an addition of the spectra of guanine and phenolic resin (FR). No supplementary peaks appear indicating no cross-linking reactions by covalent bonds between the guanine and the phenolic resin, as for instance may occur by using another nitrogen precursor as recently demonstrated [
33]. As well, a simple physical mixture between the guanine and the phenolic resin can be excluded taking into consideration the TEM and EDX results showing the formation of ordered carbon materials with uniform distribution of nitrogen in their framework.
The most plausible hypothesis to explain the formation of these materials is the creation of H-bonding between the guanine and the phenolic resin and the template. This scenario is possible since the guanine is well known to be able to create H-bondings with various compounds. The carbonyl bond (=O) of guanine may act as a hydrogen bond acceptor and may create H bonds with the –OH groups of phloroglucinol while the nitrogen groups (–NH and –NH2) may act as a hydrogen bond donor which may favor the H-bonding with the carbonyl groups (=O) of glyoxylic acid or its derivates. The as-formed phenolic resin/guanine system may further assemble by H-bondings with the micelles of Pluronic template. Two main possibilities of assembly of phenolic resin/guanine system with the template are possible. On one hand the high number of hydroxyl groups (–OH) of phloroglucinol and on the other hand the –NH and –NH2 groups of guanine, may develop H-bonds with the –O of PEO fragments of the template. In the water/ethanol solution, the PPO fragments of the template form the core of the micelles while the PEO the shell of the micelle. The phenolic resin/guanine may form H-bonding with the oxygen atoms coming from the hydrophilic PEO moieties of the pluronic micelle shell, forming a layer of resin/guanine in the surface of the micelles.
To evaluate if the guanine can self-assemble with the template we performed a supplementary synthesis where phloroglucinol was not used. Surprisingly, the resulting material is porous (
Figure 9a) and this porosity is randomly organized than compared to the carbon obtained in the presence of phloroglucinol (
Figure 9b). This demonstrates that guanine itself is able to create H-bonding and to assemble with the Pluronic template, and this assembly involves different bonds compared to phloroglucinol. In addition, guanine can be used both as nitrogen but also carbon source.
Taking into considerations these results, one can propose that the synthesis of N-doped mesoporous carbon proceed by a complex multi-component co-assembly process where the carbon precursors (the phenolic resin), the nitrogen source (guanine) and the pore agent (Pluronic F-127) are closely interacting by H-bondings. Further thermal annealing of such assembly, decompose the template and allow to obtain a mesoporous carbon with nitrogen in its structure. A general schema resuming the synthesis mechanism of assembly of phenolic resin/guanine and the template is proposed in
Figure 1.
The electrochemical performances of several N-doped carbon materials synthesized at 600 °C were evaluated in a two-electrode configuration in 0.1 M H
2SO
4 electrolyte in the potential range of 0–0.9 V using a scan rate of 20 mV s
−1. Cycling voltammetry was used to study the electrochemical behavior and a typical curve for a material prepared at 600 °C is presented in
Figure 10a. Surprisingly, the capacity value was found to be close to zero F/g, however, increasing the annealing temperature to 750 and 900 °C, the cyclic voltammetry (CV) curve shape improves and the capacitance as well, i.e., 84 F/g and 53 F/g for 750 °C and 900 °C.
To explain this behavior several important factors must be considered such as porosity and surface chemistry. Concerning the porosity, it has been seen before that this similarity between 600 °C and 750 °C, slightly higher for the latest one while for 900 °C, the porosity is significantly lower than at 600 and 750 °C (
Figure 10b). Therefore, the textural properties cannot explain why the capacitance of 600 °C sample is very low. On the other hand, the XPS showed for the 600 °C a high amount of nitrogen and oxygen groups compared to 750 and 900 °C samples (
Table 2). The high number of functional groups are supposed to have a positive effect on the wettability of carbon surface with the electrolyte as well in promoting redox reactions to improve the capacitance by pseudocapacitive effects. However, particularly the oxygen groups may be detrimental to the electronic conduction of the material. To get more insights on this aspect, EIS was performed and the Nyquist plots are shown in
Figure 10c.
A large semicircle in the high to low frequency region is seen for 600 °C sample indicating a highly resistive material, which can be accounted to the large amount of oxygen. Increasing the temperature to 750 °C, the shape of the curve changes to a semicircle in the high-to-medium frequency range, accompanied by a sloped line in the low frequency region, indicating at a first glance a better capacitive behavior and smaller diffusion resistance. At 900 °C, the semicircle become very small sign of a good electronic conductivity. From the intercept of the semicircle with the real axis, the equivalent series resistance (ESR) of the electrodes can be determined which gives information about the total resistance of the system comprising the electrode, the electrolyte, and separator. This resistance is ~13 and ~4 Ohm for 750 and 900 °C carbons, therefore, significantly much smaller than for the 600 °C sample (several thousand Ohms) proving the improvement of the electronic conductivity. To further improve the electrochemical performances, the 750 °C sample was mixed with carbon black. The CV exhibits a more rectangular CV curve indicating electric double layer capacitive behavior. The Nyquist plot show a smaller semi-circle when carbon black is used, the resistance being diminished to 6 Ohm. Therefore, the improvement in the conductivity allows to achieve a capacitance of 95 F/g for 750 °C/CB sample. A general trend of the capacitance evolution with the ERS is seen in
Figure 10d, i.e., an increase in the capacitance with the increase of the ESR for the materials treated at 600 °C and 750 °C, therefore, for materials presenting similar specific surface areas. For the 900 °C carbon, even if the ERS is lower, the delivered capacitance is much lower than the other materials and this can be related to its lower specific surface area (
Table 1).
In order to compare the performances of such materials with those reported in the literature, several electrochemical performances in H
2SO
4 were gathered in
Table 3 along with their specific surface area and nitrogen content. The selected samples were limited to material exhibiting characteristics (porosity and nitrogen content) comparable with those obtained in the present work. It can be seen that all these materials present higher capacitance than our materials even if the surface area are comparable. However, it can be noticed that no strict correlation between the BET surface area and the capacitance is observed (
Figure 10b). If the capacitance is plotted versus the nitrogen content, a nice increase trend of the capacitance with the increase of the nitrogen amount is seen (
Figure 10e) for most reported materials in the literature, suggesting an important contribution of pseudo-capacitive reactions to the overall performance. However, it can be noticed that for our materials the capacity is lower compared to other materials and in particularly for the richer N-doped sample. In this particular case, the high resistance of these materials induced by the high amounts of oxygen (
Figure 10d,f) may explain the very low capacitance. Another parameter that can be considered to explain the lower capacitance values maybe the carbon pore size. It is well known that the capacity is improved when the carbon pore size approaches the size of electrolyte ions and the maximum capacitance was demonstrated to be achieved for pore with size ~0.7 nm [
16,
17,
42]. However, in many works this useful information regarding the micropore size is often not provided (
Table 3) which makes it difficult to understand the storage mechanisms in complex doped carbons. In the present materials the micropore size is centered on ~0.6 nm, which may be considered rather small to ensure effective diffusion of electrolyte into the microporosity and the formation of the electrochemical double layer [
42].
Taking into consideration these results, it can be highlighted that several carbon characteristics (conductivity, porosity and functionalities) are impacting the electrochemical capacitance. The optimal carbon must combine firstly good electronic properties and high amounts of nitrogen but also optimal porosity. Therefore, the prepared materials are meet most of these requirements; nevertheless, their performances may be improved by enlarging the micropore size and amount. An activation step already demonstrated the utility in order to improve the porosity and to achieve higher capacitance [
8,
43] and can be implemented further for these materials.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}