

Effect of Blood Flow on Cardiac Morphogenesis and Formation of Congenital Heart Defects
 , and
, and
Abstract
:1. Introduction
2. Heart Development
2.1. Heart Tube Formation
2.2. Heart Looping
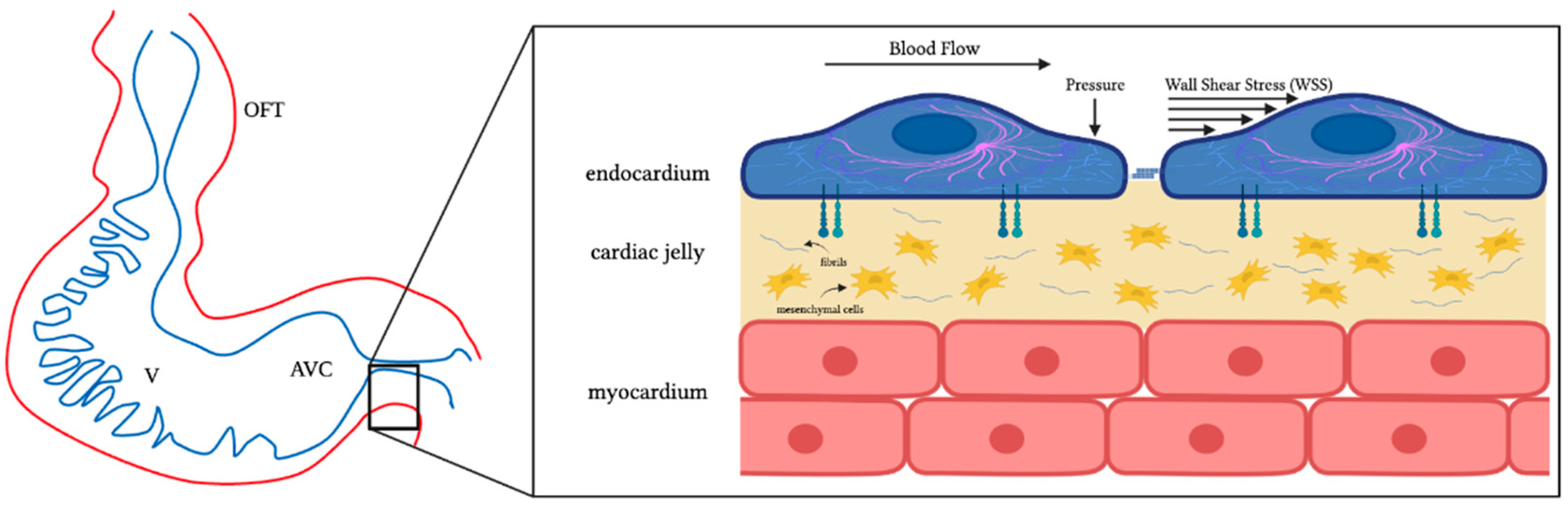
2.3. Formation and Maturation of Endocardial Cushions
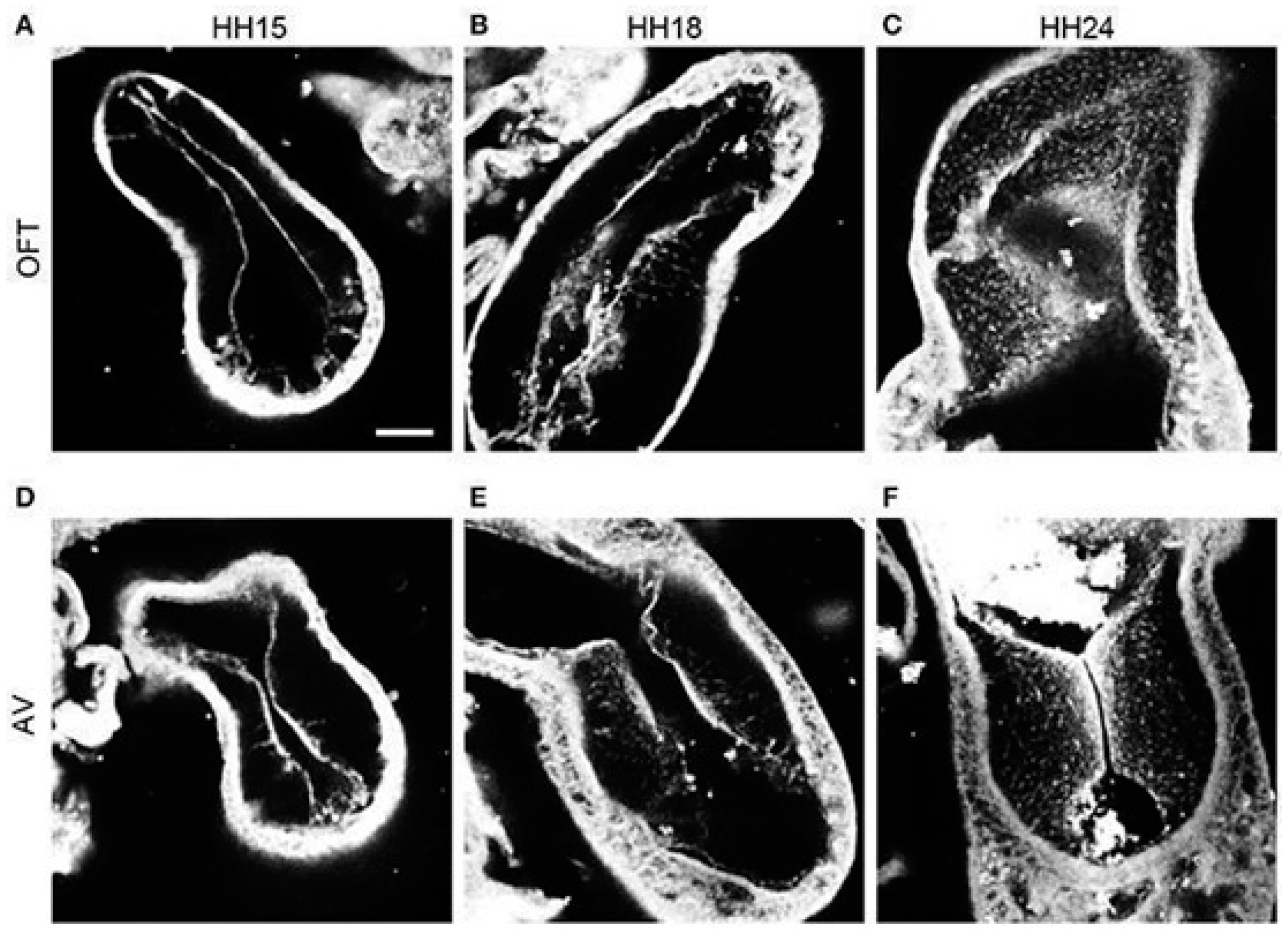
2.4. Trabeculation and Ventricular Formation
2.5. Atrial and Ventricular Septation
2.6. Alignment and Septation of the Outflow Tract
2.7. Contributon of Extra-Cardiac Cells
2.8. Conduction System
2.9. Congenital Heart Malformations
3. Animal Models of Heart Development
4. Blood Flow during Cardiac Development
4.1. Hemodynamic Stresses and Mechanotransduction

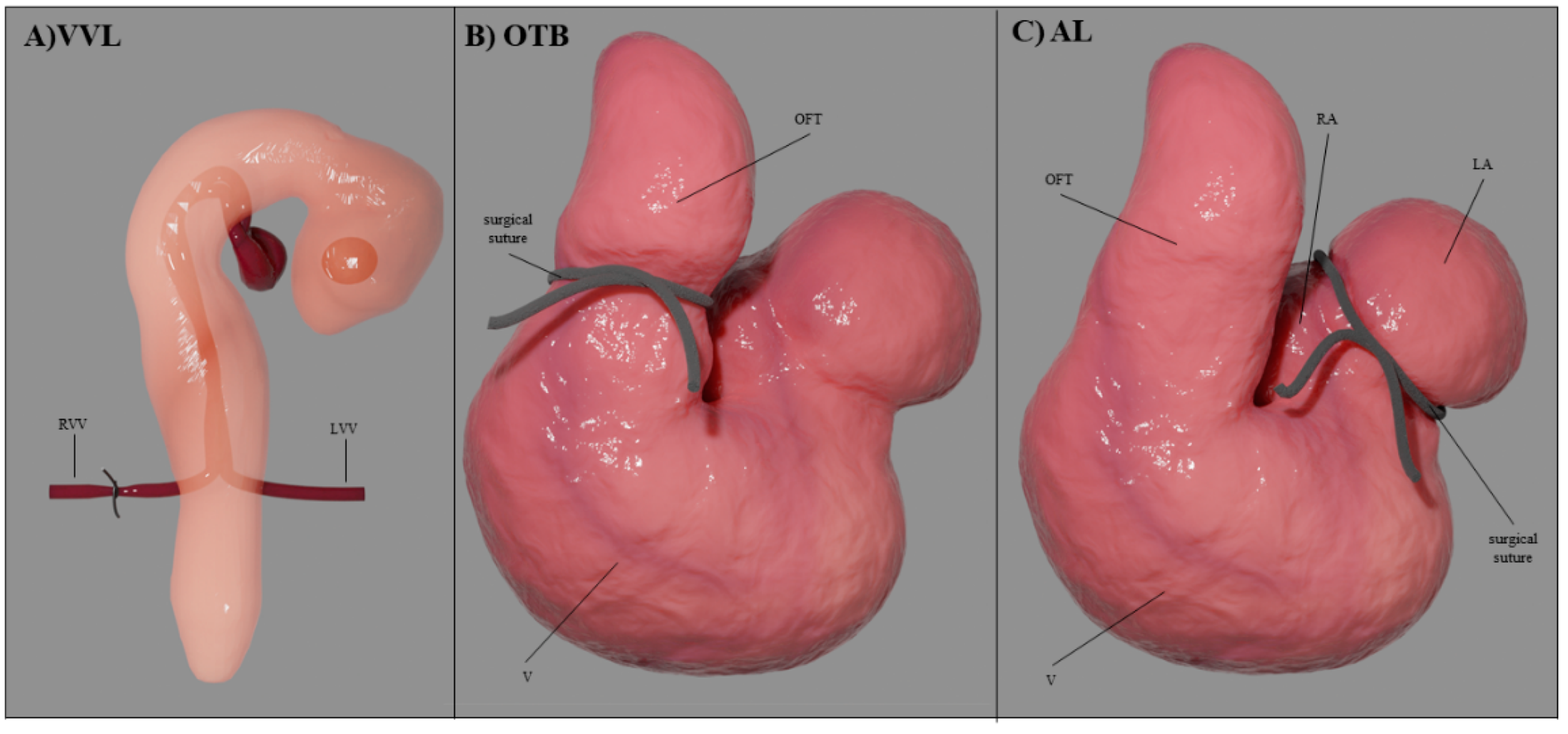
4.2. Hemodynamic Interventions in Avian Models
4.3. Computational Modeling and Analysis of Heart Hemodynamics
4.3.1. Modeling Hemodynamics during Cardiac Cushion Formation
4.3.2. Modeling the Septation Stages
5. Effect of Hemodynamics on Cardiac Development and Formation
5.1. Formation of Endocardial Cushions
5.2. Ventricular Formation
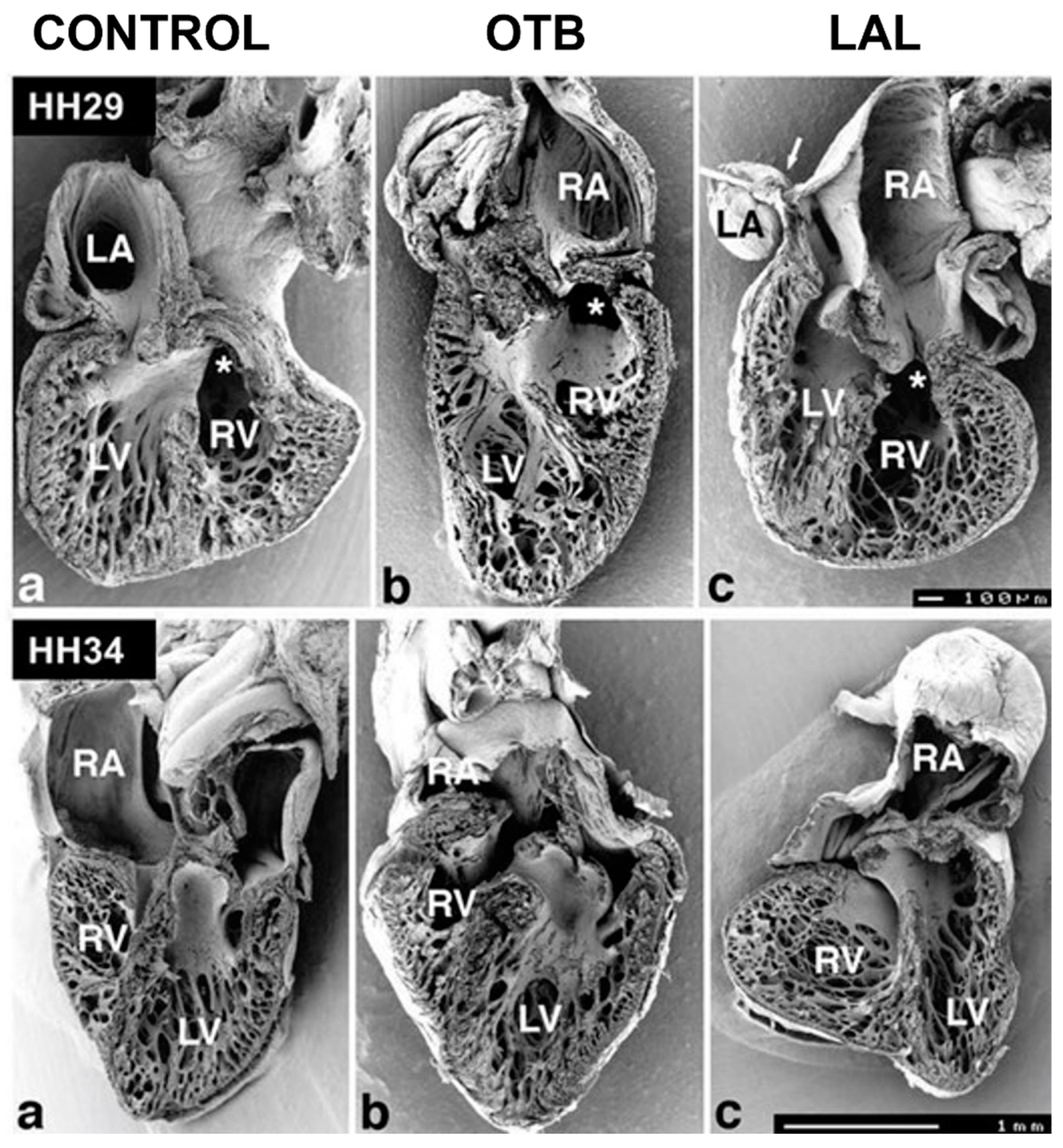

5.3. Outflow Tract and Great Vessels
5.4. Development of the Conduction System
6. Conclusions and Perspectives for Future Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Grouping | Acronym | Term |
| Cardiovascular Biology | RBC | red blood cells |
| ECM | extracellular matrix | |
| OFT | outflow tract | |
| IVS | interventricular septum | |
| AVC | atrioventricular canal | |
| APS | aorticopulmonary septum | |
| PAA | pharyngeal arch artery | |
| Endo-MT | endocardial-to-mesenchymal transition | |
| EMT | epithelial-to-mesenchymal transition | |
| HH | Hamburger–Hamilton staging | |
| Heart Conduction | HSP | His–Purkinje system |
| AV | atrioventricular node | |
| Congenital Heart Defects | CHD | congenital heart disease |
| HLHS | hypoplastic left heart syndrome | |
| CoA | coarctation of the aorta | |
| d-TGA | d-transposition of the great arteries | |
| VSD | ventricular septal defect | |
| DORV | double outlet right ventricle | |
| APW | aorticopulmonary window | |
| TOF | tetralogy of Fallout | |
| Fluid Dynamics | WSS | wall shear stress |
| P | pressure | |
| ΔP | change in pressure | |
| Q | volume flow rate | |
| FSI | fluid structure interaction | |
| CFD | computational fluid dynamics | |
| Hemodynamic Interventions | OTB | outflow tract banding |
| VVL | vitelline vein ligation | |
| AL (LAL or RAL) | (left/right) atrial ligation |
References
- Gittenberger-de Groot, A.C.; Bartelings, M.M.; Deruiter, M.C.; Poelmann, R.E. Basics of Cardiac Development for the Understanding of Congenital Heart Malformations. Pediatr. Res. 2005, 57, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L. Cardiac Development; Oxford University Press: New York, NY, USA., 2007. [Google Scholar]
- Alser, M.; Shurbaji, S.; Yalcin, H.C. Mechanosensitive Pathways in Heart Development: Findings from Chick Embryo Studies. J. Cardiovasc. Dev. Dis. 2021, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Celik, M.; Goktas, S.; Karakaya, C.; Cakiroglu, A.I.; Karahuseyinoglu, S.; Lashkarinia, S.S.; Ermek, E.; Pekkan, K. Microstructure of early embryonic aortic arch and its reversibility following mechanically altered hemodynamic load release. Am. J. Physiol.-Heart Circ. Physiol. 2020, 318, H1208–H1218. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, B.C.W.; Hierck, B.P.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Development-related changes in the expression of shear stress responsive genesKLF-2, ET-1, andNOS-3 in the developing cardiovascular system of chicken embryos. Dev. Dyn. 2004, 230, 57–68. [Google Scholar] [CrossRef]
- Dyer, L.A.; Rugonyi, S. Fetal Blood Flow and Genetic Mutations in Conotruncal Congenital Heart Disease. J. Cardiovasc. Dev. Dis. 2021, 8, 90. [Google Scholar] [CrossRef]
- Granados-Riveron, J.T.; Brook, J.D. Formation, Contraction, and Mechanotransduction of Myofribrils in Cardiac Development: Clues from Genetics. Biochem. Res. Int. 2012, 2012, 504906. [Google Scholar] [CrossRef]
- Hove, J.R.; Köster, R.W.; Forouhar, A.S.; Acevedo-Bolton, G.; Fraser, S.E.; Gharib, M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 2003, 421, 172–177. [Google Scholar] [CrossRef]
- Midgett, M.; Rugonyi, S. Congenital heart malformations induced by hemodynamic altering surgical interventions. Front. Physiol. 2014, 5, 287. [Google Scholar] [CrossRef]
- Midgett, M.; Thornburg, K.; Rugonyi, S. Blood flow patterns underlie developmental heart defects. Am. J. Physiol. Circ. Physiol. 2017, 312, H632–H642. [Google Scholar] [CrossRef]
- Deruiter, M.C.; Poelmann, R.E.; Vanderplas-De Vries, I.; Mentink, M.M.T.; Gittenberger-De Groot, A.C. The development of the myoeardium and endoeardium in mouse embryos Fusion of two heart tubes? Anat. Embryol. 1992, 185, 461–473. [Google Scholar] [CrossRef]
- Ivanovitch, K.; Esteban, I.; Torres, M. Growth and Morphogenesis during Early Heart Development in Amniotes. J. Cardiovasc. Dev. Dis. 2017, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Stalsberg, H.; DeHaan, R.L. The Precardiac Areas and Formation of the Tubular Heart in the Chick Embryo. Dev. Biol. 1968, 19, 128–159. [Google Scholar] [CrossRef]
- Sugi, Y.; Markwald, R.R. Formation and Early Morphogenesis of Endocardial Endothelial Precursor Cells and the Role of Endoderm. Dev. Biol. 1996, 175, 66–83. [Google Scholar] [CrossRef]
- Abu-Issa, R.; Kirby, M.L. Heart field: From mesoderm to heart tube. Annu. Rev. Cell Dev. Biol. 2007, 23, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Männer, J.; Yelbuz, T.M. Functional morphology of the cardiac jelly in the tubular heart of vertebrate embryos. J. Cardiovasc. Dev. Dis. 2019, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Männer, J. Cardiac Looping in the Chick Embryo: A Morphological Review with Special Reference to Terminological and Biomechanical Aspects of the Looping Process. Anat. Rec. 2000, 259, 248–262. [Google Scholar] [CrossRef]
- Männer, J. The anatomy of cardiac looping: A step towards the understanding of the morphogenesis of several forms of congenital cardiac malformations. Clin. Anat. 2009, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Waldo, K.L.; Kumiski, D.H.; Wallis, K.T.; Stadt, H.A.; Hutson, M.R.; Platt, D.H.; Kirby, M.L. Conotruncal myocardium arises from a secondary heart field. Development 2001, 128, 3179–3188. [Google Scholar] [CrossRef] [PubMed]
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90. [Google Scholar] [CrossRef]
- Person, A.D.; Klewer, S.E.; Runyan, R.B. Cell Biology of Cardiac Cushion Development. Int. Rev. Cytol. 2005, 243, 287–335. [Google Scholar]
- Midgett, M.; Chivukula, V.K.; Dorn, C.; Wallace, S.; Rugonyi, S. Blood flow through the embryonic heart outflow tract during cardiac looping in HH13–HH18 chicken embryos. J. R. Soc. Interface 2015, 12, 20150652. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, M.G.; Fitzharris Timothy, P. Origin of cushion tissue in the developing chick heart: Cinematographic recordings of in situ formation. Science 1980, 207, 1359–1360. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Lin, C.Y.; Chen, C.H.; Zhou, B.; Chang, C.P. Partitioning the heart: Mechanisms of cardiac septation and valve development. Development 2012, 139, 3277–3299. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, B.P.T.; Krawitz, S.A.; Gaussin, V. Atrioventricular valve development during late embryonic and postnatal stages involves condensation and extracellular matrix remodeling. Dev. Biol. 2007, 302, 208–217. [Google Scholar] [CrossRef]
- Goddard, L.M.; Duchemin, A.-L.; Ramalingan, H.; Wu, B.; Chen, M.; Bamezai, S.; Yang, J.; Li, L.; Morley, M.P.; Wang, T.; et al. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Dev. Cell 2017, 43, 274–289.e5. [Google Scholar] [CrossRef]
- Midgett, M.; López, C.S.; David, L.; Maloyan, A.; Rugonyi, S. Increased Hemodynamic Load in Early Embryonic Stages Alters Endocardial to Mesenchymal Transition. Front. Physiol. 2017, 8, 56. [Google Scholar] [CrossRef]
- Wu, M. Mechanisms of Trabecular Formation and Specification During Cardiogenesis. Pediatr. Cardiol. 2018, 39, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, D.; McQuinn, T. Embryogenesis of the Heart Muscle. Heart. Fail. Clin. 2008, 4, 235–245. [Google Scholar] [CrossRef]
- Sedmera, D.; Pexieder, T.; Vuillemin, M.; Thompson, R.P.; Anderson, R.H. Developmental Patterning of the Myocardium. Anat. Rec. 2000, 258, 319–337. [Google Scholar] [CrossRef]
- Waldo, K.; Miyagawa-Tomita, S.; Kumiski, D.; Kirby, M.L. Cardiac Neural Crest Cells Provide New Insight into Septation of the Cardiac Outflow Tract: Aortic Sac to Ventricular Septal Closure. Dev. Biol. 1998, 196, 129–144. [Google Scholar] [CrossRef]
- Tang, W.; Martik, M.L.; Li, Y.; Bronner, M.E. Cardiac neural crest contributes to cardiomyocytes in amniotes and heart regeneration in zebrafish. Elife 2019, 8, e47929. [Google Scholar] [CrossRef] [PubMed]
- Quijada, P.; Trembley, M.A.; Small, E.M. The Role of the Epicardium during Heart Development and Repair. Circ. Res. 2020, 126, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Vigmond, E.J.; Stuyvers, B.D. Modeling our understanding of the His-Purkinje system. Prog. Biophys. Mol. Biol. 2016, 120, 179–188. [Google Scholar] [CrossRef]
- Reckova, M.; Rosengarten, C.; Dealmeida, A.; Stanley, C.P.; Wessels, A.; Gourdie, R.G.; Thompson, R.P.; Sedmera, D. Hemodynamics is a key epigenetic factor in development of the cardiac conduction system. Circ. Res. 2003, 93, 77–85. [Google Scholar] [CrossRef]
- Pierpont, M.E.M.; Moller, J.H. The Genetics of Cardiovascular Disease; Martinus Nijhoff Publishing: Boston, MA, USA, 1987. [Google Scholar]
- Kheradvar, A.; Zareian, R.; Kawauchi, S.; Goodwin, R.L.; Rugonyi, S. Animal Models for Heart Valve Research and Development. Drug Discov. Today Dis. Models 2017, 24, 55–62. [Google Scholar] [CrossRef]
- Pinson, C.W.; Morton, M.J.; Thornburg, K.L. Mild pressure loading alters right ventricular function in fetal sheep. Circ. Res. 1991, 68, 947–957. [Google Scholar] [CrossRef]
- O’Tierney, P.F.; Anderson, D.F.; Faber, J.J.; Louey, S.; Thornburg, K.L.; Giraud, G.D. Reduced systolic pressure load decreases cell-cycle activity in the fetal sheep heart. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 299, R573–R578. [Google Scholar] [CrossRef]
- Reller, M.D.; Morton, M.J.; Giraud, G.D.; Wu, D.E.; Thornburg, K.L. Severe right ventricular pressure loading in fetal sheep augments global myocardial blood flow to submaximal levels. Circulation 1992, 86, 581–588. [Google Scholar] [CrossRef]
- Edwards, A.; Menahem, S.; Veldman, A.; Schranz, D.; Chan, Y.; Nitsos, I.; Wong, F. Fetal cardiac catheterization using a percutaneous transhepatic access technique: Preliminary experience in a lamb model. Ultrasound Obstet. Gynecol. 2013, 42, 58–63. [Google Scholar] [CrossRef]
- Wong, F.Y.; Veldman, A.; Sasi, A.; Teoh, M.; Edwards, A.; Chan, Y.; Graupner, O.; Enzensberger, C.; Axt-Fliedner, R.; Black, M.J.; et al. Induction of left ventricular hypoplasia by occluding the foramen ovale in the fetal lamb. Sci. Rep. 2020, 10, 880. [Google Scholar] [CrossRef]
- Liu, K.; Yu, W.; Tang, M.; Tang, J.; Liu, X.; Liu, Q.; Li, Y.; He, L.; Zhang, L.; Evans, S.M.; et al. Dual genetic tracing system identifies diverse and dynamic origins of cardiac valve mesenchyme. Development 2018, 145, dev167775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleaver, O. Mouse models of vascular development and disease. Curr. Opin. Hematol. 2021, 28, 179–188. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.E.; Tzima, E. Pulling on my heartstrings. Curr. Opin. Hematol. 2016, 23, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, D. Pathways to embryonic heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1578–H1579. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lopez, A.; Larin, K.V.; Overbeek, P.; Larina, I.V. Live four-dimensional optical coherence tomography reveals embryonic cardiac phenotype in mouse mutant. J. Biomed. Opt. 2015, 20, 090501. [Google Scholar] [CrossRef]
- Hoog, T.G.; Fredrickson, S.J.; Hsu, C.-W.; Senger, S.M.; Dickinson, M.E.; Udan, R.S. The effects of reduced hemodynamic loading on morphogenesis of the mouse embryonic heart. Dev. Biol. 2018, 442, 127–137. [Google Scholar] [CrossRef]
- Rödel, C.J.; Abdelilah-Seyfried, S. A zebrafish toolbox for biomechanical signaling in cardiovascular development and disease. Curr. Opin. Hematol. 2021, 28, 198–207. [Google Scholar] [CrossRef]
- Vedula, V.; Lee, J.; Xu, H.; Kuo, C.-C.J.; Hsiai, T.K.; Marsden, A.L. A method to quantify mechanobiologic forces during zebrafish cardiac development using 4-D light sheet imaging and computational modeling. PLoS Comput. Biol. 2017, 13, e1005828. [Google Scholar] [CrossRef]
- Hsu, J.J.; Vedula, V.; Baek, K.I.; Chen, C.; Chen, J.; Chou, M.I.; Lam, J.; Subhedar, S.; Wang, J.; Ding, Y.; et al. Contractile and hemodynamic forces coordinate Notch1b-mediated outflow tract valve formation. JCI Insight 2019, 4, e124460. [Google Scholar] [CrossRef]
- Steed, E.; Faggianelli, N.; Roth, S.; Ramspacher, C.; Concordet, J.-P.; Vermot, J. klf2a couples mechanotransduction and zebrafish valve morphogenesis through fibronectin synthesis. Nat. Commun. 2016, 7, 11646. [Google Scholar] [CrossRef]
- Bulk, A.; Bark, D.; Johnson, B.; Garrity, D.; Dasi, L.P. Mechanisms influencing retrograde flow in the atrioventricular canal during early embryonic cardiogenesis. J. Biomech. 2016, 49, 3162–3167. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.; Eberth, J.; Goodwin, R.; Potts, J. Altered Hemodynamics in the Embryonic Heart Affects Outflow Valve Development. J. Cardiovasc. Dev. Dis. 2015, 2, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.R.; Ware, S.M. Genetics and Genetic Testing in Congenital Heart Disease. Clin. Perinatol. 2015, 42, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Delgado, L.; Mercader, N. Interplay between cardiac function and heart development. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 1707–1716. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Zhang, Y.-L.; Frangos, J.A. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15463–15468. [Google Scholar] [CrossRef]
- Lehoux, S.; Castier, Y.; Tedgui, A. Molecular mechanisms of the vascular responses to haemodynamic forces. J. Intern. Med. 2006, 259, 381–392. [Google Scholar] [CrossRef]
- Makino, A.; Prossnitz, E.R.; Bünemann, M.; Wang, J.M.; Yao, W.; Schmid-Schönbein, G.W. G protein-coupled receptors serve as mechanosensors for fluid shear stress in neutrophils. Am. J. Physiol.-Cell Physiol. 2006, 290, C1633–C1639. [Google Scholar] [CrossRef]
- Aikawa, R.; Nagai, T.; Kudoh, S.; Zou, Y.; Tanaka, M.; Tamura, M.; Akazawa, H.; Takano, H.; Nagai, R.; Komuro, I. Integrins Play a Critical Role in Mechanical Stress–Induced p38 MAPK Activation. Hypertension 2002, 39, 233–238. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.N.; Yan, W.; Chen, K.; Guo, L.; Tummalapali, L.; Dedhar, S.; St-Arnaud, R.; Wu, C.; Sepulveda, J.L. Role of the integrin-linked kinase/PINCH1/alpha-parvin complex in cardiac myocyte hypertrophy. Lab. Investig. 2005, 85, 1342–1356. [Google Scholar] [CrossRef]
- Jacot, J.G.; Martin, J.C.; Hunt, D.L. Mechanobiology of cardiomyocyte development. J. Biomech. 2010, 43, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salameh, A.; Wustmann, A.; Karl, S.; Blanke, K.; Apel, D.; Rojas-Gomez, D.; Franke, H.; Mohr, F.W.; Janousek, J.; Dhein, S. Cyclic Mechanical Stretch Induces Cardiomyocyte Orientation and Polarization of the Gap Junction Protein Connexin43. Circ. Res. 2010, 106, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, V.; Hamilton, H.L. A series of normal stages in the development of the chick embryo. Dev. Dyn. 1992, 195, 231–272. [Google Scholar] [CrossRef] [PubMed]
- Stekelenburg-De Vos, S.; Steendijk, P.; Ursem, N.T.C.; Wladimiroff, J.W.; Delfos, R.; Poelmann, R.E. Systolic and Diastolic Ventricular Function Assessed by Pressure-Volume Loops in the Stage 21 Venous Clipped Chick Embryo. Pediatric. Res. 2005, 57, 16–21. [Google Scholar] [CrossRef]
- Broekhuizen, M.L.A.; Hogers, B.; DeRuiter, M.C.; Poelmann, R.E.; Gittenberger-De Groot, A.C.; Wladimiroff, J.W. Altered hemodynamics in chick embryos after extraembryonic venous obstruction. Ultrasound Obstet. Gynecol. 1999, 13, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.B.; Rosenquist, G.C. Spectrum of cardiovascular anomalies following cardiac loop constriction in the chick embryo. Birth Defects Orig. Artic. Ser. 1978, 14, 431–442. [Google Scholar] [PubMed]
- Keller, B.B.; Yoshigi, M.; Tinney, J.P. Ventricular-vascular uncoupling by acute conotruncal occlusion in the stage 21 chick embryo. Am. J. Physiol.-Heart Circ. Physiol. 1997, 273, H2861–H2866. [Google Scholar] [CrossRef]
- Salman, H.E.; Alser, M.; Shekhar, A.; Gould, R.A.; Benslimane, F.M.; Butcher, J.T.; Yalcin, H.C. Effect of left atrial ligation-driven altered inflow hemodynamics on embryonic heart development: Clues for prenatal progression of hypoplastic left heart syndrome. Biomech. Model. Mechanobiol. 2021, 20, 733–750. [Google Scholar] [CrossRef]
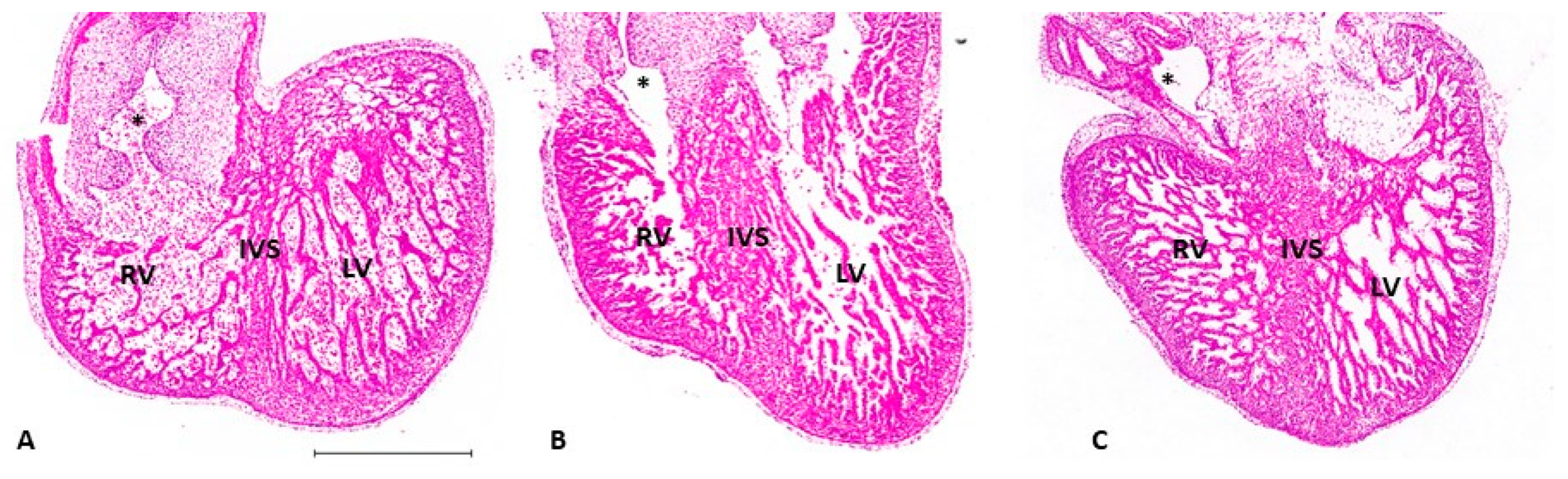
- Sedmera, D.; Pexieder, T.; Rychterova, V.; Hu, N.; Clark, E.B. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat. Rec. 1999, 254, 238–252. [Google Scholar] [CrossRef]
- De Almeida, A.; McQuinn, T.; Sedmera, D. Increased Ventricular Preload Is Compensated by Myocyte Proliferation in Normal and Hypoplastic Fetal Chick Left Ventricle. Circ. Res. 2007, 100, 1363–1368. [Google Scholar] [CrossRef]
- Courchaine, K.; Rugonyi, S. Quantifying blood flow dynamics during cardiac development: Demystifying computational methods. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, S.; Raut, S.S.; Jana, A.; Biederman, R.W.; Doyle, M.; Muluk, S.C.; Finol, E.A. Fluid-Structure Interaction Modeling of Abdominal Aortic Aneurysms: The Impact of Patient-Specific Inflow Conditions and Fluid/Solid Coupling. J. Biomech. Eng. 2013, 135, 81001. [Google Scholar] [CrossRef]
- Salman, H.E.; Yalcin, H.C. Computational Modeling of Blood Flow Hemodynamics for Biomechanical Investigation of Cardiac Development and Disease. J. Cardiovasc. Dev. Dis. 2021, 8, 14. [Google Scholar] [CrossRef]
- Taber, L.A.; Zhang, J.; Perucchio, R. Computational Model for the Transition from Peristaltic to Pulsatile Flow in the Embryonic Heart Tube. J. Biomech. Eng. 2007, 129, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Moghadam, M.E.; Kung, E.; Cao, H.; Beebe, T.; Miller, Y.; Roman, B.; Lien, C.-L.; Chi, N.C.; Marsden, A.; et al. Moving Domain Computational Fluid Dynamics to Interface with an Embryonic Model of Cardiac Morphogenesis. PLoS ONE 2013, 8, e72924. [Google Scholar] [CrossRef] [PubMed]
- Courchaine, K.; Gray, M.; Beel, K.; Thornburg, K.; Rugonyi, S. 4-D Computational Modeling of Cardiac Outflow Tract Hemodynamics over Looping Developmental Stages in Chicken Embryos. J. Cardiovasc. Dev. Dis. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
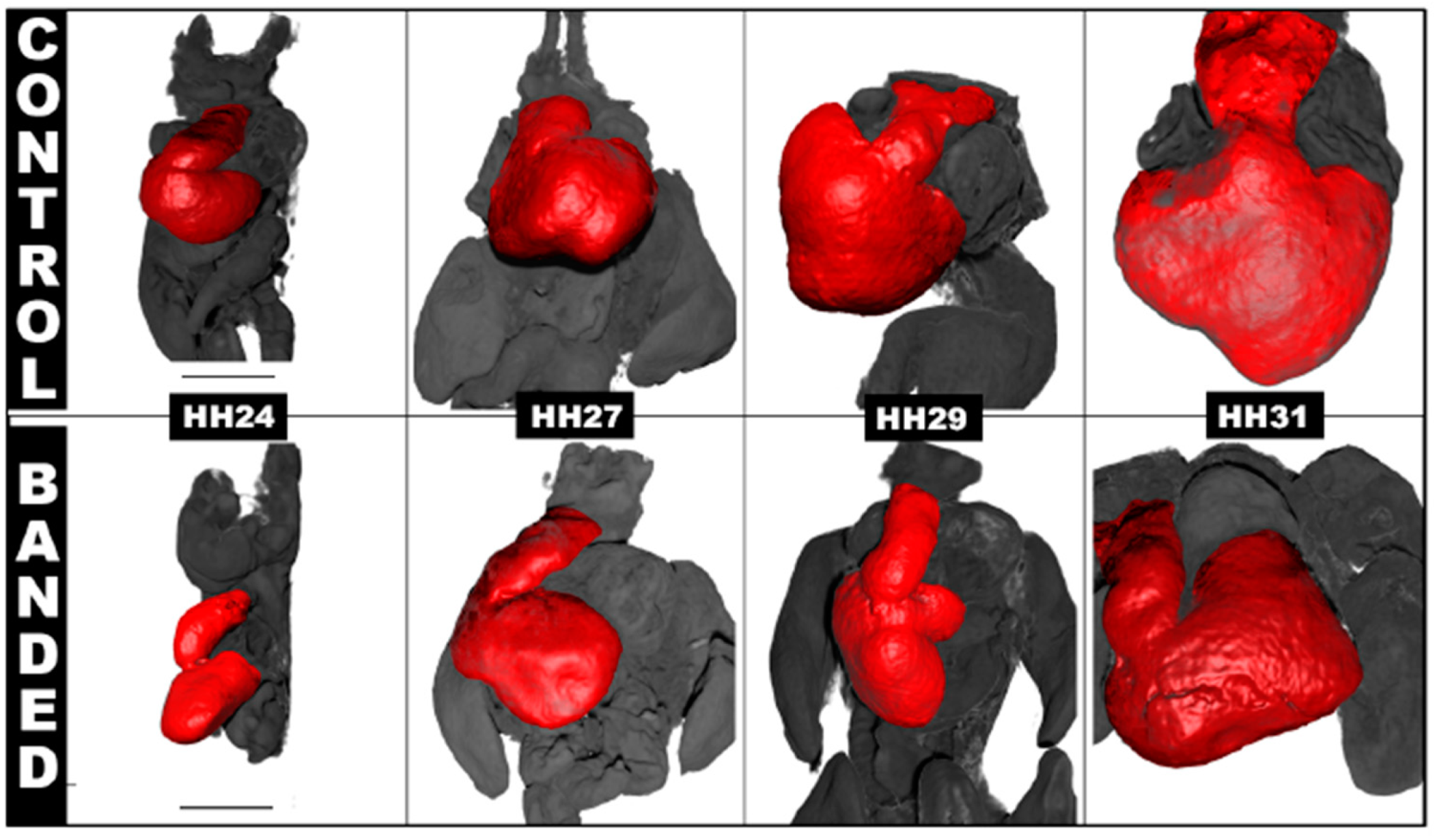
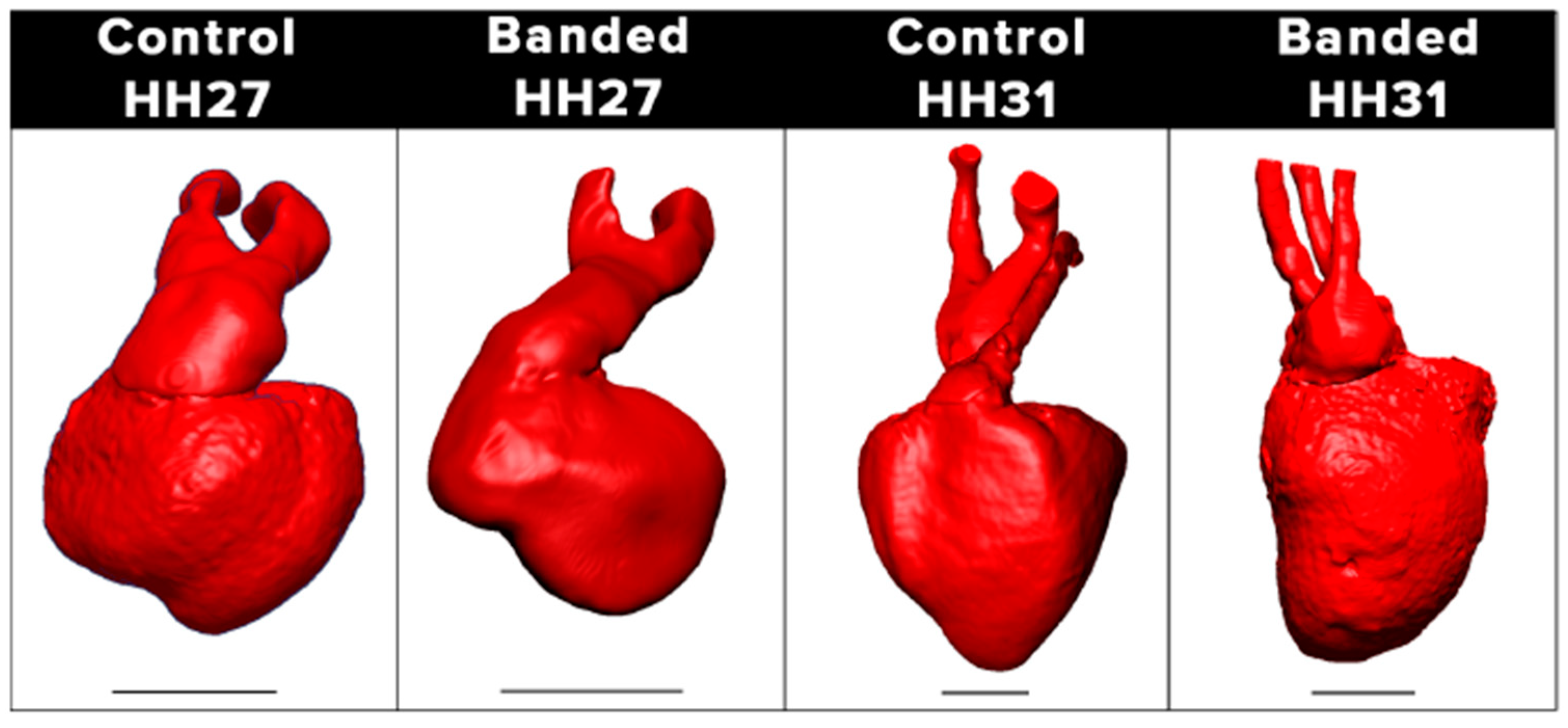
- Chivukula, V.; Goenezen, S.; Liu, A.; Rugonyi, S. Effect of Outflow Tract Banding on Embryonic Cardiac Hemodynamics. J. Cardiovasc. Dev. Dis. 2015, 3, 1. [Google Scholar] [CrossRef]
- Ho, S.; Tan, G.X.Y.; Foo, T.J.; Phan-Thien, N.; Yap, C.H. Organ Dynamics and Fluid Dynamics of the HH25 Chick Embryonic Cardiac Ventricle as Revealed by a Novel 4D High-Frequency Ultrasound Imaging Technique and Computational Flow Simulations. Ann. Biomed. Eng. 2017, 45, 2309–2323. [Google Scholar] [CrossRef]
- Ho, S.; Chan, W.X.; Phan-Thien, N.; Yap, C.H. Organ Dynamics and Hemodynamic of the Whole HH25 Avian Embryonic Heart, Revealed by Ultrasound Biomicroscopy, Boundary Tracking, and Flow Simulations. Sci. Rep. 2019, 9, 18072. [Google Scholar] [CrossRef]
- Wiputra, H.; Lim, G.L.; Chia, D.A.K.; Mattar, C.N.Z.; Biswas, A.; Yap, C.H. Methods for fluid dynamics simulations of human fetal cardiac chambers based on patient-specific 4D ultrasound scans. J. Biomech. Sci. Eng. 2016, 11, 15–00608. [Google Scholar] [CrossRef]
- Bharadwaj, K.N.; Spitz, C.; Shekhar, A.; Yalcin, H.C.; Butcher, J.T. Computational fluid dynamics of developing avian outflow tract heart valves. Ann. Biomed. Eng. 2012, 40, 2212–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgett, M.; Goenezen, S.; Rugonyi, S. Blood flow dynamics reflect degree of outflow tract banding in Hamburger–Hamilton stage 18 chicken embryos. J. R. Soc. Interface 2014, 11, 20140643. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.L.; Parnall, M.; Loughna, S. Effect of altered haemodynamics on the developing mitral valve in chick embryonic heart. J. Mol. Cell Cardiol. 2017, 108, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Rennie, M.Y.; Stovall, S.; Carson, J.P.; Danilchik, M.; Thornburg, K.L.; Rugonyi, S. Hemodynamics modify collagen deposition in the early embryonic chicken heart outflow tract. J. Cardiovasc. Dev. Dis. 2017, 4, 24. [Google Scholar] [CrossRef]
- Clark, E.B.; Frommelt, P.; Vandekieft, G.K.; Dummett, J.L.; Tomanek, R.J.; Hu, N. Effect of increased pressure on ventricular growth in stage 21 chick embryos. Am. J. Physiol. 1989, 257, 55–61. [Google Scholar] [CrossRef]
- Tobita, K.; Schroder, E.A.; Tinney, J.P.; Garrison, J.B.; Keller, B.B. Regional passive ventricular stress-strain relations during development of altered loads in chick embryo. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, 2386–2396. [Google Scholar] [CrossRef]
- Hutchins, G.M.; Bulkley, B.H.; Moore, G.W.; Piasio, M.A.; Lohr, F.T. Shape of the human cardiac ventricles. Am. J. Cardiol. 1978, 41, 646–654. [Google Scholar] [CrossRef]
- Hogers, B.; Gittenberger-De Groot, A.C.; Deruiter, M.C.; Mentink, M.M.T.; Poelmann, R.E. Cardiac inflow malformations are more lethal and precede cardiac outflow malformations. Chick embryonic venous clip model. In The Role of Blood Flow in Normal and Abnormal Heart Development; Ponsen & Looijen BV: Wageningen, The Netherlands, 1998; pp. 79–100. [Google Scholar]
- de Jong, F.; Opthof, T.; Wilde, A.A.; Janse, M.J.; Charles, R.; Lamers, W.H.; Moorman, A.F. Persisting zones of slow impulse conduction in developing chicken hearts. Circ. Res. 1992, 71, 240–250. [Google Scholar] [CrossRef]
- Attin, M.; Clusin, W.T. Basic Concepts of Optical Mapping Techniques in Cardiac Electrophysiology. Biol. Res. Nurs. 2009, 11, 195–207. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Tworetzky, W.; Wilkins-Haug, L.; Jennings, R.W.; Van Der Velde, M.E.; Marshall, A.C.; Marx, G.R.; Colan, S.D.; Benson, C.B.; Lock, J.E.; Perry, S.B. Balloon Dilation of Severe Aortic Stenosis in the Fetus. Circulation 2004, 110, 2125–2131. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.-T.; Marini, D.; Seed, M.; Sun, L. Maternal hyperoxygenation in congenital heart disease. Transl. Pediatr. 2021, 10, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Landis, B.J.; Levey, A.; Levasseur, S.M.; Glickstein, J.S.; Kleinman, C.S.; Simpson, L.L.; Williams, I.A. Prenatal Diagnosis of Congenital Heart Disease and Birth Outcomes. Pediatr. Cardiol. 2013, 34, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.; Glickstein, J.S.; Kleinman, C.S.; Levasseur, S.M.; Chen, J.; Gersony, W.M.; Williams, I.A. The impact of prenatal diagnosis of complex congenital heart disease on neonatal outcomes. Pediatr. Cardiol. 2010, 31, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Holland, B.J.; Myers, J.A.; Woods, C.R. Prenatal diagnosis of critical congenital heart disease reduces risk of death from cardiovascular compromise prior to planned neonatal cardiac surgery: A meta-analysis. Ultrasound Obstet. Gynecol. 2015, 45, 631–638. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hemodynamic Intervention | Stage Performed | Physiological Response |
|---|---|---|
| VVL | HH17-18 | acute ↓Q → ↓stroke volume → ↓WSS |
| OTB | HH17-24 | ↓OFT diameter with ~ constant Q → ↓WSS ↑ventricular blood pressure → ↑contractile force → ↑σ |
| AL | HH21-24 | ↓Q in ventricle (and flow imbalance) → ↓stroke volume → ↓WSS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinidad, F.; Rubonal, F.; Rodriguez de Castro, I.; Pirzadeh, I.; Gerrah, R.; Kheradvar, A.; Rugonyi, S. Effect of Blood Flow on Cardiac Morphogenesis and Formation of Congenital Heart Defects. J. Cardiovasc. Dev. Dis. 2022, 9, 303. https://doi.org/10.3390/jcdd9090303
Trinidad F, Rubonal F, Rodriguez de Castro I, Pirzadeh I, Gerrah R, Kheradvar A, Rugonyi S. Effect of Blood Flow on Cardiac Morphogenesis and Formation of Congenital Heart Defects. Journal of Cardiovascular Development and Disease. 2022; 9(9):303. https://doi.org/10.3390/jcdd9090303
Chicago/Turabian StyleTrinidad, Fernando, Floyd Rubonal, Ignacio Rodriguez de Castro, Ida Pirzadeh, Rabin Gerrah, Arash Kheradvar, and Sandra Rugonyi. 2022. "Effect of Blood Flow on Cardiac Morphogenesis and Formation of Congenital Heart Defects" Journal of Cardiovascular Development and Disease 9, no. 9: 303. https://doi.org/10.3390/jcdd9090303