
Gelation upon the Mixing of Amphiphilic Graft and Triblock Copolymers Containing Enantiomeric Polylactide Segments through Stereocomplex Formation



Abstract
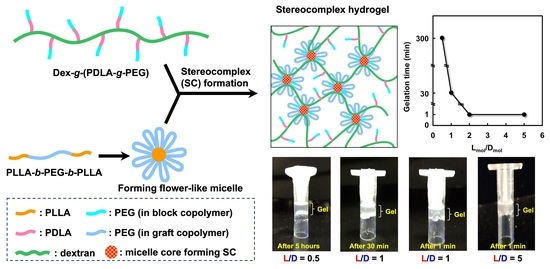
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of the Copolymers
2.2. Gelation Behavior of the Mixture Solution
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Synthesis of PLLA-b-PEG-b-PLLA Triblock Copolymers
4.3. Synthesis of Dex-g-(PLLA-b-PEG) and Dex-g-(PDLA-b-PEG)
4.4. DLS Measurement of Copolymer Solutions
4.5. Gelation Behavior
4.6. Mechanical Strength of the Hydrogel
4.7. Wide-Angle X-ray Diffraction Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Park, M.H.; Joo, M.K.; Choi, B.G.; Jeong, B. Biodegradable thermogels. Accounts Chem. Res. 2012, 45, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ding, J.D. Injectable hydrogels as unique biomedical materials. Chem. Soc. Rev. 2008, 37, 1473–1481. [Google Scholar] [CrossRef]
- Yang, J.A.; Yeom, J.; Hwang, B.W.; Hoffman, A.S.; Hahn, S.K. In situ-forming injectable hydrogels for regenerative medicine. Prog. Polym. Sci. 2014, 39, 1973–1986. [Google Scholar] [CrossRef]
- Huynh, C.T.; Nguyen, M.K.; Lee, D.S. Injectable block copolymer hydrogels: Achievements and future challenges for biomedical applications. Macromolecules 2011, 44, 6629–6636. [Google Scholar] [CrossRef]
- Nagahama, K.; Takahashi, A.; Ohya, Y. Biodegradable polymers exhibiting temperature-responsive sol-gel transition as injectable biomedical materials. React. Funct. Polym. 2013, 73, 979–985. [Google Scholar] [CrossRef]
- Nagahama, K.; Ouchi, T.; Ohya, Y. Temperature-induced hydrogels through self-assembly of cholesterol-substituted star PEG-b-PLLA copolymers: An injectable scaffold for tissue engineering. Adv. Funct. Mater. 2008, 18, 1220–1231. [Google Scholar] [CrossRef]
- Yoshizaki, Y.; Ii, M.; Takai, H.; Mayumi, N.; Fujiwara, S.; Kuzuya, A.; Ohya, Y. Cellular therapy for myocardial ischemia using a temperature-responsive biodegradable injectable polymer system with adipose-derived stem cells. Sci. Technol. Adv. Mater. 2021, 22, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Yeon, B.; Park, M.H.; Moon, H.J.; Kim, S.J.; Cheon, Y.W.; Jeong, B. 3D culture of adipose-tissue-derived stem cells mainly leads to chondrogenesis in poly(ethylene glycol)-poly(l-alanine) diblock copolymer thermogel. Biomacromolecules 2013, 14, 3256–3266. [Google Scholar] [CrossRef]
- Choi, B.G.; Park, M.H.; Cho, S.H.; Joo, M.K.; Oh, H.J.; Kim, E.H.; Park, K.; Han, D.K.; Jeong, B. In situ thermal gelling polypeptide for chondrocytes 3D culture. Biomaterials 2010, 31, 9266–9272. [Google Scholar] [CrossRef]
- Choi, S.; Baudys, M.; Kim, S.W. Control of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic rats. Pharm. Res. 2004, 21, 827–831. [Google Scholar] [CrossRef]
- Huynh, D.P.; Im, G.J.; Chae, S.Y.; Lee, K.C.; Lee, D.S. Controlled release of insulin from pH/temperature-sensitive injectable pentablock copolymer hydrogel. J. Control. Release 2009, 137, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yu, L.; Liu, X.J.; Chen, C.; Chen, Q.H.; Ding, J.D. A long-acting formulation of a polypeptide drug exenatide in treatment of diabetes using an injectable block copolymer hydrogel. Biomaterials 2013, 34, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ni, J.; Chen, L.; Yu, L.; Xu, J.W.; Ding, J.D. Biodegradable and thermoreversible PCLA-PEG-PCLA hydrogel as a barrier for prevention of postoperative adhesion. Biomaterials 2011, 32, 4725–4736. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, Y.; Nagata, T.; Fujiwara, S.; Takai, S.; Jin, D.; Kuzuya, A.; Ohya, Y. Postoperative adhesion prevention using a biodegradable temperature-responsive injectable polymer system and concomitant effects of chymase inhibitor. ACS Appl. Bio Mater. 2021, 4, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Choe, J.W.; Kwon, G.Y.; Cho, D.Y.; Sohn, D.S.; Kim, S.W.; Woo, Y.C.; Lee, C.J.; Kang, H. The effects of barrier materials on reduction of pericardial adhesion formation in rabbits: A comparative study of a hyaluronan-based solution and a temperature sensitive poloxamer solution/gel material. J. Surg. Res. 2011, 166, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Xu, W.; Shen, W.J.; Cao, L.P.; Liu, Y.; Li, Z.S.; Ding, J. Poly(lactic acid-co-glycolic acid)-poly(ethylene glycol)-poly(lactic acid-co-glycolic acid) thermogel as a novel submucosal cushion for endoscopic submucosal dissection. Acta Biomater. 2014, 10, 1251–1258. [Google Scholar] [CrossRef]
- Kan, P.; Lin, X.Z.; Hsieh, M.F.; Chang, K.Y. Thermogelling emulsions for vascular embolization and sustained release of drugs. J. Biomed. Mater. Res. B 2005, 75, 185–192. [Google Scholar] [CrossRef]
- Chen, X.L.; Huang, L.; Sun, H.J.; Cheng, S.Z.D.; Zhu, M.Q.; Yang, G. Stimuli-responsive nanocomposite: Potential injectable embolization Agent. Macromol. Rapid Commun. 2014, 35, 579–584. [Google Scholar] [CrossRef]
- Weng, L.H.; Rostambeigi, N.; Zantek, N.D.; Rostamzadeh, P.; Bravo, M.; Carey, J. An in situ forming biodegradable hydrogel-based embolic agent for interventional therapies. Acta Biomater. 2013, 9, 8182–8191. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Lee, D.S.; Kim, S.W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388, 860–862. [Google Scholar] [CrossRef]
- Yoshida, Y.; Takahashi, A.; Kuzuya, A.; Ohya, Y. Instant preparation of a biodegradable injectable polymer formulation exhibiting a temperature-responsive sol-gel transition. Polym. J. 2014, 46, 632–635. [Google Scholar] [CrossRef]
- Shim, W.S.; Kim, S.W.; Lee, D.S. Sulfonamide-based pH- and temperature-sensitive biodegradable block copolymer hydrogels. Biomacromolecules 2006, 7, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk Wolthuis, W.N.E.; Hoogeboom, J.A.M.; van Steenbergen, M.J.; Tsang, S.K.Y.; Hennink, W.E. Degradation and release behavior of dextran-based hydrogels. Macromolecules 1997, 30, 4639–4645. [Google Scholar] [CrossRef]
- Mellott, M.B.; Searcy, K.; Pishko, M.V. Release of protein from highly cross-linked hydrogels of poly(ethylene glycol) diacrylate fabricated by UV polymerization. Biomaterials 2001, 22, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Shin, Y.M.; Joung, Y.K.; Shin, H.; Park, K.D. In situ forming hydrogels based on tyramine conjugated 4-arm-PPO-PEO via enzymatic oxidative reaction. Biomacromolecules 2010, 11, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.P.; Chu, C.R.; Payne, K.A.; Marra, K.G. Injectable in situ forming biodegradable chitosan-hyaluronic acid based hydrogels for cartilage tissue enjineering. Biomaterials 2009, 30, 2499–2506. [Google Scholar] [CrossRef]
- Phelps, E.A.; Enemchukwu, N.O.; Fiore, V.F.; Sy, J.C.; Murthy, N.; Sulchek, T.A. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv. Mater. 2012, 24, 64–70. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Schwartz, M.P.; Halevi, A.E.; Nuttelman, C.R.; Bowman, C.N.; Anseth, K.S. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv. Mater. 2009, 21, 5005–5010. [Google Scholar] [CrossRef]
- DeForest, C.A.; Polizzotti, B.D.; Anseth, K.S. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater. 2009, 8, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cao, X.D.; Li, Y.L.; Zeng, L.; Yuan, B.; Chen, X.F. An injectable hyaluronic acid/PEG hydrogel for cartilage tissue engineering formed by integrating enzymatic cross-linking and Diels-Alder “click chemistry”. Polym. Chem. 2014, 5, 1082–1090. [Google Scholar] [CrossRef]
- Ikada, Y.; Jamshidi, K.; Tsuji, H.; Hyon, S.-H. Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 1987, 20, 904–906. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y. Stereocomplex formation between enatiomeric poly(lactic acid)s. 6. Binary blends from copolymers. Macromolecules 1992, 25, 5719–5723. [Google Scholar] [CrossRef]
- Tsuji, H.; Tamai, K.; Kimura, K.; Kubota, A.; Takahashi, A.; Kuzuya, A.; Ohya, Y. Stereocomplex- and homo-crystallization of blends from 2-armed poly(l-lactide) and poly(d-lactide) with identical and opposite chain directional architectures and of 2-armed stereo diblock poly(lactide). Polymer 2016, 96, 167–181. [Google Scholar] [CrossRef]
- DeSantis, P.; Kovacs, A.J. Molecular conformation of poly(S-laictide). Biopolymers 1968, 6, 209–306. [Google Scholar]
- Okihara, T.; Tsuji, M.; Kawaguchi, A.; Katayama, K.; Tsuji, H.; Hyon, S.-H.; Ikada, Y. Crystal structure of sterocomplex of poly(l-alanine-lactide) and poly(d-lactide). J. Macromol. Sci. Part B Phys. 1991, 30, 119–140. [Google Scholar] [CrossRef]
- De Jong, S.J.; De Smedt, S.C.; Wahls, M.W.C.; Demeester, J.; Kettenes-van den Bosch, J.J.; Hennink, W.E. Novel self-assembled hydrogels by stereocomplex formation in aqueous solution of enantiomeric lactic acid oligomers grafted to dextran. Macromolecules 2000, 33, 3680–3686. [Google Scholar] [CrossRef]
- De Jong, S.J.; van Eerdenbrugh, B.; van Nostrum, C.F.; Kettenes-van de Bosch, J.J.; Hennink, W.E. Physically cross-linked dextran hydrogels by stereocomplex formation of lactic acid oligomers: Degradation and protein release behavior. J. Control. Release 2001, 71, 261–275. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.J.; De Smedt, S.C.; Demeester, J.; van Nostrum, C.F.; Kettenes-van den Bosch, J.J.; Hennink, W.E. Biodegradable hydrogels based on stereocomplex formation between lactic acid oligomers grafted to dextran. J. Control. Release 2001, 72, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Mukose, T.; Yamaoka, T.; Yamane, H.; Sakurai, S.; Kimura, Y. Novel thermo-responsive formation of a hydrogel by stereo-complexation between PLLA-PEG-PLLA and PDLA-PEG-PDLA block copolymers. Macromol. Biosci. 2001, 1, 204–208. [Google Scholar] [CrossRef]
- Slager, J.; Domb, A.J. Biopolymer stereocomplexes. Adv. Drug Deliv. Rev. 2003, 55, 549–583. [Google Scholar] [CrossRef]
- Tsuji, H. Poly(lactide) stereocomplexes: Formation, structure, properties, degradation, and applications. Macromol. Biosci. 2005, 5, 569–597. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code Name | Mn of PEG (g/mol) | Mn of PL (D) LA (g/mol) 1 | DP of PL(D)LA 1 | Mw/Mn 2 | |
|---|---|---|---|---|---|
| MeO-PEG-b-PDLA | b-D | 1000 | 2800 | 19 | 1.09 |
| MeO-PEG-b-PLLA | b-L | 1000 | 2900 | 20 | 1.11 |
| PLLA-b-PEG-b-PLLA | tri-L | 4600 | 1200 | 8.1 | 1.07 |
| Code Name | Mn × 10−4 (g/mol) 1 | Mw/Mn 2 | No. of Side Chains 3 | |
|---|---|---|---|---|
| Dex-g-(PLLA-b-PEG) | gb-L | 16.6 | 2.80 | 4.0 |
| Dex-g-(PDLA-b-PEG) | gb-D | 17.0 | 2.82 | 5.3 |
| Sample | tri-L (wt%) | gb-D (wt%) | gb-L (wt%) | L/D (Block/Graft) 2 | Sol or Gel | Gelation Time (min) | Storage Modulus after 24 h (Pa) |
|---|---|---|---|---|---|---|---|
| gb-L/gb-D | 0 | 3.6 | 4.4 | 1 | Gel | 51 | 5.0 |
| tri-L/gb-D(0.5) | 0.5 | 8.0 | 0 | 0.5 | Gel | 300 | 8.4 |
| tri-L/gb-D(1) | 1.0 | 8.0 | 0 | 1 | Gel | 30 | 51.6 |
| tri-L/gb-D(2) | 2.0 | 8.0 | 0 | 2 | Gel | <1 | 105.0 |
| tri-L/gb-D(5) | 5.0 | 8.0 | 0 | 5 | Gel | <1 | 92.3 |
| tri-L/gb-L(5) | 5.0 | 0 | 10.1 | 0 (5) | Sol | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohya, Y.; Yoshida, Y.; Kumagae, T.; Kuzuya, A. Gelation upon the Mixing of Amphiphilic Graft and Triblock Copolymers Containing Enantiomeric Polylactide Segments through Stereocomplex Formation. Gels 2024, 10, 139. https://doi.org/10.3390/gels10020139
Ohya Y, Yoshida Y, Kumagae T, Kuzuya A. Gelation upon the Mixing of Amphiphilic Graft and Triblock Copolymers Containing Enantiomeric Polylactide Segments through Stereocomplex Formation. Gels. 2024; 10(2):139. https://doi.org/10.3390/gels10020139
Chicago/Turabian StyleOhya, Yuichi, Yasuyuki Yoshida, Taiki Kumagae, and Akinori Kuzuya. 2024. "Gelation upon the Mixing of Amphiphilic Graft and Triblock Copolymers Containing Enantiomeric Polylactide Segments through Stereocomplex Formation" Gels 10, no. 2: 139. https://doi.org/10.3390/gels10020139