
Recent Update on PCSK9 and Platelet Activation Experimental Research Methods: In Vitro and In Vivo Studies

 , and
, and
Abstract
:1. Introduction
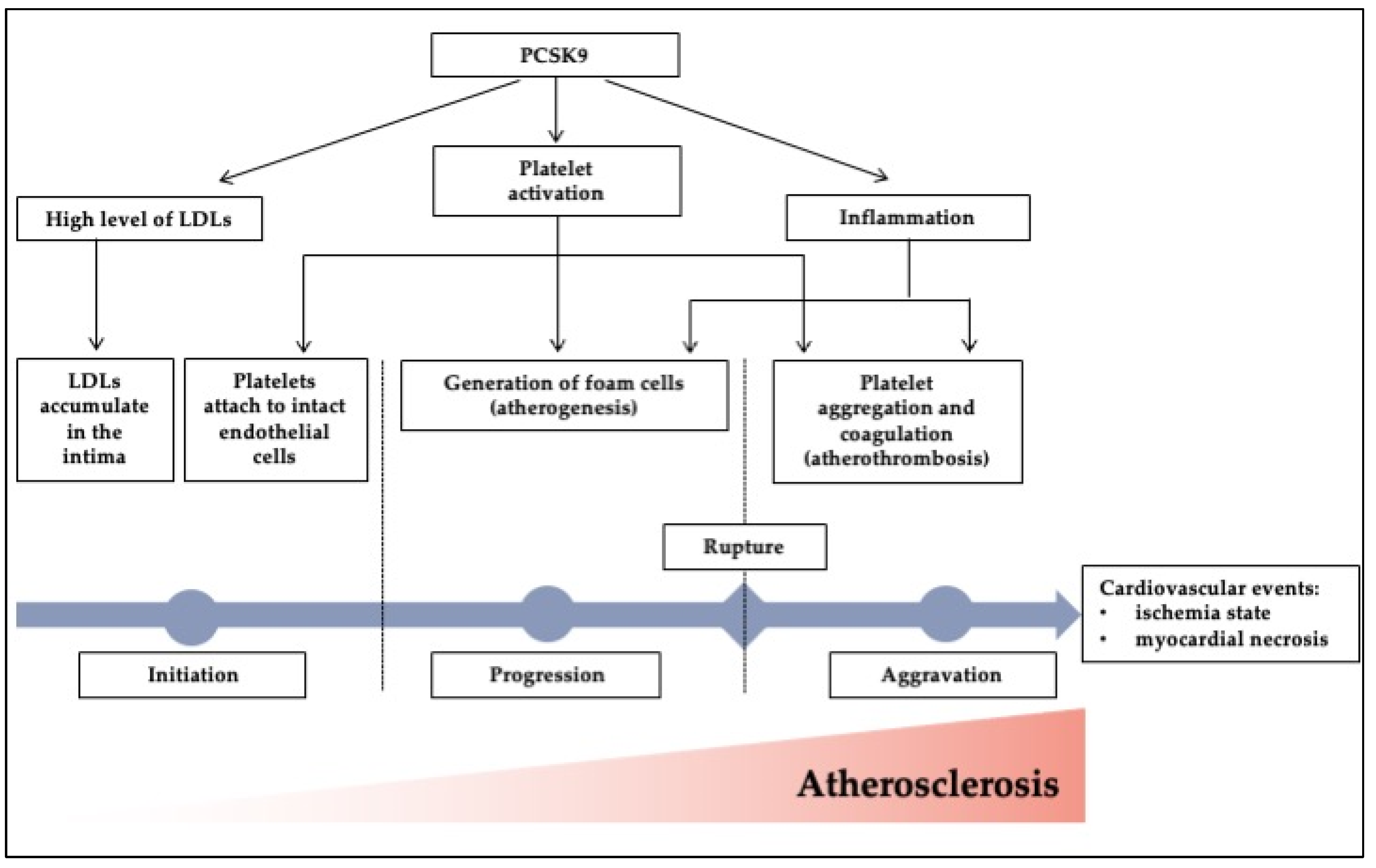
2. Atherosclerosis, Platelet Activation, Myocardial Infarction (MI), and PCSK9
3. In Vitro and In Vivo Model of Study on PCSK9 and Platelet Activation
3.1. In Vitro Studies Using Isolated Platelets Model
3.2. In Vivo Studies Using Animal Models
3.2.1. Mouse
3.2.2. Rabbit
4. Other Potential Model for PCSK9 and Platelet Activation Research
4.1. Mouse Aortic Endothelial Cells (MAEC) and Mouse Cardiac Endothelial Cell (MCEC) Lines
4.2. Mouse
4.3. Rat
4.4. Rabbit
4.5. Porcine
4.6. Non-Human Primate
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andreadou, I.; Tsoumani, M.; Vilahur, G.; Ikonomidis, I.; Badimon, L.; Varga, Z.V.; Ferdinandy, P.; Schulz, R. PCSK9 in Myocardial Infarction and Cardioprotection: Importance of Lipid Metabolism and Inflammation. Front. Physiol. 2020, 11, 602497. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Awan, Z.; Chrétien, M.; Mbikay, M. PCSK9: A key modulator of cardiovascular health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Ouadda, A.B.D.; Spahis, S.; Sane, A.T.; Garofalo, C.; Grenier, É.; Emonnot, L.; Yara, S.; Couture, P.; Beaulieu, J.-F.; et al. PCSK9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis 2013, 227, 297–306. [Google Scholar] [CrossRef]
- Lopez, E.O.; Ballard, B.D.; Jan, A. Cardiovascular Disease; [Updated 2021 Aug 11]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK535419/ (accessed on 15 December 2021).
- Liao, J.; Huang, W.; Liu, G. Animal models of coronary heart disease. J. Biomed. Res. 2015, 30, 3–10. [Google Scholar] [CrossRef]
- Badimon, L.; Padró, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart Journal. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; Copyright © 2022-2021; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Lindemann, S.; Krämer, B.; Seizer, P.; Gawaz, M. Platelets, inflammation and atherosclerosis. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 203–211. [Google Scholar] [CrossRef]
- World Health Organization. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 15 November 2021).
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Abifadel, M.; Rabès, J.-P.; Devillers, M.; Munnich, A.; Erlich, D.; Junien, C.; Varret, M.; Boileau, C. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum. Mutat. 2009, 30, 520–529. [Google Scholar] [CrossRef]
- Seidah, N.G. The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol. Glob. Cardiol. Sci. Pract. 2017, 2017, e201702. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence Variations inPCSK9,Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Cupido, A.J.; Laufs, U. Gene Therapy Targeting PCSK9. Metabolites 2022, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular biology of PCSK9: Its role in LDL metabolism. Trends Biochem. Sci. 2007, 32, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.-C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and Its Natural Mutants: Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004, 279, 48865–48875. [Google Scholar] [CrossRef]
- Cameron, J.; Holla, Ø.L.; Ranheim, T.; Kulseth, M.A.; Berge, K.E.; Leren, T.P. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum. Mol. Genet. 2006, 15, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and inlivers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.D.; Fazio, S. PCSK9 and Atherosclerosis—Lipids and Beyond. J. Atheroscler. Thromb. 2017, 24, 462–472. [Google Scholar] [CrossRef]
- Puteri, M.U.; Azmi, N.U.; Kato, M.; Saputri, F.C. PCSK9 Promotes Cardiovascular Diseases: Recent Evidence about Its Association with Platelet Activation-Induced Myocardial Infarction. Life 2022, 12, 190. [Google Scholar] [CrossRef]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a Scavenger Receptor Involved in Immunity, Metabolism, Angiogenesis, and Behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef]
- Cammisotto, V.; Pastori, D.; Nocella, C.; Bartimoccia, S.; Castellani, V.; Marchese, C.; Scavalli, A.S.; Ettorre, E.; Viceconte, N.; Violi, F.; et al. PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation. Antioxidants 2020, 9, 296. [Google Scholar] [CrossRef]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Sabouri-Rad, S.; Gotto, A.M.; Pirro, M.; Banach, M.; Awan, Z.; Barreto, G.E.; Sahebkar, A. PCSK9 and inflammation: A review of experimental and clinical evidence. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 237–245. [Google Scholar] [CrossRef]
- Petersen-Uribe, A.; Kremser, M.; Rohlfing, A.-K.; Castor, T.; Kolb, K.; Dicenta, V.; Emschermann, F.; Li, B.; Borst, O.; Rath, D.; et al. Platelet-Derived PCSK9 Is Associated with LDL Metabolism and Modulates Atherothrombotic Mechanisms in Coronary Artery Disease. Int. J. Mol. Sci. 2021, 22, 11179. [Google Scholar] [CrossRef] [PubMed]
- Navarese, E.P.; Kolodziejczak, M.; Winter, M.-P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Bradwin, G.; Rose, L.M. Plasma proprotein convertase subtilisin/kexin type 9 levels and the risk of first cardiovascular events. Eur. Heart J. 2016, 37, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Bryda, E.C. The Mighty Mouse: The impact of rodents on advances in biomedical research. Mo. Med. 2013, 110, 207–211. [Google Scholar] [PubMed]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef]
- Camera, M.; Rossetti, L.; Barbieri, S.S.; Zanotti, I.; Canciani, B.; Trabattoni, D.; Ruscica, M.; Tremoli, E.; Ferri, N. PCSK9 as a Positive Modulator of Platelet Activation. J. Am. Coll. Cardiol. 2018, 71, 952–954. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Grin, P.M.; Khan, M.; Prat, A.; Zhou, J.; Fox-Robichaud, A.E.; Seidah, N.G.; Liaw, P.C. Differential Expression of PCSK9 Modulates Infection, Inflammation, and Coagulation in a Murine Model of Sepsis. Shock 2016, 46, 672–680. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Q.; Wang, J.; Guo, C.; Kleiman, K.; Meng, H.; Knight, J.S.; Eitzman, D.T. Proprotein convertase subtilisin/kexin type 9 (PCSK9) Deficiency is Protective Against Venous Thrombosis in Mice. Sci. Rep. 2017, 7, 14360. [Google Scholar] [CrossRef]
- Halim, S.A.; Ghafar, N.A.; Jubri, Z.; Das, S. Induction of Myocardial Infarction in Experimental Animals: A Review. J. Clin. Diagnostic Res. 2018, 12, AE01–AE05. [Google Scholar] [CrossRef]
- Wang, J.; Bo, H.; Meng, X.; Wu, Y.; Bao, Y.; Li, Y. A simple and fast experimental model of myocardial infarction in the mouse. Tex. Heart Inst. J. 2006, 33, 290–293. [Google Scholar] [PubMed]
- Al-Mashhadi, R.H.; Sørensen, C.B.; Kragh, P.M.; Christoffersen, C.; Mortensen, M.B.; Tolbod, L.P.; Thim, T.; Du, Y.; Li, J.; Liu, Y.; et al. Familial Hypercholesterolemia and Atherosclerosis in Cloned Minipigs Created by DNA Transposition of a Human PCSK9 Gain-of-Function Mutant. Sci. Transl. Med. 2013, 5, 166ra1. [Google Scholar] [CrossRef]
- Fan, J.; Kitajima, S.; Watanabe, T.; Xu, J.; Zhang, J.; Liu, E.; Chen, Y.E. Rabbit models for the study of human atherosclerosis: From pathophysiological mechanisms to translational medicine. Pharmacol. Ther. 2015, 146, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Keyamura, Y.; Nagano, C.; Kohashi, M.; Niimi, M.; Nozako, M.; Koyama, T.; Itabe, H.; Yoshikawa, T. Dietary Cholesterol Atherogenic Changes in Juvenile Rabbits. Biol. Pharm. Bull. 2015, 38, 785–788. [Google Scholar] [CrossRef]
- El-Seweidy, M.M.; Sarhan Amin, R.; Atteia, H.H.; El-Zeiky, R.R.; Al-Gabri, N.A. Dyslipidemia induced inflammatory status, platelet activation and endothelial dysfunction in rabbits: Protective role of 10-Dehydrogingerdione. Biomed. Pharmacother. 2019, 110, 456–464. [Google Scholar] [CrossRef]
- Elseweidy, M.M.; Amin, R.S.; Atteia, H.H.; El-Zeiky, R.R.; Al-Gabri, N.A. New Insight on a Combination of Policosanol and 10-Dehydrogingerdione Phytochemicals as Inhibitors for Platelet Activation Biomarkers and Atherogenicity Risk in Dyslipidemic Rabbits: Role of CETP and PCSK9 Inhibition. Appl. Biochem. Biotechnol. 2018, 186, 805–815. [Google Scholar] [CrossRef]
- Sun, H.; Krauss, R.M.; Chang, J.T.; Teng, B.-B. PCSK9 deficiency reduces atherosclerosis, apolipoprotein B secretion, and endothelial dysfunction. J. Lipid Res. 2018, 59, 207–223. [Google Scholar] [CrossRef]
- Denis, M.; Marcinkiewicz, J.; Zaid, A.; Gauthier, D.; Poirier, S.; Lazure, C.; Seidah, N.G.; Prat, A. Gene Inactivation of Proprotein Convertase Subtilisin/Kexin Type 9 Reduces Atherosclerosis in Mice. Circulation 2012, 125, 894–901. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Li, S.; Xu, R.-X.; Sun, J.; Tang, Y.; Li, J.-J. Proprotein convertase subtilisin/kexin type 9 expression is transiently up-regulated in the acute period of myocardial infarction in rat. BMC Cardiovasc. Disord. 2014, 14, 192. [Google Scholar] [CrossRef]
- Palee, S.; McSweeney, C.M.; Maneechote, C.; Moisescu, D.M.; Jaiwongkam, T.; Kerdphoo, S.; Chattipakorn, S.C.; Chattipakorn, N. PCSK9 inhibitor improves cardiac function and reduces infarct size in rats with ischaemia/reperfusion injury: Benefits beyond lipid-lowering effects. J. Cell. Mol. Med. 2019, 23, 7310–7319. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Zhang, T.; Zha, Y.; Liang, J.; Cheng, Y. Construction of PCSK9 point mutation rabbits using CRISPR/Cas9. J. Zhejiang Univ. Med. Sci. 2021, 50, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Hedayat, A.F.; Park, K.-H.; Kwon, T.-G.; Woollard, J.R.; Jiang, K.; Carlson, D.F.; Lerman, A.; Lerman, L.O. Peripheral vascular atherosclerosis in a novel PCSK9 gain-of-function mutant Ossabaw miniature pig model. Transl. Res. 2018, 192, 30–45. [Google Scholar] [CrossRef]
- Yuan, F.; Guo, L.; Park, K.H.; Woollard, J.R.; Taek-Geun, K.; Jiang, K.; Melkamu, T.; Zang, B.; Smith, S.L.; Fahrenkrug, S.C.; et al. Ossabaw Pigs with a PCSK9 Gain-of-Function Mutation Develop Accelerated Coronary Atherosclerotic Lesions: A Novel Model for Preclinical Studies. J. Am. Heart Assoc. 2018, 7, e006207. [Google Scholar] [CrossRef]
- Frank-Kamenetsky, M.; Grefhorst, A.; Anderson, N.N.; Racie, T.S.; Bramlage, B.; Akinc, A.; Butler, D.; Charisse, K.; Dorkin, R.; Fan, Y.; et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA 2008, 105, 11915–11920. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Smith, J.; Breton, C.; Clark, P.; Zhang, J.; Ying, L.; Che, Y.; Lape, J.; Bell, P.; Calcedo, R.; et al. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat. Biotechnol. 2018, 36, 717–725. [Google Scholar] [CrossRef]
- Wang, L.; Breton, C.; Warzecha, C.C.; Bell, P.; Yan, H.; He, Z.; White, J.; Zhu, Y.; Li, M.; Buza, E.L.; et al. Long-term stable reduction of low-density lipoprotein in nonhuman primates following in vivo genome editing of PCSK9. Mol. Ther. 2021, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef]
- Ibáñez, B.; Heusch, G.; Ovize, M.; Van de Werf, F. Evolving Therapies for Myocardial Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef]

{kind=link}
| Study | Model (Cell/Animal) | Method | Results |
|---|---|---|---|
| In vitro | Purified human platelets; cultivated human liver cells (HepG2) | Platelet isolation, SDS-PAGE Western blot assay, platelet aggregation assay, thrombus formation assay, monocyte migration, and monocyte differentiation assay | PCSK9 was expressed and released by platelets that promote atherothrombosis and inflammation process during atherosclerosis progression [26]. |
| Purified human platelets | Platelet isolation, co-immunoprecipitation and phosphorylation assay by SDS-PAGE Western blot assay, platelet aggregation assay, and thrombus formation assay | High levels of PCSK9 in the circulation are linked to enhanced platelet activation and thrombus formation, which involves the molecular activation of CD36 and NOX2 [23]. | |
| Purified human and mice platelets | Platelet isolation, co-immunoprecipitation or phosphorylation assay by SDS-PAGE Western blot assay, and platelet aggregation assay | Specific binding of PCSK9 and CD36, which stimulates Src, ERK5, and JNK, enhancing ROS production and promoting the activation of the p38/cPLA2/COX-1/TXA2 signaling cascades [21]. | |
| In vivo | Mouse | Establishment of PCSK9-/- mice, FeCl3 injury-induced carotid artery thrombosis; | Platelet activation was impeded when PCSK9 was downregulated [31]. |
| PCSK9-expressing mice, sepsis-induced hypercoagulation; | Upregulation of PCSK9 is positively correlated with blood coagulation [32]. | ||
| PCSK9-/- mice inferior vena cava ligation; | PCSK9-/- mice developed significantly smaller venous thrombus than wild-type mice [33]. | ||
| LDLR-/- mice, ischemia-induced microthrombosis and MI expansion by establishing animal model of MI, CD36-/-mice | The activation of CD36 platelets via PCSK9 exacerbates microvascular blockage and promotes MI [21] | ||
| Rabbit | Hyperlipidemic rabbits induced by high-fat diet of 0.5% w/w cholesterol for 90 days; | A novel natural-derived cholesteryl ester transfer protein (CETP) inhibitor, 10-dehydrogingerdione, suppressed PCSK9 expression and functions in hyperlipidemic rabbits, which was accompanied by a reduction in cellular adhesion inflammatory molecules, platelet activation, and endothelial dysfunction markers when compared to atorvastatin treatment [39]. | |
| Hyperlipidemic rabbits induced by high-fat diet of 0.5% w/w cholesterol for 90 days; | Daily administration of policosanol and/or 10-dehydrogingerdione at a dose of 10 mg/kg bw inhibited CETP, increased HDL-C, decreased PCSK9, and decreased platelet activation and inflammation markers such sCD40L, sP-selectin, and interferon-gamma (IFN-γ) [40]. |
| Cells/Animals | Model of Study | Results |
|---|---|---|
| Cell-based | Mouse aortic endothelial cells (MAEC) and mouse cardiac endothelial cell (MCEC) line; | Cells treated with LDb-LDL (containing PCSK9) induced greater expression of pro-inflammatory and pro-autophagy genes, which are important for atherogenic properties, indicating the key role of PCSK9 in promoting atherosclerosis [41]. |
| Mouse | Transgenic Apolipoprotein E (ApoE) knockout (KO) mouse; | Direct relationship between PCSK9 and the development of atherosclerosis in an ApoE-deficient mouse background [42]. |
| Rat | Acute myocardial infarction (AMI) model; | Plasma PCSK9 concentration was significantly raised 12 h after AMI, which was subsequently verified by increased liver PCSK9 mRNA levels [43] |
| acute cardiac ischemia/reperfusion (I/R) model; | PCSK9 inhibitor treatment before ischemia protects the heart from the injuries incurred by I/R [44]. | |
| Rabbit | Spontaneous hyperlipidemic rabbits (Watanabe heritable hyperlipidemic (WHHL); | WHHL rabbits are genetically defective in LDLR function and can develop hyperlipidemia even on a regular standard diet. Some rabbit WHHL has been linked to coronary artery disease and myocardial infarction [37]. |
| PCSK9 point mutation rabbits utilizing the CRISPR/Cas9 technology; | Established a rabbit model of PCSK9S386A point mutation, which serves as an animal model for investigating the molecular mechanisms of impaired PCSK9 function and developing effective and reliable diagnosis and treatment measures [45]. | |
| Porcine | Ossabaw-PCSK9 GOF pig animal model; | PCSK9-GOF pigs saw an increase in cholesterol, triglycerides, and blood pressure levels at 3 and 6 months. LDL, total cholesterol, and triglyceride levels in Ossabaw-PCSK9 pigs showed a significant increase compared to the control group. Peripheral arteries in the PCSK9-GOF group showed medial thickening and plaque formation in PCSK9-GOF was higher than control [36,46]. |
| D374Y-PCSK9 Yucatan transgenic pigs; | Yucatan minipig D374Y-PCSK9 transgene produces hypercholesterolemia. The liver LDLR level was reduced by 90% compared to the control group; moreover, this transgenic model has lower HDL levels compared to other pig strains. Thickening of the aorta reaches 80% so that it can be used for further studies of atherosclerosis [36]. | |
| Non-human primate | Cynomolgus macaques; | siRNA-mediated PCSK9 inhibition reduces plasma LDL-c but not HDL-c in NHPs. [48]. |
| Rhesus macaques; | Adeno-associated virus carrying the genetic code for a meganuclease targeting genetic alterations, thus resulting in PCSK9 protein reduction. The researchers found dose-dependent reductions in PCSK9 levels of up to 84%, as well as LDL-c reductions of up to 60%. [49,50]. | |
| Cynomolgus macaques; | Knocking down PCSK9′s gene expression and function in the liver of the cynomolgus monkey, with over 60% editing achieved. According to their findings, PCSK9 levels in the blood have been reduced by nearly 90% [51]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puteri, M.U.; Azmi, N.U.; Ridwan, S.; Iqbal, M.; Fatimah, T.; Rini, T.D.P.; Kato, M.; Saputri, F.C. Recent Update on PCSK9 and Platelet Activation Experimental Research Methods: In Vitro and In Vivo Studies. J. Cardiovasc. Dev. Dis. 2022, 9, 258. https://doi.org/10.3390/jcdd9080258
Puteri MU, Azmi NU, Ridwan S, Iqbal M, Fatimah T, Rini TDP, Kato M, Saputri FC. Recent Update on PCSK9 and Platelet Activation Experimental Research Methods: In Vitro and In Vivo Studies. Journal of Cardiovascular Development and Disease. 2022; 9(8):258. https://doi.org/10.3390/jcdd9080258
Chicago/Turabian StylePuteri, Meidi Utami, Nuriza Ulul Azmi, Salbiah Ridwan, Muhammad Iqbal, Tresni Fatimah, Tri Diana Puspita Rini, Mitsuyasu Kato, and Fadlina Chany Saputri. 2022. "Recent Update on PCSK9 and Platelet Activation Experimental Research Methods: In Vitro and In Vivo Studies" Journal of Cardiovascular Development and Disease 9, no. 8: 258. https://doi.org/10.3390/jcdd9080258